")
Back to Journals » The Application of Clinical Genetics » Volume 13
Molybdenum Cofactor Deficiency: Mega Cisterna Magna in Two Consecutive Pregnancies and Review of the Literature
Authors Alonzo Martínez MC , Cazorla E , Cánovas E, Anniuk K, Cores AE, Serrano AM
Received 30 November 2019
Accepted for publication 16 January 2020
Published 30 January 2020 Volume 2020:13 Pages 49—55
DOI https://doi.org/10.2147/TACG.S239917
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
MC Alonzo Martínez,1 E Cazorla,1 E Cánovas,1 K Anniuk,1 AE Cores,2 AM Serrano1
1Department of Obstetrics and Gynecology, Hospital Universitario de Torrevieja, Torrevieja, Alicante, Spain; 2Department of Radiology, Hospital Universitario de Torrevieja, Torrevieja, Alicante, Spain
Correspondence: E Cazorla
Servicio de Obstetricia y Ginecología, Hospital Universitario de Torrevieja, Ctra. Torrevieja a San Miguel de las Salinas CV 95 Partida de la Ceñuela, Torrevieja 03186, Alicante, Spain
Tel +34 629679956
Fax +34 965721368
Email [email protected]
Abstract: The molybdenum cofactor deficiency is an autosomal recessive disease, characterized by rapidly progressive and severe neurological damage that mimics a hypoxic-ischemic encephalopathy due to the accumulation of toxic metabolites that cause rapid neurodegeneration after the delivery. It is eventually lethal, in a similar way to the rare isolated sulfite oxidase deficiency. This serious pathology usually causes death in the immediate neonatal period in the more severe variants. We report a case of two consecutive pregnancies with enlarged cisterna magna as the only prenatal pathological finding since 26 weeks of gestation (WG) and the subsequent death of the newborns in the first week after birth. After the second pregnancy, we reached the diagnosis of molybdenum cofactor deficiency due to MOCS1 gene mutation. According to the cases reported in the literature, this is the case with the earliest neuroimage prenatal findings.
Keywords: inborn error of metabolism, molybdenum cofactor, prenatal diagnosis, sulfite, cPMP, hypoxic-ischemic encephalopathy
Introduction
The molybdenum cofactor is necessary for the catalytic activity of four enzymes in humans: xanthine dehydrogenase, sulfite oxidase, aldehyde oxidase and mitochondrial Amidoxime Reducing Component (mARC).1,2 Mutations in the genes that encode this cofactor lead to a very rare disease with an unknown prevalence and around 150 cases have been described so far.3
It is an autosomal recessive disease, characterized by a rapidly progressive severe neurological damage. It mimics a hypoxic-ischemic encephalopathy, resulting from the accumulation of toxic metabolites, mostly sulfite and S-sulfocysteine (SSC), which causes rapid neurodegeneration after the delivery being eventually lethal, similar to the rare isolated sulfite oxidase deficiency.1,4–7 Therefore, it is a very serious pathology that usually causes death in the immediate neonatal period. Patients who overcome it show a severe mental disability, prostration in bed and very poor quality of life.1,4
Below we present the case of a patient with two consecutive pregnancies with fetal diagnosis of mega cisterna magna (MCM) as the only prenatal pathological finding, with early neonatal death of both siblings after the first week of life and diagnosis of molybdenum cofactor deficiency after the second pregnancy.
Clinical Report
A 25-year-old patient, nulliparous, black race, with no relevant background except consanguinity with her couple, was attended in our Hospital for her first gestation. The follow-up of the pregnancy was uneventful, with serological and ultrasound controls at 20 and 32 WG within normality, without growth restrictions or microcephaly. Aneuploidy screening was not performed because of delay in obstetric control. Eutocic delivery was attended at 38 WG and a female of 3130 g was born, Apgar score 9/10/10 and umbilical arterial pH 7.35. At birth, she had rough facial features, small and low-set auricular pavilions, sunken nasal bridge, broad forehead and important nuchal fold. At 30 hrs after birth, she debuted with repeated episodes of apnea, desaturation and bradycardia that required intubation and mechanical ventilation, hypotension, metabolic acidosis and increased C-reactive protein (CRP). Subsequently, she developed tonic-clonic seizures and myoclonus resistant to anticonvulsant treatment, as well as periods of areflexia-hyperreflexia. An empirical treatment test for early neonatal epileptic encephalopathies with biotin, pyridoxine and folic acid was performed without success. The metabolic study revealed an increase in lactic acid in cerebrospinal fluid suspected of mitochondrial pathology. Magnetic resonance imaging (MRI) of the brain showed, from a morphological point of view, hypoplastic cerebellum with MCM. From a functional approach, there was a signal alteration in diffusion-weighted imaging (DWI) with relatively asymmetric cortico-subcortical involvement and implication of deep brain structures with thalamic, striatal and corticospinal tract involvement. There was hypersignal in DWI and hyposignal in the apparent diffusion coefficient (ADC) maps. The blood tests did not show parameters of infection and the cultures were negative. She presented cardiorespiratory arrest and secondary death to convulsive status refractory to treatment on the tenth day of life. The diagnosis of severe neonatal epileptogenic encephalopathy of probable inborn error of metabolism (IEM) origin was established. The parents rejected neonatal necropsy and karyotype.
One year later, the patient came to the consultation with a new spontaneous pregnancy and it was controlled in the High Obstetric Risk consultation due to her history of early neonatal death. At 20 WG, the morphological ultrasound was normal. However, at 26 WG showed MCM and cavum vergae. At 29 WG MRI showed the same findings with enlargement of MCM (Figure 1). At 33 WG, persistence of MCM (17mm) and dilated III ventricle (9mm) was observed in ultrasound. No growth restrictions or microcephaly were found. After eutocic delivery at 38 WG (male newborn, Apgar score 9/10/10, 3042 g, umbilical arterial pH 7.40), he began a progressive neonatal deterioration similar to the previous one. From a morphological point of view, the most remarkable MRI findings were MCM and cavum vergae, and from a functional approach, signal impairment in structures such as thalamus, brainstem and basal ganglia. Also, there was diffuse subcortical involvement with hypersignal in T2 weighted images, all initially labeled as hypoxic-ischemic encephalopathy.
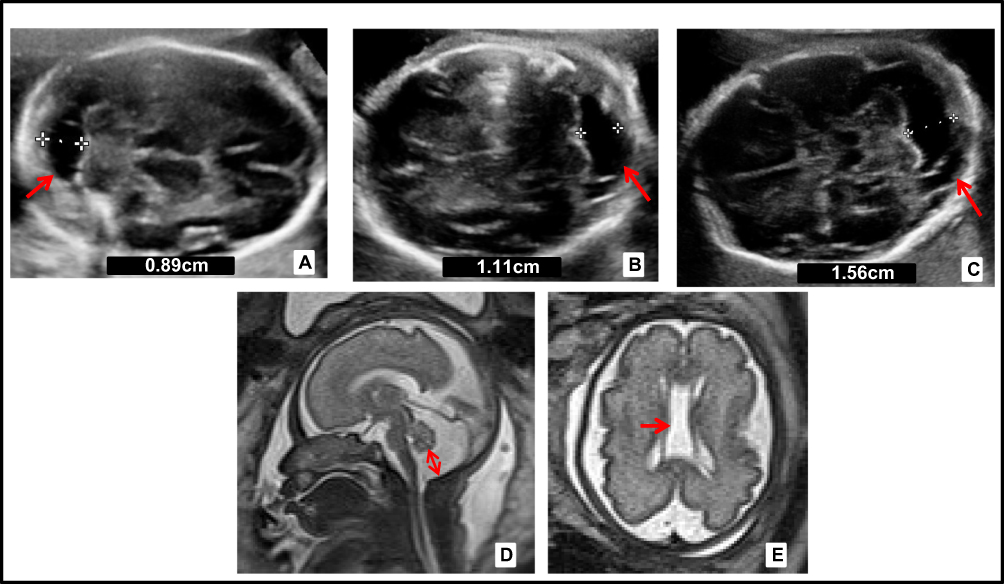 |
Figure 1 Prenatal neurosonography of the second pregnancy. (A) Ultrasound at 21 WG, cisterna magna of 8.9 mm (arrow). (B) Ultrasound at 26+4 WG, mega cisterna magna (MCM) 11mm (arrow). (C) Ultrasound at 28 WG, MCM of 15.6mm (arrow). (D, E) Magnetic resonance imaging (MRI) in T2_HASTE at 29 WG, MCM of 17mm and presence of cavum vergae (arrows). White crosses indicate the measurements of the cisterna magna. The numeric value is included in each figure. |
The blood test highlighted hypouricemia (0,2mg/dl) and the urine test showed very high excretion of sulfocysteine (125 mmol/mol creatinine) and xanthines (711 mcrmol/mmol creatinine). Finally, he presented desaturation, sudden bradycardia resistant to treatment and the neonate died 11 days after birth. The subsequent metabolic and genetic study showed a combined deficiency of xanthine dehydrogenase and sulfite oxidase (molybdenum cofactor deficiency) caused by homozygous mutation in the MOCS1 gene (c.721delC/p.L241fs_246X). Therefore, presence of mutation of the MOCS1 gene in heterozygosis in both parents was confirmed.
Months later, the patient was pregnant again. The ultrasound parameters of the control at 20 WG were within normal. Finally, the patient accepted the diagnostic amniocentesis being negative for the molybdenum cofactor deficiency. After eutocic delivery in 38 WG, a completely normal girl was born.
Discussion
The enzymes that depend on the presence of the molybdenum cofactor are xanthine dehydrogenase, sulfite oxidase, aldehyde oxidase and mitochondrial amidoxime reducing component (mARC).1,2 The first one is involved in the purines degradation and the uric acid formation whose deficit would only cause the formation of xanthine stones.1,5 The second one intervenes in an essential step in the degradation of sulfurized amino acids such as cysteine and methionine (sulfite-sulfate),2 whose alteration is probably the cause of a serious encephalopathy (Figure 2). The enzyme aldehyde oxidase and mARC do not intervene in metabolic pathways relevant to the neonate survival, and pathologies due to isolated alterations of these enzymes have not been described in the literature.1
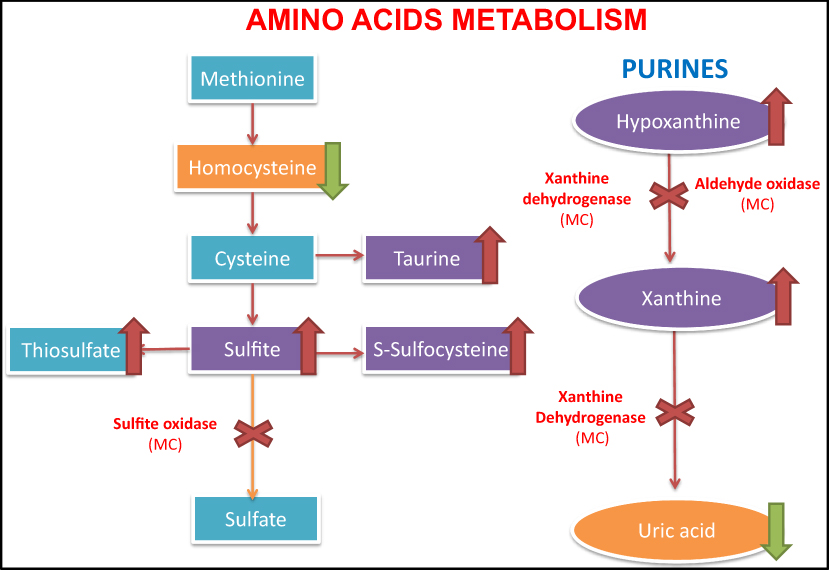 |
Figure 2 Metabolic pathway with enzymes dependent on the molybdenum cofactor. Left, the sulfur-containing amino acid metabolism, with the accumulation of sulfite and s-sulfocysteine due to sulfite oxidase deficiency (dependent molybdenum cofactor enzyme). On the right, the purine metabolism with accumulation of hypoxanthine and xanthine in blood, as well as the decrease of uric acid levels in the blood. |
Genetically, three types of molybdenum cofactor deficiency can be distinguished. Type A, MOCS1 gene mutation (more than 60% of the cases),4,8 which under normal conditions gives rise to an intermediate metabolite of the synthesis chain of the molybdenum cofactor, cPMP (cyclic Piranopterin Monophosphate). Type B, MOCS2 or MOCS3 gene mutation which participates in the formation of the molybdopterin (MPT).2,3 Finally, Type C is caused by GPHN gene mutation that intervenes in the last step prior to the formation of the cofactor and only one case described in the literature (Figure 3).2,9–11
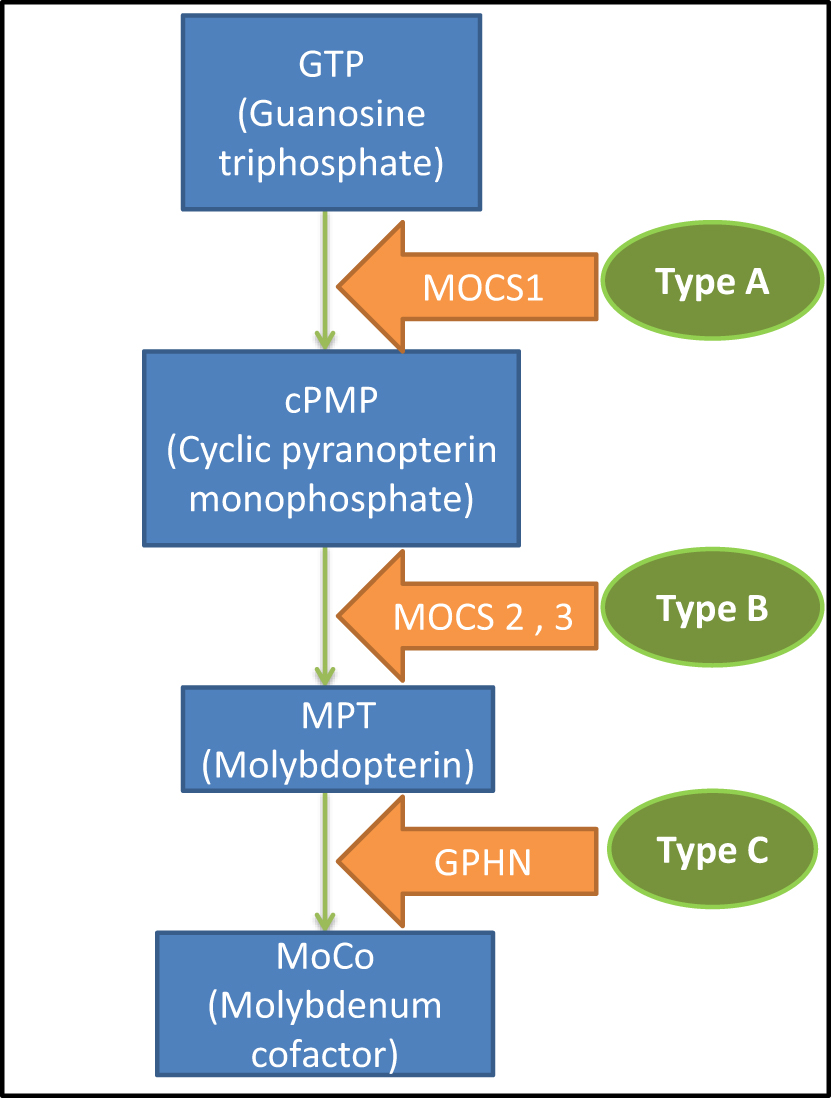 |
Figure 3 Biosynthesis of the molybdenum cofactor and the disease classification. |
There is a classic form of the disease observed in the first weeks of life and an atypical one characterized by a late onset.4,6,12,13 In the classical variant, patients manifest a crude facial phenotype with depressed hyperteloric eyes, elongated palpebral fissures and late-onset lens dislocation.1,2,5 In addition, they have thickened lips and broad philtrum, thick cheeks, small nose and microcephaly.2,5,6 They usually debut with seizures that do not yield with medical treatment, spasticity and feeding difficulties. The estimated survival is between a week and a few months. In the few cases that exceed the month of life, patients present very severe mental disability, severe psychomotor delay, tetraparesis, bed rest and very poor quality of life.1,2,5,6
There have been 13 cases described in the literature of the atypical form of the disease.14 Normally debuts later in life after other illness or infection process that triggers the symptoms of the disease with variable manifestations like psychomotor delay and extrapyramidal and pyramidal symptoms.14 A residual molybdenum cofactor enzymatic function can usually be found and all three MOCS genes had been involved in this variant.12–14 Probably, there are other similar cases that have not been diagnosed due to their mild clinical symptoms that can be confused with other pathologies.
At the beginning of pregnancy, diagnosis can be made by a chorionic biopsy, detecting the sulfite oxidase deficiency in the chorionic villi.2,5 Amniocentesis is useful to detect high levels of SSC in amniotic fluid and to search the specific fetal DNA mutations.2
The findings in the neuroimaging are heterogeneous, they do not have a characteristic chronology nor are they pathognomonic. The most frequent alterations are bilateral ventriculomegaly, MCM and microgyria due to the significant cerebral atrophy observed in these patients.1,4,6 In addition, multiple subcortical cysts, basal ganglia lesions with involvement of the caudate and the thalamus,2,3,5,15 cerebellar hypoplasia,2,6,15 dysgenesis of the corpus callosum,4,5 cerebral and diffuse cytotoxic edema and the neuroradiological walnut aspect of the brain have been observed.4–6 After the delivery, cortico-subcortical lesions can be observed with non-uniform distribution on DWI, showing different dates of brain injuries due to the accumulation of the toxic metabolites.4 This MRI finding can help us in the differential diagnosis with hypoxic-ischemic encephalopathy, in which the increased signal on DWI is seen throughout all cortical and subcortical areas.4
The finding of MCM is observed in 1/100 newborns and may be a variant of normality. However, we should rule out vermian hypoplasia, Dandy–Walker malformation, presence of Blake’s cyst, posterior fossa arachnoid cyst, ischemic lesions, cytomegalovirus infection and 18 trisomy, mainly. After discarding these possibilities, we must think about molybdenum cofactor deficiency, especially if there is a history of consanguinity or siblings who died in the immediate neonatal period with seizures refractory to treatment.
In the literature, there are few cases diagnosed prenatally and, mostly, by antecedents of previously affected siblings, so it is difficult to determine when the brain insult begins. In 2011, Cami-Nawi et al presented a case with a mutation in the MOCS1 gene where alterations in brain ultrasound are observed from 35 WG with numerous subcortical cavities, ventriculomegaly, dysgenesis of the corpus callosum and a hypoplastic cerebellum with MCM.15 In 2018, Lubout et al also reported data from the prenatal MRI of two cases with MOCS1 gene mutation that suggested a possible early brain injury at 32 WG consisting in MCM and smaller cerebellum in one of them and at 36 WG, a mild enlargement of lateral ventricles in both cases.16 On the other hand, in a series of 8 cases published by Vijayakumar K in 2011, microcephaly, MCM, ventriculomegaly, acute symmetrical subthalamic lesions and in globus pallidi, chronic hemispheric infarctions related to prenatal lesions and other findings were observed in postnatal imaging tests.17 In the first gestation of our case, ultrasound controls until 32 WG were normal. However, in the second pregnancy, changes were observed from 26 WG with MCM and presence of cavum vergae (Figure 1). Unlike what was initially believed, both our findings and those presented in the literature suggest a placental filtration deficit of toxic metabolites, reflected in neuroimaging by ultrasound and MRI from the beginning of the third trimester although less evidently than postnatally. In addition, cerebral damage by sulfite seems to be greater with the increase of consumption of adenosine triphosphate (ATP) after delivery.4
In the immediate postnatal period, we should suspect the presence of this pathology mainly if there are seizures refractory to treatment. For the preliminary diagnosis, it is possible to look for the presence of different elevated metabolites in urine such as sulfite and its secondary products, thiosulfite and SSC,1,6,9 xanthine and taurine. These products accumulate due to a deficiency of the enzymes dependent on the molybdenum cofactor (Figure 2).1,6 We can perform a rapid laboratory analysis of sulfites using a test strip but with a high percentage of false negatives, caused mainly by taking the sample very early after birth or by delays in the study of the sample after its emission.1,2,5,6,9 Another alternative is the use of high-performance liquid chromatography (HPLC) with a high sensitivity for the detection of SSC in urine, useful for the diagnosis and patients monitoring.9 In peripheral blood, low levels of uric acid and increase of xanthines are indicative of xanthine dehydrogenase deficiency.1,5,9 On the other hand, hypohomocysteinemia, the increase in urine of sulfite and its derivatives along with a normal or low xanthine and normal uric acid in blood and urine guide us to the isolated deficiency of sulfite oxidase with a similar phenotype.7,9 Therefore, the molybdenum cofactor deficiency will cause a combined enzymatic alteration, where all the mentioned analytical parameters could be seen. The definitive postnatal diagnosis is reached by the detection of a low cofactor activity or a sulfite oxidase deficiency in a skin fibroblast culture.2,6 Differential diagnosis should be made with isolated deficiency of sulfite oxidase, neonatal hypoxic-ischemic encephalopathy, hyperekplexia and amino acid intolerance.1,2,6 For the first one, the clinical presentation and neuroimaging are indistinguishable from molybdenum cofactor deficiency and it is necessary both genetic testing and measurement of the metabolites described above.7,9 The differential diagnosis with neonatal hypoxic-ischemic encephalopathy can be made through genetic testing for MOCS genes and the neuroimaging findings mentioned above. The hyperekplexia debuts with an exaggerated startle reaction to stimuli and spasticity that subsides with sleep, no seizures, no dimorphic features and genetic analysis and electroencephalogram is advisable.18 Finally, amino acid intolerance should be ruled out with tandem mass spectrometry and other laboratory tests that indicate abnormal quantities of amino acids in blood and urine.19
In the past, numerous treatments have shown no benefit such as tetrahydrobiopterin, D-penicillamine, food intake restriction containing sulfur amino acids, sulfate supplementation and the direct administration of molybdenum cofactor.2,6 In cases where the MOCS1 gene is altered (Type A, more than 60% of the cases), there is a deficit of cPMP, the immediate more stable precursor of molybdenum cofactor, that has provided promising results in the treatment of the disease. Recently, it has been seen that administering cPMP as substitution therapy does not improve the brain damage already produced but prevents its progressive worsening.1,2,8 In 2012, Hitzert et al published a case diagnosed prenatally, induction of labor 36 WG and treatment of the newborn a few hours after with cPMP through a central line with good results.8 After a daily treatment with cPMP, the patient manifested an almost normal neurodevelopment.8 Lubout et al published a case where the treatment is administered shortly after termination of pregnancy in 39 WG, presenting at 41 months an inferior Bayley scale development (18–26 months).16 However, cPMP is available in very few centers worldwide so few patients have access to it currently. At the end of 2017, Kumar et al published an article demonstrating that the use of antagonists of N-methyl-D-aspartate receptor (NMDA-R) such as memantine may help to delay neuronal death until the initiation of cPMP treatment in cases of the MOCS1 gene mutation.20
The molybdenum cofactor deficiency type C by mutation of the GPHN gene is one of the rarest and most severe of all (only one case described in the literature) and it has been demonstrated in cell cultures that the repeated administration of high doses of inorganic molybdenum can be effective only in the treatment of the disease caused by this gene.1,11 On the other hand, the response to a low-methionine diet has been poor in the case presented by Mayr et al (MOCS1 mutation, atypical variant),12 but successful in the case reported by Huijimans et al (MOCS3 mutation, atypical variant) with an improvement of symptoms.13
In some articles, labor induction is recommended before 37 WG or when lesions are observed in neurosonography for early initiation of treatment with cPMP.16 However, in some cases like ours, these alterations can be as early as in 26 WG, when ending a pregnancy with extreme prematurity usually has serious consequences.
Conclusion
IEM are rare and potentially serious diseases with little information available because of the low number of cases diagnosed in the world. For this reason, the management of this type of pathologies must be centralized in tertiary hospitals with specific units.
This entity must be considered before the finding of seizures resistant to medical treatment with clinical and radiology reminiscent of perinatal hypoxic-ischemic encephalopathy, especially in the context of consanguinity. In addition, we must reject the intolerance to amino acids and hyperekplexia.1,2,6
The genetic counselling of the parents is essential to avoid new cases in the future. In our case, the diagnosis is delayed until the death of the second sibling due to rejection by the parents of necropsy and genetic diagnosis of the first newborn.
Obtaining an intrauterine genetic diagnosis is even more important after the discovery of the molybdenum cofactor precursor cPMP therapy and its good results in the neurodevelopment of the newborn if it is administered immediately after delivery. The timing of the induction of labor should be carefully evaluated due to the growing evidence of the brain alteration onset before the pregnancy comes to term. According to the literature published up to the present time, this is the case where lesions in neurosonography have been evidenced earlier.
Compliance with Ethical Standards
This study was approved by the Ethics Committee of Torrevieja-Vinalopó (Comité Ético de Investigación (CEIm) Torrevieja-Vinalopó, registry number CC1-201219). Written informed consent was obtained for publication of the case details and accompanying images.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Reiss J, Hahnewald R. Molybdenum cofactor deficiency: mutations in GPHN, MOCS1 and MOCS2. Hum Mutat. 2011;32(1):10–18. doi:10.1002/humu.21390
2. Orphanet [homepage on the Internet]. Encephalopathy due to sulfite oxidase deficiency; 2012. Available from: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Ing=ES&Expert=833.
3. Durmaz MS, Özbakir B. Molybdenum cofactor deficiency: neuroimaging findings. Radiol Case Rep. 2018;13(3):592–595. doi:10.1016/j.radcr.2018.02.025
4. Higuchi R, Sugimoto T, Tamura A, et al. Early features in neuroimaging of two siblings with molybdenum cofactor deficiency. Pediatrics. 2014;133(1):e267–e271. doi:10.1542/peds.2013-0935
5. Omim [homepage on the Internet]. Molybdenum cofactor deficiency, complementation group A, MOCODA; 2013. Available from: http://www.omim.org/entry/252150.
6. Palacios A, García MT, Sánchez J, Nogales A, Puche A, Ugarte M. Déficit del cofactor Molibdeno como causa de encefalopatía epiléptica precoz [Molybdenum cofactor deficiency as a cause of early epileptic encephalopathy]. An Pediatr. 2008;69(2):187–189. doi:10.1157/13124906
7. Claerhout H, Witters P, Régal L, et al. Isolated sulfite oxidase deficiency. J Inherit Metab Dis. 2018;41(1):101–108. doi:10.1007/s10545-017-0089-4
8. Hitzert MM, Bos AF, Bergman KA, et al. Favorable outcome in a newborn with molybdenum cofactor type A deficiency treated with cPMP. Pediatrics. 2012;130(4):e1005–e1010. doi:10.1542/peds.2011-3330
9. Belaidi AA, Arjune S, Santamaria-Araujo JA, Sass JO, Schwarz G. Molybdenum cofactor deficiency: a new HPLC method for fast quantification of S-Sulfocysteine in urine and serum. JIMD Rep. 2012;5:35–43.
10. Mendel RR. The molybdenum cofactor. J Biol Chem. 2013;288:13165–13172. doi:10.1074/jbc.R113.455311
11. Reiss J, Gross-Hardt S, Christensen E, Schmidt P, Mendel RR, Schwarz G. A mutation in the gene for the neurotransmitter receptor-clustering protein gephyrin causes a novel form of molybdenum cofactor deficiency. Am J Hum Genet. 2001;68(1):208–213. doi:10.1086/316941
12. Mayr SJ, Sass JO, Vry J, et al. A mild case of molybdenum cofactor deficiency defines an alternative route of MOCS1 protein maturation. J Inherit Metab Dis. 2018;41(2):187–196. doi:10.1007/s10545-018-0138-7
13. Huijmans JGM, Schot R, Klerk JBC, et al. Molybdenum cofactor deficiency: identification of a patient with homozygote mutation in the MOCS3 gene. Am J Med Genet. 2017;173(6):1601–1606. doi:10.1002/ajmg.a.38240
14. Scelsa B, Gasperini S, Righini A, et al. Mild phenotype in Molybdenum cofactor deficiency: a new patient and review of the literature. Mol Genet Genomic Med. 2019;7(6):e657. doi:10.1002/mgg3.657
15. Carmi-Nawi N, Malinger G, Mandel H, Ichida K, Lerman-Sagie T, Lev D. Prenatal brain disruption in molybdenum cofactor deficiency. J Child Neurol. 2011;26(4):460–464. doi:10.1177/0883073810383017
16. Lubout CMA, Derks TGJ, Meiners L, et al. Molybdenum cofactor deficiency type A: prenatal monitoring using MRI. Eur J Paediatr Neurol. 2018;22(3):536–540. doi:10.1016/j.ejpn.2017.11.006
17. Vijayakumar K, Gunny R, Grunewald S, et al. Clinical neuroimaging features and outcome in molybdenum cofactor deficiency. Pediatr Neurol. 2011;45(4):246–252. doi:10.1016/j.pediatrneurol.2011.06.006
18. GeneReviews® [homepage on the Internet]. Hereditary Hyperekplexia. Seattle (WA): University of Washington; 2017. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1260/.
19. Reid Sutton V. Inborn errors of metabolism: classification. UpToDate. 2019. Available from: https://www.uptodate.com/contents/inborn-errors-of-metabolism-classification#H15. Accessed 22 January 2020.
20. Kumar A, Dejanovic B, Hetsch F, et al. S-sulfocysteine/NMDA receptor-dependent signaling underlies neurodegeneration in molybdenum cofactor deficiency. J Clin Invest. 2017;127(12):4365–4378. doi:10.1172/JCI89885
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.