")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 8
Molecular Targets of Ferroptosis in Hepatocellular Carcinoma
Authors Liao H, Shi J, Wen K , Lin J, Liu Q, Shi B, Yan Y, Xiao Z
Received 23 June 2021
Accepted for publication 11 August 2021
Published 24 August 2021 Volume 2021:8 Pages 985—996
DOI https://doi.org/10.2147/JHC.S325593
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Manal Hassan
Hao Liao,1,2,* Juanyi Shi,1,2,* Kai Wen,1,2 Jianhong Lin,1,2 Qinghua Liu,1,2 Bingchao Shi,1,2 Yongcong Yan,1,2 Zhiyu Xiao1,2
1Department of Hepatobiliary Surgery, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, 510120, People’s Republic of China; 2Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, 510120, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yongcong Yan; Zhiyu Xiao
Department of Hepatobiliary Surgery, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Yanjiang West Road #107, Guangzhou, 510120, People’s Republic of China
Tel +86-15800039176; +86-13682283695
Fax +86 20 34071073
Email [email protected]; [email protected]
Abstract: Ferroptosis is a special form of regulatory cell death caused by the accumulation of intracellular iron and lipid peroxidation. Here, we summarize the research progress on ferroptosis in hepatocellular carcinoma (HCC), trace the development of the concept of ferroptosis and its key regulatory factors, and discuss the application value of ferroptosis in the treatment of HCC from different perspectives. We believe that exploring the relationship between ferroptosis and HCC and clarifying the metabolism and expression of ferroptosis-specific genes and molecules will accelerate the development of novel ferroptosis-related molecules as HCC markers and therapeutic targets. We hope to provide a theoretical basis for better diagnosis and treatment to effectively improve the prognosis of patients with HCC.
Keywords: hepatocellular carcinoma, ferroptosis, iron, regulatory cell death, lipid peroxidation, sorafenib
Introduction
According to statistics, there were approximately 905,677 new cases of hepatocellular carcinoma (HCC) and 830,180 HCC deaths in 2020.1 The World Health Organization predicts that more than 1 million patients will die of HCC in 2030.2 Although surgical treatment based on radical surgery has greatly improved patient prognosis, the five-year survival rate of HCC is only 12%.3 In addition, although clinical first-line drugs such as sorafenib have a decisive therapeutic effect on advanced HCC, the median survival time is only 8 to 11 months.4 Therefore, therapeutic options for HCC are urgently needed to provide patients with earlier intervention, thereby reducing the morbidity and mortality of patients with HCC.
Ferroptosis is a special form of nonapoptotic cell death caused by lipid peroxidation and iron accumulation.5 In 2012, Dixon et al5 found that RSL and erastin mediate regulatory cell death that is different from traditional necrosis and apoptosis. Based on this finding, the concept of ferroptosis was proposed as an iron-dependent nonapoptotic form of cell death. Morphologically, ferroptotic cells have no nuclear pyknosis, nuclear fragmentation or nuclear dissolution. These cells do not display necrosis,6 shrinkage, chromatin agglutination, apoptotic body formation or cytoskeleton disintegration during cell death.7 Rather, these cells display cell membrane rupture and blistering, mitochondrial atrophy, increased membrane density, reduced cristae, and lack of chromatin agglutination in the nucleus.8
Although the function of ferroptosis is not yet fully understood, based on its connection with many tumors, the induction of ferroptosis may be the key to the treatment of HCC. At present, it has been found to play a pivotal role in HCC progression and it appears that many drugs used to treat HCC may exert their pharmacologic actions via ferroptosis. Many scholars also believe that ferroptosis can be a key factor in the diagnosis, treatment and prognosis of HCC. To this end, we reviewed the regulatory mechanisms, pathways and applications of ferroptosis in HCC to improve patient outcomes in HCC.
Regulation Mechanism of Ferroptosis in HCC
Recently, many studies have clearly introduced the basic principles of ferroptosis in the treatment of HCC. We will briefly outline the regulatory mechanisms related to ferroptosis from the perspective of the two major characteristics of ferroptosis: iron and lipid peroxidation (Figure 1).
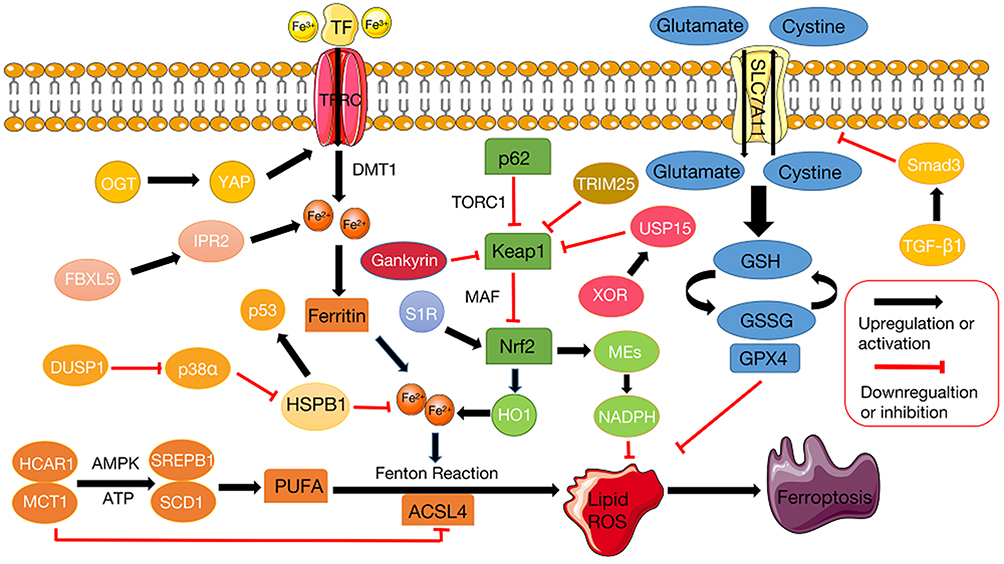 |
Figure 1 Regulation pathways and key molecular mechanisms of ferroptosis. The initiation and response of ferroptosis occurs because Fe2+ enters the cell with transferrin and promotes the high expression of PUFAs in the form of Fe3+ to mediate lipid peroxidation, resulting in disorder of the GSH system and suppression of its core regulatory enzyme GPX4. The key regulator of the Xc- system SLC7A11 mediates the exchange of glutamate and cysteine at a 1:1 molar ratio. Within the cell, glutamate and cysteine ultimately synthesize GSH to assist GPX4 in maintaining intracellular redox homeostasis. HSPB1 and Nrf2 pathways regulate labile iron pools and increase ROS resulting from the Fenton reaction. ACSL4 catalyzes the biosynthesis of PUFAs, thereby mediating lipid peroxidation. PUFA polyunsaturated fatty acids, GSH glutathione, GPX4 glutathione peroxidase 4, SLC7A11 solute carrier family 7 member 11, HSPB1 Heat shock protein beta-1, ROS reactive oxygen species, NRF2 Nuclear factor erythroid 2-related factor 2, ACSL4 acyl-CoA synthetase long chain family member 4 (ACSL4). Key regulator of ferroptos is (black), potential regulator of ferroptosis is (white). |
Iron in the Liver
Iron is a metal with redox activity that can participate in the formation of radicals through the Fenton reaction and cause the accumulation of reactive oxygen species (ROS). It also plays a vital role in sensitizing cells to ferroptosis.9,10 Under normal circumstances, iron inside and outside of the cell maintains a subtle balance through the iron transport system.11 Iron storage is an important function of the liver and iron overload in the liver is particularly closely related to the occurrence of liver-related disorders, such as HCC,7,12–17 hereditary hemoglobinosis,18,19 and liver fibrosis.14,19
Ferroportin (FPN) is a transmembrane ferritin that mediates the transport of iron from inside to outside the cell. In fact, it is the only known iron export protein that regulates iron efflux in mammals.20 Research has confirmed that the liver-derived hormone hepcidin can maintain iron homeostasis in the liver by negatively regulating FPN.21 Moreover, hepcidin can significantly delay the progression of liver failure and serve as a prognostic indicator.22
At present, many studies have investigated the maintenance of iron homeostasis in the liver as an effective measure to prevent HCC and have unearthed many potential key targets related to the disruption of iron homeostasis. Poly rC binding protein 1 (PCBP1) is a multifunctional protein that affects the binding of iron to receptors in mammalian cells. PCBP1 deficiency results in iron and lipid metabolism disorders and eventually leads to lipid peroxidation and steatosis in the liver.23 Transferrin is a specific and high-affinity iron-binding protein synthesized by the liver that plays a vital role in iron homeostasis.24 Some scholars have found that it is closely related to the occurrence and development of liver cirrhosis by mediating iron homeostasis.25 In addition, further studies have confirmed that transferrin receptor 1 (TRF1) affects liver iron uptake by minutely adjusting the expression of hepcidin.26 Iron homeostasis is also affected by F-box and leucine-rich repeat protein 5 (FBXL5) and iron regulatory protein 2 (IRP2). IRP2 is an RNA-binding protein that controls the production of iron accumulation-related proteins, while FBXL5 negatively regulates IRP2. The deficiency of FBXL5 leads to disorders of iron homeostasis in the liver and the whole body, mediating the occurrence of steatohepatitis.27 Muto et al28 discovered a new tumor suppressor molecule, FBXL5, through a mouse model. Insufficient FBXL5 will cause iron metabolism disorders, causing a series of stress and compensatory responses in the liver and tissues. In addition, the depletion of the FBXL5 gene is not only related to the chemical carcinogen DEN but also related to the overexpression of the hepatitis C virus (HCV) core antigen, which participates in the progression of HCC.
Iron in HCC
Since iron can transfer energy through the conversion between the two oxidation states of iron (Fe3+) and ferrous iron (Fe2+), it exerts considerable influence on cell energy production and metabolism.29 Previous studies have shown that serum iron levels are associated with the progression of hepatitis B virus (HBV)-related HCC. Serum iron levels <15.1 μmol/l in conjunction with higher AFP levels are high-risk HBV-related HCC patients.30
Abnormal iron homeostasis is one of the salient features of HCC, suggesting that we can treat HCC by maintaining iron homeostasis. Artesunate (ART) is clinically used as an antimalarial drug, but it can induce ROS-dependent ferroptosis by maintaining labile iron pools (LIPs).31 Specifically, ART promotes the accumulation of LIPs in the endoplasmic reticulum (ER) by acidifying lysosomes to promote the lysosomal degradation of the iron-storage protein ferritin. This can lead to the overproduction of ROS and severe compromise of the ER, which may result in cell death.31 In addition, Shang et al32 found that ceruloplasmin is negatively related to ferroptosis and participates in the regulation of ferroptosis by compromising iron homeostasis inside the cell. Divalent metal-ion transporter-1 (DMT1) is a transmembrane iron transporter that has a significant role in intestinal iron absorption.33 Previous work has shown that both transferrin-bound iron and free iron are absorbed by hepatocytes by means of DMT1. Hoki et al34 revealed that DMT1 is associated with increased mitochondrial oxidative phosphorylation and glycolysis, and it may affect the mitochondrial respiratory chain by interfering with iron homeostasis. O-GlcNAcylation is a reversible posttranslational modification catalyzed by O-GlcNAc transferase.35 Some researchers have found that oxyglycosylation can increase the transcription level of transferrin receptor (TFRC) by promoting the expression of yes-associated protein, thus elevating liver iron concentration in HCC and increasing the vulnerability of cells to ferroptosis.36
In addition to directly inhibiting tumor proliferation through the RAF/MEK/ERK signaling pathway, sorafenib can block vascular endothelial growth factor receptor and platelet-derived growth factor receptor, thus inhibiting angiogenesis.37 Previous works have found that iron deficiency restricts cancer cell proliferation and induces angiogenesis through the hypoxia inducible factor-1 and vascular endothelial growth factor (VEGF) pathways.38 Urano et al13 proved that the combined use of iron inhibitors and antiangiogenic drugs can enhance the tumor killing effect of sorafenib by inducing cell cycle arrest and apoptosis. Therefore, the combined use of sorafenib and deferasirox may be efficacious in the treatment of HCC.
PUFAs in HCC
Previous studies have shown that polyunsaturated fatty acids (PUFAs) can inhibit liver inflammation and reduce the formation of tumor necrosis factor-α (TNF-α).39 In 2009, Lim et al40 found that PUFAs inhibited the growth of HCC by simultaneously inhibiting cyclooxygenase-2 (COX-2) and β-catenin. Specifically, PUFAs have been shown to induce the key rate-limiting enzyme of prostaglandin E2 catabolism, 15-hydroxyprostaglandin dehydrogenase (15-PGDH). 15-PGDH acts as a prostaglandin-degrading enzyme to antagonize COX-2, and it also inhibits tumor growth.40 In 2011, Weylandt et al41 found that PUFAs may exert significant anti-TNF-α effects and suppress the expression of COX-2 through its hydroxylated metabolites 18-hydroxyeicosapentaenoic acid and 17-hydroxydocosahexaenoic acid. Downstream effects include the inhibition of inflammation and tumor formation.
It has been shown that PUFAs can prevent the occurrence of HCC. For example, eating fish rich in saturated fatty acids reduces the likelihood of developing HCC. It is also effective for patients infected with HBV/HCV.42 Understanding how immunonutrient agonists are linked to a high-fat diet, the immune system, and the microbial balance of the gut microbiota is essential for understanding the etiology of HCC.43 In addition to affecting the changes between PUFAs, monounsaturated fatty acids and iron homeostasis, nutritional agonists such as quinoa and salvia can also mediate immune regulation against HCC through innate immunity that affects the enterohepatic circulation.43 This also inspired us to improve the antitumor effect through diet.
In 2020, Leineweber et al44 found that sorafenib may mediate antitumor effects by its effect on PUFAs. In particular, sorafenib is an effective inhibitor of soluble epoxide hydrolase (sEH). sEH is able to catalyze the conversion of arachidonic acid (AA) and omega-3 docosahexaenoic acid (DHA) into the corresponding glycols. AA epoxy eicosapentaenoic acid (EETs) may promote tumor growth and metastasis, while DHA 19,20-epoxydocosapentaenoic acid (19,20-EDP) has the opposite effect. It has been speculated that although the effect on EETs may affect the therapeutic effect of sorafenib, supplementation with DHA in HCC treatment with sorafenib can increase the level of 19,20-EDP and enhance its antitumor effect. Therefore, it is reasonable to hypothesize that although its effect on EETs may affect the overall therapeutic response to sorafenib, supplementation with DHA in the treatment of HCC with sorafenib can reverse the effect of EETs and potentiate sorafenib’s antitumor capability.44
Ferroptosis Regulators in HCC
In recent years, the role of ferroptosis in HCC has been confirmed, and research on ferroptosis has been conducted. Here, we will review the literature on ferroptosis including the main regulators that are involved (Figure 1).
ACSL4 in HCC
In recent years, researchers have confirmed that ferroptosis is a peroxidation reaction activated by PUFAs catalyzed and synthesized by long chain fatty acyl-CoA ligase 4 (ACSL4).45,46 In HCC, ACSL4 can stimulate the expression of lipase through the c-Myc/sterol regulatory element binding protein 1 (SREBP1) pathway and regulate lipid production to participate in proliferation and metastasis.47 In addition to fat, lactic acid (another important metabolic mediator) is also indispensable in the process of ferroptosis. In recent years, several studies have confirmed that lactic acid is a negative regulator of ferroptosis, and these studies have investigated the effects of fat and lactic acid in ferroptosis.48 Hydroxycarboxylic acid receptor 1 /monocarboxylate transporter 1 in HCC can mediate the uptake of lactic acid. The production of ATP, inactivation of AMP-activated protein kinase, increased expression of SREBP1 and stream stearoyl-coenzyme A desaturase-1 follows, generating PUFAs that induce resistance to ferroptosis.48
Moreover, at the metabolic level, the role of ferroptosis at the transcriptional level of HCC is also of great importance. In 2014 and 2020, Cui et al49 and Qin et al50 found that microRNA-205 and microRNA-211-5p target the 3ʹUTR of ACSL4, leading to cell fat and cholesterol metabolism disorders and to the growth, invasion and metastasis of HCC.
Many research teams have elaborated on the relationship between sorafenib and ACSL4 and proposed that the expression of ACSL4 can be used to predict the therapeutic effect of sorafenib in HCC. Feng et al51 suggested that the presence of ACSL4 is negatively associated with sorafenib and that it can be used as a predictor of the therapeutic effect of sorafenib. Xia et al52 further confirmed that silencing ACSL4 and inducing GADD45B at the same time can increase the synergistic tumor-killing function of aspirin and sorafenib in HCC. This evidence indicates that ACSL4 is a promising biomarker for HCC, especially in sorafenib-induced ferroptosis.
GPX4 in HCC
Glutathione peroxidase 4 (GPX4) cycles glutathione (GSH) between the reduced and oxidized states and converts lipid hydroperoxides to lipid alcohols. This process prevents the formation of Fe2+-dependent toxic lipid ROS.53,54
GPX4 can be used as a prognostic and typing marker for HCC55 and can also eliminate peroxides in membranes and lipoproteins. Inducing GPX4 in the treatment of chronic hepatitis C may reduce viral infectivity and the development of HCC.56 GPX4 has been proven to cause lipid peroxidation-mediated ferroptosis, while vitamin E is a fat-soluble antioxidant responsible for protecting membranes against oxidative damage, which suggests that we can prevent GPX4-induced ferroptosis by supplementation with vitamin E.57
At the transcriptional level, GPX4 is also an integral part of ferroptosis. It has been reported that circ‐interleukin‐4 receptor can act as a molecular sponge of microRNA-541-3p to interact with GPX4. This interaction is accompanied by an increase in iron levels and oxidative stress and targeted inhibition of ferroptosis.58
GPX4 has also been studied at the metabolic level. Cells with cysteine mutations are highly sensitive to peroxide-induced ferroptosis. GPX4 requires the biosynthesis of selenocysteine for its activity. Active GPX4 provides strong resistance to the irreversible peroxidation of cancer cells.59 Selenium, as an important component of GPX4, can regulate the expression of prognostic tumor markers, such as activator protein 1, interleukin-8 and VEGF, by coacting with lipid peroxides. Therefore, patients with low serum selenium levels may be good candidates for supplementation with selenium to prevent early HCC.60
NRF2 in HCC
Nuclear factor erythroid 2-related factor 2 (NRF2) has antioxidant elements and is regulated by Kelch-like ECH-associated protein 1 (Keap1). Its gene transcription is partially under the control of ROS.61,62
Most of the current research on NRF2 in HCC involves the p62-Keap1-NRF2 axis. In 2013, Ichimura et al63 pointed out that although the Keap1-NRF2 system and autophagy are both involved in oxidative stress, metabolism, and innate immunity, the interaction between these two pathways has not been fully elucidated. They reported that the autophagy adaptor protein p62 is phosphorylated in a target of rapamycin complex 1-dependent manner to promote its binding to Keap1, which means that the Keap1-NRF2 system and autophagy are coupled and interdependent. In addition, the accumulation of phosphorylated p62 continues to activate NRF2, which can promote the tumorigenesis of HCC.63 In 2016, Sun et al64 elucidated the role of the p62-Keap1-NRF2 axis in protecting HCC from ferroptosis. The authors reported that when exposed to compounds that cause ferroptosis, such as erastin, sorafenib, and buthionine sulfoximine, p62 can downregulate the expression of Keap1 to prevent the degradation of NRF2 and chemoattract NRF2 toward accumulation in the nucleus. In addition, a mutual effect between NRF2 and v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog initiates the transcription of heme oxygenase 1.64 Yang et al65 found that gankyrin can be used as a feedback loop to maintain the homeostasis of NRF2 in the cell, and it can compete with NRF2 to bind to the Kelch domain of Keap1 and inhibit NRF2 from being degraded by proteases. Umemura et al66 also confirmed that the p62-Keap1-NRF2 axis can enable HCC progenitor cells to survive in ROS, causing them to undergo malignant mutations when mTORC1 and c-Myc are activated. Therefore, targeting the p62-Keap1-NRF2 axis may help prevent chronic liver disease from progressing to HCC and reduce postoperative recurrence of resectable HCC.
NRF2 is involved not only in HCC but also in liver cancer stem cell (CSCs). In 2020, Sun et al67 found that xanthine oxidoreductase is expressed at low levels in human HCC tissues and CSCs and that it mediates the formation of HCC and chemotherapy resistance by inhibiting the proliferation of CSCs. Specifically, XOR and ubiquitin-specific peptidase 15 synergistically promote the deubiquitination and expression of Keap1, leading to the ubiquitination and accumulation of ROS in CSCs through the degradation of NRF2.67
In recent years, NRF2 activity in HCC has been investigated at the subcellular level. Tripartite motif-containing 25 (TRIM25) is the most effective gene for tumor cells to resist ER stress. Studies have found that TRIM25 promotes tumor cell survival by inducing Keap1 ubiquitination and degradation, thereby activating NRF2 signaling and reducing ROS levels during ER stress.68 Furthermore, malic enzymes (MEs) in mitochondria have been shown to be involved in the production of reduced NADPH. Since NAPDH can neutralize the oxidative effect of ROS, HCC can activate MEs through NRF2 transcription to resist cell damage to ROS.69
The expression of Sigma-1 receptor (S1R) has been proposed to contribute to cell protection by inhibiting ischemia-induced ROS production.70–72 Related articles have reported that S1R can regulate ROS through NRF2. When NRF2 is activated and causes ROS accumulation, the level of S1R is elevated to protect HCC against sorafenib-induced ferroptosis.73
To clarify the role of NRF2 in HCC, in addition to human HCC cells, many studies have started with animal models and attempted further exploration. In 2014, Petrelli et al74 used a rat model to illustrate the molecular mechanisms involved in the HCC drug resistance process through miRNA and gene expression profiles and provided evidence that NRF2 is carcinogenic in the precancerous/premalignant stage. In summary, NRF2 is positively correlated with the progression of HCC, and microRNA-200A has a negative regulatory effect on NRF2. When NRF2 is suppressed, the precancerous lesions in the rat model disappear.74 Moreover, the relationship between mutations of CTNNB1 and NRF2 has also been described in some articles. The CTNNB1 gene mutation is the second most common gene mutation in HCC, accounting for approximately 26–38% of all cases.75,76 In 2015, Zavattari et al77 studied a drug-resistant rat hepatocyte model and found that the CTNNB1 mutation occurred late and only appeared in fully advanced HCC, while the NRF2 mutation occurred early and may mediate the transformation of precancerous lesions to HCC through clonal amplification. In 2021, Tao et al78 found that although CTNNB1 mutations are not uncommon in HCC, CTNNB1 mutations alone will not lead to the occurrence of HCC. They appear to have a synergistic effect with NRF2 mutations to promote the occurrence of HCC.
SLC7A11 in HCC
Solute carrier family 7 member 11 (SLC7A11) has a profound effect on stabilizing the level of GSH in cells. It can indirectly inactivate GPX4 by reducing the uptake of cysteine and the biosynthesis of GSH, thereby limiting the synthesis of GSH, increasing the ROS of lipids and ultimately causing ferroptosis.79
Studies have found that the RNA-binding protein DAZAP1 binds to the 3ʹ noncoding region of SLC7A11 to regulate its stability and govern the role of SLC7A11/GPX4 in ferroptosis, so that sorafenib-induced ferroptosis is inhibited.80
In 2020, Kim et al81 proposed that SLC7A11 is regulated by transforming growth factor-β1 (TGF-β1), which can induce GSH consumption, increase the production of ROS, and then sensitize cells to oxidative stress. They suggested that TGF-β1 inhibits the expression of SLC7A11 by upregulating Smad3 and ultimately mediates lipid peroxidation to make HCC cells more susceptible to GPX4 inhibitors.81
As an important marker of liver cancer stem cells, CD133 has received widespread attention. Previous studies have shown that certain stemness-associated genes related to CSCs can cause ferroptosis by regulating the level of ROS.82,83 In recent years, it has been found that CD133-positive HCC cells increase resistance to ROS by upregulating GSH levels. At the same time, the SLC7A11 inhibitor sulfasalazine can specifically regulate the reduction state of CD133-positive HCC and sensitize CD133-positive HCC to chemotherapy.84 Another study showed that actinomycin D can increase the accumulation of ROS and DNA damage in HCC cells, inhibit the expression of SLC7A11 by suppressing the synthesis of CD133, and control the stemness of HCC cells. Based on these studies, we also explored SLC7A11-mediated ferroptosis characterized by elevated ROS levels as a new research perspective.
HSPB1 in HCC
Heat shock protein beta-1 (HSPB1) mainly functions as a molecular chaperone. Its phosphorylation reduces the uptake of iron and lipid peroxidation by cells, and inhibition of its expression and phosphorylation increases the anticancer properties of erastin-induced ferroptosis.85
In 2009, Guo et al86 confirmed that HSPB1 regulates the apoptosis of metastatic liver cancer by activating the IκB kinase-dependent NF-κB pathway. In the same year, Sharma et al87 discovered that although the inhibitor of HSPB1, quercetin, has certain toxicity to cells, it can enhance the proapoptotic effect of chemotherapeutics such as 5-fluorouracil and carboplatin on HCC cells by inhibiting the expression of HSPB1.
With the deepening of research in recent years, posttranslational modifications related to HSPB1 have become a focus of research. Posttranslational modifications, such as oxyglycosylation and phosphorylation, can regulate the way HSPB1 enters the nucleus and to a certain extent, regulate the intracellular stress response and provide protection for the cell.88 Moreover, the ERK signaling pathway is the main proliferation signal of HCC. Phosphorylated HSPB1 can activate the p38 MAPK-MAPK phosphatase-1 pathway, which is essential for the detection of early HCC.89
Stress-activated protein kinase p38α can reduce the accumulation of ROS in cells by reducing the expression of HSPB1. The lack of p38α is able to increase the stemness of HCC, which may lead to HCC recurrence and accelerate liver fibrosis that develops into HCC.90 In 2010, Yang et al91 suggested that microRNA-17-5p can activate the p38 MAPK pathway, which in turn facilitates the phosphorylation of HSPB1 and enhances the migration ability of HCC. In 2015, Hao et al92 found that dual specificity phosphatase 1 (DUSP1) repressed the phosphorylation of p38 MAPK, which in turn inhibited the activation of HSPB1, resulting in increased expression of p53 sites related to cell cycle arrest and apoptosis in HCC cells. DUSP1 may pass through the p38 MAPK/HSPB1/p53 positive feedback pathway to inhibit the progression of HCC. These studies provide a better understanding of the role of HSPB1 in the development of HCC.
Ferroptosis in HCC Treatment
Sorafenib has favorable efficacy in the treatment of advanced HCC; however, because of the individual responses of patients and with the emergence of tumor resistance, the efficacy of sorafenib alone may not be sufficient to prevent the progression of HCC. Therefore, in recent years, many studies explored specific mechanisms of tumor resistance to sorafenib alone or in combination with other drugs. The strategy of using more than one drug for treatment is based on enhancing the efficacy of treatment while reducing the dosage of sorafenib necessary for efficacy (Figure 2). Previously, researchers found that sorafenib can induce hepatocyte death by mediating ferroptosis.15 Specifically, after sorafenib treatment of HCC cells, mitochondrial morphology and membrane potential collapse and oxidative phosphorylation activity and ATP synthesis are reduced, thereby inducing ferroptosis and cell death.93 In addition, GSH can rescue the ferroptosis induced by sorafenib and eliminate the accumulation of mitochondrial ROS and lipid peroxides, while cysteine depletion or cysteinase inhibition has the opposite effect.93
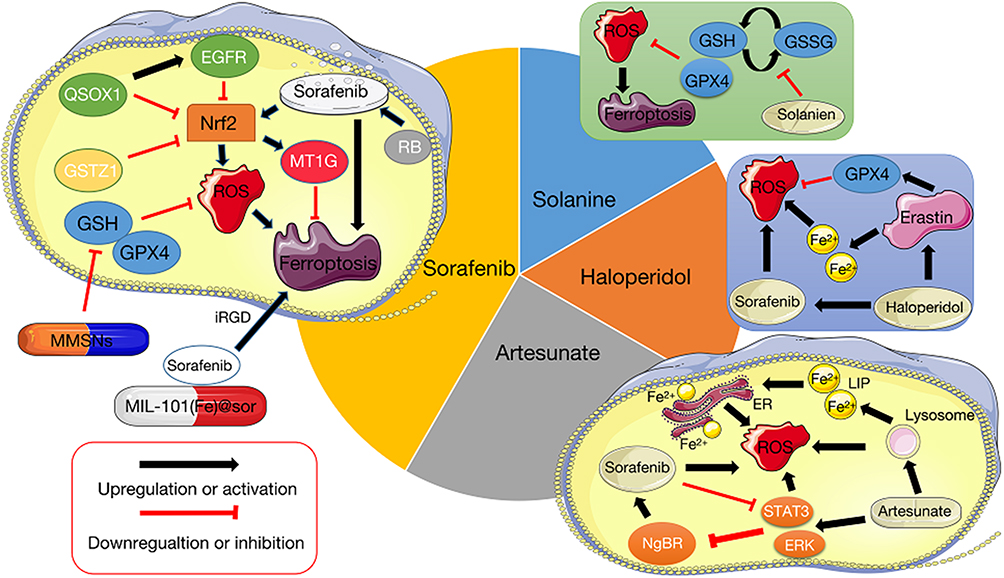 |
Figure 2 The function and possible mechanism of ferroptosis in HCC treatment. Several studies have investigated the induction of ferroptosis as a possible HCC treatment that may provide promising and effective therapy. Sorafenib has been shown to induce ferroptosis, and researchers have begun to explore the use of certain key factors to optimize sorafenib treatment, such as preparing nanoparticles that make sorafenib more susceptible to target tumors and boost its effect on intracellular redox homeostasis. Artemisinin and its derivatives are utilized in malaria treatment, but recently have been shown to be tumorigenic, apparently by interfering with iron homeostasis and the redox microenvironment. Moreover, Artemisinin acts synergistically with sorafenib. Haloperidol and solanine are also able to promote the ROS of tumor cells by influencing the intracellular GSH levels and systems. Ferroptosis-related drug and key regulator (black), potential regulator of ferroptosis (white). |
Because of recent interest in ferroptosis as a physiological phenomenon, many research teams have begun to investigate ferroptosis as an outcome in HCC treatment.94 Quiescin sulfhydryl oxidase 1 (QSOX1) contributes to the formation of disulfide bonds of multiple proteins and can also act as a tumor suppressor. This characteristic manifests itself in various cancer-associated processes, such as autophagy and extracellular matrix regulation.95,96 In 2021, Sun et al97 found that QSOX1 is a cellular pro-oxidant. In addition to enhancing sorafenib-induced ferroptosis by inhibiting NRF2 in vitro and in vivo, it can also potentiate ubiquitination-mediated epidermal growth factor receptor (EGFR) degradation and accelerate the transport of EGFR in the cell, leading to the inhibition of the activity of NRF2.
Metallothionein (MT) is a type of small molecular intracellular protein commonly expressed in eukaryotic cells that possesses a common structure including high cysteine content.98 In 2016, Houessinon et al99 found that sorafenib can induce the expression of an MT isoform, MT1G. MT1G can be used as a marker to reflect the effect of sorafenib on the redox metabolism of HCC cells. In the same year, Sun et al100 further elucidated the relevant mechanism of MT1G, identifying MT1G as a negative regulator of ferroptosis. Sorafenib regulates the expression of MT1G by activating NRF2, and MT1G promotes the resistance of tumor cells to sorafenib by inhibiting ferroptosis.
To enable sorafenib to target tumors more accurately, Liu et al101 prepared sorafenib-loaded iron-based metal-organic framework nanoparticles (MIL-101(Fe)@sor), together with a tumor-penetrating peptide iRGD that contains a tumor homing motif and a tissue penetration motif. This biomolecule conjugate was used to kill HCC cells through ferroptosis. Tang et al102 also used ferroptosis to kill HCC cells by synthesizing manganese-doped mesoporous silica nanoparticles (MMSNs). The nanoparticles cause the consumption of intracellular GSH by degrading MMSNs, which leads to the inactivation of GPX4. This in turn results in an increase in intracellular lipid peroxidation and ultimately induces ferroptosis.102
Glutathione S-transferase zeta1 (GSTZ1) is an important member of the GST superfamily. It participates in phenylalanine/tyrosine catabolism. Previous studies have shown that a lack of GSTZ1 can lead to the accumulation of the metabolite succinyl lactone, which activates the NRF2 signaling pathway.103–105 Wang et al106 found that the depletion of GSTZ1 potentiated the NRF2 pathway and elevated the level of GPX4, thus inhibiting sorafenib-induced ferroptosis. Louandre et al107 found that retinoblastoma mediates ferroptosis and kills HCC cells by participating in the toxic effects of sorafenib on HCC cells. Such toxic effects include oxidative stress and mitochondrial ROS generation.
In addition to sorafenib, several drugs commonly used in other diseases are also being attempted to treat HCC based on ferroptosis. Current studies have shown that in addition to being effective antimalarial agents, artemisinin and its derivatives also have significant anticancer effects, in which Fe2+ and ROS have an important role. This suggests that artemisinin and its derivatives display potential anticancer activities by interfering with iron homeostasis and the redox microenvironment.108–111 Jiang et al31 proposed that ART can regulate the LIPs of HCC cells and induce ROS-dependent cell death. In addition to the effects of ART itself, its combined use with sorafenib further enhances its toxic effects on tumors.112 Li et al113 suggested that sorafenib mainly causes GSH depletion and oxidative stress by mediating mitochondrial damage and SLC7A11, while ART induces ferroptosis by facilitating a series of reactions, such as lysosomal cathepsin B/L activation, ferritin degradation and lipid peroxidation. Therefore, ART is believed to sensitize HCC cells to the therapeutic effect of sorafenib. This combination therapy reduces the potential harmful toxicity of sorafenib by reducing its effective delivered dose. This should improve the biological safety and tolerability of the sorafenib treatment in vivo. Jing et al114 proposed that although sorafenib can effectively inhibit the RAF/MEK/ERK pathway, it cannot eliminate the resistance of HCC to sorafenib that is driven by the PI3K/AKT/mTOR pathway, while ART can significantly inhibit the phosphorylation of AKT and mTOR. Therefore, the combined use of the two drugs can effectively suppress the expression of these two signaling pathways, significantly affecting the progression of HCC. Furthermore, Yao et al115 also reported that ART induces ROS by activating the ERK and STAT3 signaling pathways, while sorafenib inhibits the ERK and STAT3 signaling pathways combined with ART to synergistically limit the progression of HCC. Neurite outgrowth inhibitor protein B receptor (NgBR) is a human apoptosis-inducing gene that regulates cell apoptosis by promoting ER stress, intracellular calcium disturbance and the caspase-7 pathway.116,117 He et al118 found that NgBR activates the MEK/ERK pathway to make HCC cells resistant to sorafenib, and ART increases the sensitivity of HCC cells to sorafenib by inhibiting NgBR.
Bai et al119 has identified haloperidol as an antipsychotic medicine that significantly elevates the levels of Fe2+, GSH and lipid peroxidation in cells, and it significantly promotes ferroptosis induced by erastin and sorafenib. Jin et al120 found that solanine significantly decreases the expression of GPX4 and GSH synthase, it compromises the GSH redox system induced by GPX4, and it ultimately promotes ferroptosis in HCC cells.
Conclusion and Prospects
In this review, we assessed the development of ferroptosis-related concepts and focused on the core regulatory factors of ferroptosis and treatment methods based on ferroptosis. By reviewing the evolution of ferroptosis in HCC and analyzing its application in the treatment of HCC, we can better understand ferroptosis and its regulatory factors. In contrast to other published reviews on ferroptosis and liver cancer, our review enumerates the known key regulators of ferroptosis and emphasizes and identifies potential recently identified regulators of ferroptosis in HCC. This provides clinicians with numerous choices in the treatment of HCC. Additionally, individualized treatment for specific targets can also result in better prognosis for patients. Nanoparticles such as MMSNs and MIL-101(Fe)@sor prepared in the tumor microenvironment have also been developed to act on tumor cells, capitalizing on their ability to induce ferroptosis in tumor cells. Therefore, we have reason to believe that ferroptosis therapy may be effective in the treatment of advanced HCC.
Further study and research are needed to optimize HCC treatment and improve patient prognosis. Some questions that need to be addressed are as follows. Are there additional key regulatory factors for ferroptosis? Are there any side effects of an effective dose of sorafenib to induce ferroptosis? Has the exact mechanism by which lipid peroxidation induces ferroptosis been fully elucidated? What role does the immune system have in the ferroptosis phenomenon noted in tumor treatment; specifically, what is the mechanism of action? Can the ferroptosis process be reversed? How can tumor cells be targeted more accurately and effectively for eradication? Can drugs that induce ferroptosis be developed and used clinically? What are the side effects of drugs that induce ferroptosis and how may these side effects be minimized?
Consent for Publication
All authors approved the final manuscript.
Funding
This work was funded by National Natural Science Foundation of China (No. 82073045) and China Postdoctoral Science Foundation (2020TQ0384).
Disclosure
Yongcong Yan and Zhiyu Xiao are co-correspondence authors for this study. All authors declare no conflicts of interest.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Villanueva A. Hepatocellular Carcinoma. N Engl J Med. 2019;380(15):1450–1462. doi:10.1056/NEJMra1713263
3. Zheng R, Qu C, Zhang S, et al. Liver cancer incidence and mortality in China: temporal trends and projections to 2030. Chin J Cancer Res. 2018;30(6):571–579. doi:10.21147/j.issn.1000-9604.2018.06.01
4. Llovet JM, Montal R, Sia D, et al. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol. 2018;15(10):599–616. doi:10.1038/s41571-018-0073-4
5. Dixon SJ, Lemberg K, Lamprecht M, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
6. D’Arcy MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int. 2019;43(6):582–592. doi:10.1002/cbin.11137
7. Doll S, Conrad M. Iron and ferroptosis: a still ill-defined liaison. IUBMB Life. 2017;69(6):423–434. doi:10.1002/iub.1616
8. Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–379. doi:10.1038/cdd.2015.158
9. Tang D, Chen X, Kang R, et al. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–125. doi:10.1038/s41422-020-00441-1
10. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell. 2019;35(6):830–849. doi:10.1016/j.ccell.2019.04.002
11. Liang C, Zhang X, Yang M, et al. Recent progress in ferroptosis inducers for cancer therapy. Adv Mater. 2019;31(51):e1904197. doi:10.1002/adma.201904197
12. Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer. 2013;13(5):342–355. doi:10.1038/nrc3495
13. Urano S, Ohara T, Noma K, et al. Iron depletion enhances the effect of sorafenib in hepatocarcinoma. Cancer Biol Ther. 2016;17(6):648–656. doi:10.1080/15384047.2016.1177677
14. Pietrangelo A. Iron in NASH, chronic liver diseases and HCC: how much iron is too much? J Hepatol. 2009;50(2):249–251. doi:10.1016/j.jhep.2008.11.011
15. Louandre C, Ezzoukhry Z, Godin C, et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer. 2013;133(7):1732–1742. doi:10.1002/ijc.28159
16. Dongiovanni P, Fracanzani AL, Cairo G, et al. Iron-dependent regulation of MDM2 influences p53 activity and hepatic carcinogenesis. Am J Pathol. 2010;176(2):1006–1017. doi:10.2353/ajpath.2010.090249
17. Recalcati S, Correnti M, Gammella E, et al. Iron metabolism in liver cancer stem cells. Front Oncol. 2019;9:149. doi:10.3389/fonc.2019.00149
18. Shen J, Sheng X, Chang Z, et al. Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep. 2014;7(1):180–193. doi:10.1016/j.celrep.2014.02.042
19. Poliklinik MK. Survival and causes of death in hemochromatosis observations in 163 patients. Ann N Y Acad Sci. 1988:526:245–257.
20. Trujillo-Alonso V, Pratt EC, Zong H, et al. FDA-approved ferumoxytol displays anti-leukaemia efficacy against cells with low ferroportin levels. Nat Nanotechnol. 2019;14(6):616–622. doi:10.1038/s41565-019-0406-1
21. Schwartz AJ, Das NK, Ramakrishnan SK, et al. Hepatic hepcidin/intestinal HIF-2α axis maintains iron absorption during iron deficiency and overload. J Clin Invest. 2019;129(1):336–348. doi:10.1172/JCI122359
22. Spivak I, Arora J, Meinzer C, et al. Low serum hepcidin is associated with reduced short-term survival in adults with acute liver failure. Hepatology. 2019;69(5):2136–2149. doi:10.1002/hep.30486
23. Protchenko O, Baratz E, Jadhav S, et al. Iron chaperone poly rC binding protein 1 protects mouse liver from lipid peroxidation and steatosis. Hepatology. 2021;73(3):1176–1193. doi:10.1002/hep.31328
24. Yu Y, Jiang LJ, H. W, et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood. 2020;136(6):726–739. doi:10.1182/blood.2019002907
25. Parrow NL, Maria Feola YL, Guerra A, et al. Lobe specificity of iron binding to transferrin modulates murine erythropoiesis and iron homeostasis. Blood. 2019;134(17):1373–1384. doi:10.1182/blood.2018893099
26. Carine Fillebeen EC, Wagner J, Katsarou A, et al. Transferrin receptor 1 controls systemic iron homeostasis by fine-tuning hepcidin expression to hepatocellular iron load. Blood. 2019;133(4):344–355. doi:10.1182/blood-2018-05-850404
27. Moroishi T, Nishiyama M, Takeda Y, et al. The FBXL5-IRP2 axis is integral to control of iron metabolism in vivo. Cell Metab. 2011;14(3):339–351. doi:10.1016/j.cmet.2011.07.011
28. Muto Y, Moroishi T, Ichihara K, et al. Disruption of FBXL5-mediated cellular iron homeostasis promotes liver carcinogenesis. J Exp Med. 2019;216(4):950–965. doi:10.1084/jem.20180900
29. Andrews NC. Disorders of iron metabolism. N Engl J Med. 1999;341(26):1986–1995. doi:10.1056/NEJM199912233412607
30. Wei Y, Ye W, Zhao W. Serum iron levels decreased in patients with HBV-related hepatocellular carcinoma, as a risk factor for the prognosis of HBV-related HCC. Front Physiol. 2018;9:66. doi:10.3389/fphys.2018.00066
31. Jiang Z, Wang Z, Chen L, et al. Artesunate induces ER-derived-ROS-mediated cell death by disrupting labile iron pool and iron redistribution in hepatocellular carcinoma cells. Am J Cancer Res. 2021;11(3):691–711.
32. Shang Y, Luo M, Yao F, et al. Ceruloplasmin suppresses ferroptosis by regulating iron homeostasis in hepatocellular carcinoma cells. Cell Signal. 2020;72:109633. doi:10.1016/j.cellsig.2020.109633
33. Hiromi Gunshin BM, Berger UV, Gunshin Y, et al. Cloning and characterization of a mammalian proton coupled metal-ion transporter. Nature. 1997;388(6641):482–488. doi:10.1038/41343
34. Hoki T, Katsuta E, Yan L, et al. Low DMT1 expression associates with increased oxidative phosphorylation and early recurrence in hepatocellular carcinoma. J Surg Res. 2019;234:343–352. doi:10.1016/j.jss.2018.11.008
35. Alteen MG, Tan HY, Vocadlo DJ. Monitoring and modulating O-GlcNAcylation: assays and inhibitors of O-GlcNAc processing enzymes. Curr Opin Struct Biol. 2021;68:157–165. doi:10.1016/j.sbi.2020.12.008
36. Zhu G, Murshed A, Li H, et al. O-GlcNAcylation enhances sensitivity to RSL3-induced ferroptosis via the YAP/TFRC pathway in liver cancer. Cell Death Discov. 2021;7(1):83. doi:10.1038/s41420-021-00468-2
37. Liu L, Cao Y, Chen C, et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66(24):11851–11858. doi:10.1158/0008-5472.CAN-06-1377
38. Ohara T, Noma K, Urano S, et al. A novel synergistic effect of iron depletion on antiangiogenic cancer therapy. Int J Cancer. 2013;132(11):2705–2713. doi:10.1002/ijc.27943
39. Schmocker C, Weylandt KH, Kahlke L, et al. Omega-3 fatty acids alleviate chemically induced acute hepatitis by suppression of cytokines. Hepatology. 2007;45(4):864–869. doi:10.1002/hep.21626
40. Lim K, Han C, Dai Y, et al. Omega-3 polyunsaturated fatty acids inhibit hepatocellular carcinoma cell growth through blocking β-catenin and cyclooxygenase-2. Mol Cancer Ther. 2009;8(11):3046–3055. doi:10.1158/1535-7163.MCT-09-0551
41. Weylandt KH, Krause LF, Gomolka B, et al. Suppressed liver tumorigenesis in fat-1 mice with elevated omega-3 fatty acids is associated with increased omega-3 derived lipid mediators and reduced TNF-alpha. Carcinogenesis. 2011;32(6):897–903. doi:10.1093/carcin/bgr049
42. Sawada N, Inoue M, Iwasaki M, et al. Consumption of n-3 fatty acids and fish reduces risk of hepatocellular carcinoma. Gastroenterology. 2012;142(7):1468–1475. doi:10.1053/j.gastro.2012.02.018
43. Laparra Llopis JM, Brown D, Saiz B. Chenopodium quinoa and salvia hispanica provide immunonutritional agonists to ameliorate hepatocarcinoma severity under a high-fat diet. Nutrients. 2020;12(7):1946. doi:10.3390/nu12071946
44. Leineweber CG, Pietzner A, Zhang IW, et al. Assessment of the effect of sorafenib on omega-6 and omega-3 epoxyeicosanoid formation in patients with hepatocellular carcinoma. Int J Mol Sci. 2020;21(5):1875. doi:10.3390/ijms21051875
45. Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. 2019;15(12):1137–1147. doi:10.1038/s41589-019-0408-1
46. Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–98. doi:10.1038/nchembio.2239
47. Chen J, Ding C, Chen Y, et al. ACSL4 reprograms fatty acid metabolism in hepatocellular carcinoma via c-Myc/SREBP1 pathway. Cancer Lett. 2021;502:154–165. doi:10.1016/j.canlet.2020.12.019
48. Zhao Y, Li M, Yao X, et al. HCAR1/MCT1 regulates tumor ferroptosis through the lactate-mediated AMPK-SCD1 activity and its therapeutic implications. Cell Rep. 2020;33(10):108487. doi:10.1016/j.celrep.2020.108487
49. Cui M, Xiao Z, Sun B, et al. Involvement of cholesterol in hepatitis B virus X protein-induced abnormal lipid metabolism of hepatoma cells via up-regulating miR-205-targeted ACSL4. Biochem Biophys Res Commun. 2014;445(3):651–655. doi:10.1016/j.bbrc.2014.02.068
50. Qin X, Zhang J, Lin Y, et al. Identification of MiR-211-5p as a tumor suppressor by targeting ACSL4 in hepatocellular carcinoma. J Transl Med. 2020;18(1):326. doi:10.1186/s12967-020-02494-7
51. Feng J, Lu PZ, Zhu GZ, et al. ACSL4 is a predictive biomarker of sorafenib sensitivity in hepatocellular carcinoma. Acta Pharmacol Sin. 2021;42(1):160–170. doi:10.1038/s41401-020-0439-x
52. Xia H, Lee KW, Chen J, et al. Simultaneous silencing of ACSL4 and induction of GADD45B in hepatocellular carcinoma cells amplifies the synergistic therapeutic effect of aspirin and sorafenib. Cell Death Discov. 2017;3(1):17058. doi:10.1038/cddiscovery.2017.58
53. Labunskyy VM, Hatfield DL, Gladyshev VN. Selenoproteins: molecular pathways and physiological roles. Physiol Rev. 2014;94(3):739–777. doi:10.1152/physrev.00039.2013
54. Forcina GC, Dixon SJ. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics. 2019;19(18):e1800311. doi:10.1002/pmic.201800311
55. Guerriero E, Capone F, Accardo M, et al. GPX4 and GPX7 over-expression in human hepatocellular carcinoma tissues. Eur J Histochem. 2015;59(4):2540. doi:10.4081/ejh.2015.2540
56. Brault C, Lévy P, Duponchel S, et al. Glutathione peroxidase 4 is reversibly induced by HCV to control lipid peroxidation and to increase virion infectivity. Gut. 2016;65(1):144–154. doi:10.1136/gutjnl-2014-307904
57. Yang WS, SriRamaratnam R, Welsch M, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331. doi:10.1016/j.cell.2013.12.010
58. Xu Q, Zhou L, Yang G, et al. CircIL4R facilitates the tumorigenesis and inhibits ferroptosis in hepatocellular carcinoma by regulating the miR-541-3p/GPX4 axis. Cell Biol Int. 2020;44(11):2344–2356. doi:10.1002/cbin.11444
59. Ingold I, Berndt C, Schmitt S, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell. 2018;172(3):409–422. doi:10.1016/j.cell.2017.11.048
60. Rohr-Udilova N, Sieghart W, Eferl R, et al. Antagonistic effects of selenium and lipid peroxides on growth control in early hepatocellular carcinoma. Hepatology. 2012;55(4):1112–1121. doi:10.1002/hep.24808
61. Itoh K, Chiba T, Takahashi S, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236(2):313–322. doi:10.1006/bbrc.1997.6943
62. Itoh K, Wakabayashi N, Katoh Y, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13(1):76–86. doi:10.1101/gad.13.1.76
63. Ichimura Y, Waguri S, Sou YS, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51(5):618–631. doi:10.1016/j.molcel.2013.08.003
64. Sun X, Ou Z, Chen R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63(1):173–184. doi:10.1002/hep.28251
65. Yang C, Tan YX, Yang GZ, et al. Gankyrin has an antioxidative role through the feedback regulation of Nrf2 in hepatocellular carcinoma. J Exp Med. 2016;213(5):859–875. doi:10.1084/jem.20151208
66. Umemura A, He F, Taniguchi K, et al. p62, upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell. 2016;29(6):935–948. doi:10.1016/j.ccell.2016.04.006
67. Sun Q, Zhang Z, Lu Y, et al. Loss of xanthine oxidoreductase potentiates propagation of hepatocellular carcinoma stem cells. Hepatology. 2020;71(6):2033–2049. doi:10.1002/hep.30978
68. Liu Y, Tao S, Liao L, et al. TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat Commun. 2020;11(1):348. doi:10.1038/s41467-019-14190-2
69. Lee D, Zhang MS, Tsang FHC, et al. Adaptive and constitutive activations of malic enzymes confer liver cancer multi-layered protection against ROS. Hepatology. 2021. doi:10.1002/hep.31761
70. Sanju Narayanan RB, Mesangeau C, Poupaert JH, McCurdy CR. Early development of sigma-receptor ligands. Future Med Chem. 2011;3(1):79–94. doi:10.4155/fmc.10.279
71. Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131(3):596–610. doi:10.1016/j.cell.2007.08.036
72. Hayashi T, Fujimoto M. Detergent-resistant microdomains determine the localization of sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol Pharmacol. 2010;77(4):517–528. doi:10.1124/mol.109.062539
73. Bai T, Lei P, Zhou H, et al. Sigma-1 receptor protects against ferroptosis in hepatocellular carcinoma cells. J Cell Mol Med. 2019;23(11):7349–7359. doi:10.1111/jcmm.14594
74. Petrelli A, Perra A, Cora D, et al. MicroRNA/gene profiling unveils early molecular changes and nuclear factor erythroid related factor 2 (NRF2) activation in a rat model recapitulating human hepatocellular carcinoma (HCC). Hepatology. 2014;59(1):228–241. doi:10.1002/hep.26616
75. Schulze K, Imbeaud S, Letouzé E, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47(5):505–511. doi:10.1038/ng.3252
76. Wheeler DA, Roberts LR; Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169(7):1327–1341. doi:10.1016/j.cell.2017.05.046
77. Zavattari P, Perra A, Menegon S, et al. Nrf2, but not β-catenin, mutation represents an early event in rat hepatocarcinogenesis. Hepatology. 2015;62(3):851–862. doi:10.1002/hep.27790
78. Tao J, Krutsenko Y, Moghe A, et al. Nuclear factor erythroid 2–related factor 2 and β-Catenin coactivation in hepatocellular cancer: biological and therapeutic implications. Hepatology. 2021. doi:10.1002/hep.31730
79. Wenyu Lin CW, Liu G, Bi C, Wang X, Zhou Q, Jin H. SLC7A11:xCTin cancer- biological functions and therapeutic implications. Am J Cancer Res. 2020;10(10):3106–3126.
80. Wang Q, Guo Y, Wang W, et al. RNA binding protein DAZAP1 promotes HCC progression and regulates ferroptosis by interacting with SLC7A11 mRNA. Exp Cell Res. 2021;399(1):112453. doi:10.1016/j.yexcr.2020.112453
81. Kim DH, Kim WD, Kim SK, et al. TGF-β1-mediated repression of SLC7A11 drives vulnerability to GPX4 inhibition in hepatocellular carcinoma cells. Cell Death Dis. 2020;11(5):406. doi:10.1038/s41419-020-2618-6
82. Kim HM, Haraguchi N, Ishii H, et al. Increased CD13 expression reduces reactive oxygen species, promoting survival of liver cancer stem cells via an epithelial-mesenchymal transition-like phenomenon. Ann Surg Oncol. 2012;19(Suppl 3):S539–48. doi:10.1245/s10434-011-2040-5
83. Ishimoto T, Nagano O, Yae T, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell. 2011;19(3):387–400. doi:10.1016/j.ccr.2011.01.038
84. Song Y, Jang J, Shin TH, et al. Sulfasalazine attenuates evading anticancer response of CD133-positive hepatocellular carcinoma cells. J Exp Clin Cancer Res. 2017;36(1):38. doi:10.1186/s13046-017-0511-7
85. Sun X, Ou Z, Xie M, et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene. 2015;34(45):5617–5625. doi:10.1038/onc.2015.32
86. Guo K, Kang NX, Li Y, et al. Regulation of HSP27 on NF-κB pathway activation may be involved in metastatic hepatocellular carcinoma cells apoptosis. BMC Cancer. 2009;9(1):100. doi:10.1186/1471-2407-9-100
87. Sharma A, Upadhyay AK, Bhat MK. Inhibition of Hsp27 and Hsp40 potentiates 5-fluorouracil and carboplatin mediated cell killing in hepatoma cells. Cancer Biol Ther. 2009;8(22):2106–2113. doi:10.4161/cbt.8.22.9687
88. Guo K, Gan L, Zhang S, et al. Translocation of HSP27 into liver cancer cell nucleus may be associated with phosphorylation and O-GlcNAc glycosylation. Oncol Rep. 2012;28(2):494–500. doi:10.3892/or.2012.1844
89. Matsushima-Nishiwaki R, Takai S, Adachi S, et al. Phosphorylated heat shock protein 27 represses growth of hepatocellular carcinoma via inhibition of extracellular signal-regulated kinase. J Biol Chem. 2008;283(27):18852–18860. doi:10.1074/jbc.M801301200
90. Sakurai T, Kudo M, Umemura A, et al. p38α inhibits liver fibrogenesis and consequent hepatocarcinogenesis by curtailing accumulation of reactive oxygen species. Cancer Res. 2013;73(1):215–224. doi:10.1158/0008-5472.CAN-12-1602
91. Yang F, Yin Y, Wang F, et al. miR-17-5p promotes migration of human hepatocellular carcinoma cells through the p38 mitogen-activated protein kinase-heat shock protein 27 pathway. Hepatology. 2010;51(5):1614–1623. doi:10.1002/hep.23566
92. Hao PP, Li H, Lee MJ, et al. Disruption of a regulatory loop between DUSP1 and p53 contributes to hepatocellular carcinoma development and progression. J Hepatol. 2015;62(6):1278–1286. doi:10.1016/j.jhep.2014.12.033
93. Li Y, Xia J, Shao F, et al. Sorafenib induces mitochondrial dysfunction and exhibits synergistic effect with cysteine depletion by promoting HCC cells ferroptosis. Biochem Biophys Res Commun. 2021;534:877–884. doi:10.1016/j.bbrc.2020.10.083
94. Emma Lachaier CL, Godin C, Saidak Z, et al. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014;34(11):6417–6422.
95. Zhang XF, Wang J, Jia HL, et al. Core fucosylated glycan-dependent inhibitory effect of QSOX1-S on invasion and metastasis of hepatocellular carcinoma. Cell Death Discov. 2019;5(1):84. doi:10.1038/s41420-019-0164-8
96. Tal Ilani AA, Grossman I, Horowitz B, Kartvelishvily E, Cohen SR, Fass D. A secreted disulfide catalyst controls extracellular matrix composition and function. Science. 2013;341(6141):74–76. doi:10.1126/science.1238279
97. Sun J, Zhou C, Zhao Y, et al. Quiescin sulfhydryl oxidase 1 promotes sorafenib-induced ferroptosis in hepatocellular carcinoma by driving EGFR endosomal trafficking and inhibiting NRF2 activation. Redox Biol. 2021;41:101942. doi:10.1016/j.redox.2021.101942
98. Cherian MG, Jayasurya A, Bay BH. Metallothioneins in human tumors and potential roles in carcinogenesis. Mutat Res. 2003;533(1–2):201–209. doi:10.1016/j.mrfmmm.2003.07.013
99. Houessinon A, François C, Sauzay C, et al. Metallothionein-1 as a biomarker of altered redox metabolism in hepatocellular carcinoma cells exposed to sorafenib. Mol Cancer. 2016;15(1):38. doi:10.1186/s12943-016-0526-2
100. Sun X, Niu X, Chen R, et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016;64(2):488–500. doi:10.1002/hep.28574
101. Liu X, Zhu X, Qi X, et al. Co-administration of iRGD with sorafenib-loaded iron-based metal-organic framework as a targeted ferroptosis agent for liver cancer therapy. Int J Nanomedicine. 2021;16:1037–1050. doi:10.2147/IJN.S292528
102. Tang H, Chen D, Li C, et al. Dual GSH-exhausting sorafenib loaded manganese-silica nanodrugs for inducing the ferroptosis of hepatocellular carcinoma cells. Int J Pharm. 2019;572:118782. doi:10.1016/j.ijpharm.2019.118782
103. Fernandez-Canon JM, Penalva MA. Characterization of a fungal maleylacetoacetate isomerase gene and identification of its human homologue. J Biol Chem. 1998;273(1):329–337. doi:10.1074/jbc.273.1.329
104. Yang F, Li J, Deng H, et al. GSTZ 1-1 deficiency activates NRF 2/IGF1R axis in HCC via accumulation of oncometabolite succinylacetone. EMBO J. 2019;38(15):e101964. doi:10.15252/embj.2019101964
105. Li J, Wang Q, Yang Y, et al. GSTZ1 deficiency promotes hepatocellular carcinoma proliferation via activation of the KEAP1/NRF2 pathway. J Exp Clin Cancer Res. 2019;38(1):438. doi:10.1186/s13046-019-1459-6
106. Wang Q, Bin C, Xue Q, et al. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021;12(5):426. doi:10.1038/s41419-021-03718-4
107. Louandre C, Marcq I, Bouhlal H, et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015;356(2):971–977. doi:10.1016/j.canlet.2014.11.014
108. Li PC, Lam E, Roos WP, et al. Artesunate derived from traditional Chinese medicine induces DNA damage and repair. Cancer Res. 2008;68(11):4347–4351. doi:10.1158/0008-5472.CAN-07-2970
109. Kong Z, Liu R, Cheng Y. Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. Biomed Pharmacother. 2019;109:2043–2053. doi:10.1016/j.biopha.2018.11.030
110. Ishikawa C, Senba M, Mori N. Evaluation of artesunate for the treatment of adult T-cell leukemia/lymphoma. Eur J Pharmacol. 2020;872:172953. doi:10.1016/j.ejphar.2020.172953
111. Efferth T. From ancient herb to modern drug: artemisia annua and artemisinin for cancer therapy. Semin Cancer Biol. 2017;46:65–83.
112. Li H, Xu K, Pian G, et al. Artesunate and sorafenib: combinatorial inhibition of liver cancer cell growth. Oncol Lett. 2019;18(5):4735–4743.
113. Li ZJ, Dai HQ, Huang XW, et al. Artesunate synergizes with sorafenib to induce ferroptosis in hepatocellular carcinoma. Acta Pharmacol Sin. 2021;42(2):301–310. doi:10.1038/s41401-020-0478-3
114. Jing W, Shuo L, Yingru X, et al. Artesunate promotes sensitivity to sorafenib in hepatocellular carcinoma. Biochem Biophys Res Commun. 2019;519(1):41–45. doi:10.1016/j.bbrc.2019.08.115
115. Yao X, Zhao CR, Yin H, et al. Synergistic antitumor activity of sorafenib and artesunate in hepatocellular carcinoma cells. Acta Pharmacol Sin. 2020;41(12):1609–1620. doi:10.1038/s41401-020-0395-5
116. Schweigreiter R, Stasyk T, Contarini I, et al. Phosphorylation-regulated cleavage of the reticulon protein Nogo-B by caspase-7 at a noncanonical recognition site. Proteomics. 2007;7(24):4457–4467. doi:10.1002/pmic.200700499
117. Teng FY, Tang BL. Nogo/RTN4 isoforms and RTN3 expression protect SH-SY5Y cells against multiple death insults. Mol Cell Biochem. 2013;384(1–2):7–19. doi:10.1007/s11010-013-1776-6
118. He W, Huang X, Berges BK, et al. Artesunate regulates neurite outgrowth inhibitor protein B receptor to overcome resistance to sorafenib in hepatocellular carcinoma cells. Front Pharmacol. 2021;12:615889. doi:10.3389/fphar.2021.615889
119. Bai T, Wang S, Zhao Y, et al. Haloperidol, a sigma receptor 1 antagonist, promotes ferroptosis in hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2017;491(4):919–925. doi:10.1016/j.bbrc.2017.07.136
120. Jin M, Shi C, Li T, et al. Solasonine promotes ferroptosis of hepatoma carcinoma cells via glutathione peroxidase 4-induced destruction of the glutathione redox system. Biomed Pharmacother. 2020;129:110282. doi:10.1016/j.biopha.2020.110282
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.