")
Back to Journals » Journal of Experimental Pharmacology » Volume 12
Molecular Targeting and Rational Chemotherapy in Acute Myeloid Leukemia
Authors Pourrajab F, Zare-Khormizi MR, Hekmatimoghaddam S , Hashemi AS
Received 17 March 2020
Accepted for publication 15 May 2020
Published 29 May 2020 Volume 2020:12 Pages 107—128
DOI https://doi.org/10.2147/JEP.S254334
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bal Lokeshwar
Fatemeh Pourrajab,1,2 Mohamad Reza Zare-Khormizi,3 Seyedhossein Hekmatimoghaddam,4,5 Azam Sadat Hashemi4,6
1Nutrition and Food Security Research Centre, Shahid Sadoughi University of Medical Sciences, Yazd, Iran; 2Department of Clinical Biochemistry and Molecular Biology, School of Medicine, Shahid Sadoughi University of Medical Sciences, Yazd, Iran; 3School of Medicine, Shahid Sadoughi University of Medical Sciences, Yazd, Iran; 4Hematology & Oncology Research Center, Shahid Sadoughi University of Medical Sciences, Yazd, Iran; 5Department of Laboratory Sciences, School of Paramedicine, Shahid Sadoughi University of Medical Sciences, Yazd, Iran; 6Department of Pediatrics, School of Medicine, Shahid Sadoughi University of Medical Sciences, Yazd, Iran
Correspondence: Seyedhossein Hekmatimoghaddam
Department of Advanced Medical Sciences and Technologies, School of Paramedicine, Shahid Sadoughi University of Medical Sciences, Yazd, Iran
Tel +0098(913)3518314
Email [email protected]
Azam Sadat Hashemi
Department of Pediatrics, School of Medicine, Shahid Sadoughi University of Medical Sciences, Yazd, Iran
Tel +0098(913)1532465
Email [email protected]
Abstract: Acute myeloid leukemia (AML) is a molecularly complex disease with multiple aberrant genetic pathways involved in its pathogenesis. Approximately one-third to one-half of patients with AML would relapse, and no standard therapy is established for relapsing and/or refractory AML (RR-AML) yet. It is unlikely that blockage of only one specific pathway will lead to prolonged remissions and cures in all fractions of the AML patients population. Nowadays, novel therapeutic agents with rational combination are being recognized which improve the cure rate for relapsed AML. These drugs and their metabolites impart unique properties in the interaction with each of the intracellular targets and metabolic enzymes whereby resulting in unique clinical activity. To date, most of the combinations have used a targeted agent combined with standard agents such as anthracyclines, cytarabine, or hypomethylating agents to improve the outcome. Rational combinations of DNA damage-inducing therapies with DNA methyltransferase and histone deacetylase inhibitors synergistically enhance the DNA damage, growth inhibition and apoptosis of myeloid cells. This review makes a thorough look at current antineoplastic agents for AML with emphasis on its genetics and molecular mechanisms of action and the role of combination regimens.
Keywords: antineoplastic agents, drug therapy, combination, genetics, leukemia, myeloid, acute, molecular targeted therapy
Outcome of Acute Myeloid Leukemia (AML)
Acute myeloid leukemia (AML) is a form of blood cancer derived from myeloid progenitor cells. It shows a rapid clinical course by infiltrating the bone marrow (BM), blood, and other tissues. The current therapy for newly diagnosed patients with AML involves induction therapy with myelo-suppressive cytotoxic agents including cytarabine and an anthracycline or anthracinedione drug, and post-remission intensification with chemotherapy or hematopoietic stem cell transplantation (HSCT).1–3 Standard induction regimens include cytarabine for 7 days with an anthracycline for 3 days (7 + 3). These standard induction regimens achieve complete remission rates (CRRs) of 60–80% in adult patients. Unfortunately, many patients (between one-third and one-half of patients) will relapse, with an estimated 5-year disease-free survival (DFS) of 20–40%. Until now, no standard therapy is recognized for patients with relapsed and/or refractory AML (RR-AML).4–6 Studies on pediatric AML cases have reported dramatically improved remission induction rates of approximately 90% and five-year event-free survival of about 50% with the advent of anthracycline-based chemotherapy regimens and improvements in supportive care.7,8 In contrast, the natural course of AML in patients older than 60 years is dismal, that is, approximately CRRs of 40–65%, relapse rates of 60–85% within 2–3 years of diagnosis, median overall survival (OS) of less than 6 months, and a 5-year OS of only 3–8%.9 Together with post-remission therapy (additional chemotherapy and/or HSCT, 5-year survival rates of <5–20% and >40% are achieved for patients older and younger than 60 years, respectively.10 About 25% of patients with AML fail to respond to initial therapy (refractory AML), and about 50% of patients who respond initially to treatment would relapse after a transient remission (relapsed AML). These RR-AML patients show a poor outcome with a median survival of less than 6 months and less than 10% 3-year overall survival rate (OSR).1,7 However, patients with acute promyelocytic leukemia (APL) fare substantially better than other subtypes of AML in response to targeted therapy based on all-trans retinoic acid (ATRA) combined with cytarabine or arsenic trioxide. They achieve CR and long-term remission rates of >90 and >80%, respectively. APL is driven by fusion proteins acting as transcription factors and involving the retinoic acid receptor alpha (RARA).10,11 Despite the advent of modern chemotherapy, the prognosis of patients with AML has remained poor, depending on the patient’s age and the presence or absence of specific somatically acquired genetic alterations. The poor prognosis and the substantially lower long-term survival rates in AML are due to either relapse or drug resistance. In light of the poor survival reported by studies, continued efforts to explore effective therapies are greatly important in RR-AML. The treatment of RR-AML remains one of the most formidable challenges in oncology today. Todays, novel therapeutic strategies are needed to improve the cure rate of patients with RR-AML wherein the duration of the first remission remains the strongest predictive factor for the outcome with salvage therapy.3–5
Combination Regimens and Salvage Chemotherapy of AML
The patients with AML who have received the standard 7 + 3 protocol as initial induction regimen and show primary refractory response (defined PR-AML) receive salvage regimen at first relapse. According to different properties of each drug, various combined chemotherapy regimens have been developed for salvage therapy after the first relapse. The CRR for those with early relapse, defined usually as relapse less than 6–12 months from induction treatment, ranges from 18% to 41%. The CRR for those with late relapse usually defined as relapse after 6–12 months of induction treatment, is 46% to 83%. The overall survival, however, is 10–29% at 3 years.1,12 However, long-term survival for children with relapsed AML ranges between 20% and 33%.7 In patients with RR-AML, various combination regimens including cytarabine, granulocyte colony-stimulating factor (G-CSF), and fludarabine (FLAG); cytarabine, G-CSF and aclarubicin or daunorubicin (CAG/DAG); cytarabine, G-CSF, and cladribine (CLAG); cytarabine, etoposide, and mitoxantrone (MEC); cytarabine plus idarubicin, daunorubicin, or mitoxantrone (IA/DA/MA); and homoharringtonine, cytarabine, and aclarubicin or daunorubicin (HAA/HAD) have been investigated as salvage therapy.1,4 In salvage therapy, regimens that contain high-dose cytarabine in combination with other agents such as mitoxantrone, etoposide, fludarabine, or cladribine have shown superior activity. In RR-AML, regimens containing CLAG or MEC have been reported to yield significant CRR of 50% and 25–60%, respectively.4 It has been reported that patients receiving the CLAG-based regimen achieved higher overall response rates (ORRs) and significant favorable OS.1,4 In a study on patients aged ≤65 years, 36% of the CLAG patients proceeded to allogeneic HSCT vs 25% of the MEC patients. In another study on patients with primary refractory AML (PR-AML) who received salvage regimens CLAG and MEC as the first re-induction therapy, the achieved CRR was 45.5% vs 22.2%, respectively. The median OSR for the refractory disease was reported 11.0 months for CLAG and 4.5 months for MEC. There are additional reports from RR-AML patients that regimens containing CLAG or MEC yield superior CRRs of 50% and 25%, respectively.4,12 Therefore, CLAG-based regimens have been introduced as a prioritized treatment option for RR-AML patients.1,4,12 Similarly, in a study in pediatric patients with RR-AML, a combination of clofarabine/cytarabine in the non-anthracycline-based treatment showed a clinically significant activity with good tolerability and overall response rate. In that study, a dose of 52 mg/m2/day for 5 days of clofarabine in combination with cytarabine was used. This salvage therapy with clofarabine/cytarabine combination yielded an acceptable response rate (complete remission plus complete remission with partial platelet recovery) of about 48% without excess toxicity in these high-risk children with RR-AML. Patients were found to have no minimal residual disease at the end of the first cycle by flow cytometric analysis, and responders had a 1-year superior OS of 100% vs 38% in nonresponders. The significance of “bridge to transplant” was demonstrated by the 3-year OS rate of 46% ± 27% for responders versus 16% ± 16% for nonresponders. The nearly 50% survival rate reported in high-risk children with RR-AML is highly encouraging and shows that this combination is an effective bridge to HSCT.8 Also, in a Phase I trial conducted on pediatric patients with RR-AML, it was concluded that the rational combinations of clofarabine with cyclophosphamide and etoposide have a superior effect than a single-agent trial of clofarabine. The overall response rate for patients treated with a single-agent trial of clofarabine was 38% vs 100% in rational combinations of clofarabine with cyclophosphamide and etoposide (Table 1).7 From a molecular point of view, clofarabine, fludarabine (in FLAG) and cladribine (in CLAG regimens) are all purine nucleoside analogues (Table 2) which possess similar structures and share the same functional mechanisms in increasing the concentration of Ara-CTP, which is the active metabolite of cytarabine and cytotoxic to leukemic cells; however, they present some differences in the interaction with DNA metabolizing enzymes. These purine nucleoside analogues are utilized during consolidation therapy for RR-AML, which act as inhibitors of DNA repair and would enhance the cytotoxicity of DNA-damaging agents such as cyclophosphamide, anthracyclines and etoposide.1,6,7 Studies demonstrate that fludarabine/cytarabine combinations also have efficacy in RR-AML patients. In this regard, combinations of fludarabine, cytarabine, and G-CSF (FLAG) or FLAG with daunorubicin in patients >10 years old who developed disease recurrence after a variety of de novo therapeutic regimens after two cycles, showed CRRs of 59% and 69%, respectively. Additionally, a combination of DNA-intercalating agent mitoxantrone and cytarabine in RR-AML children yielded an ORR of 58%.8 Based on preclinical studies, FLAM regimen consisting of flavopiridol, cytarabine, and mitoxantrone shows synergism with standard chemotherapeutic agents and yields significantly higher CRRs in comparison to 7 + 3 protocol in newly diagnosed younger adults with intermediate and poor-risk AML, although no differences in OS or event-free survival (EFS) have been reported. Herein, stronger and alternative subsequent chemotherapy has been recommended for patients with CR to maintain longer CR and better OS. An intensive post-remission therapy that included conventional chemotherapy with the combination regimen is recommended to be followed in patients who achieved CR.5,12 Accordingly, a combination of low-dose cytarabine and an alkaloid ester called homoharringtonine (HHT) with G-CSF (CHG regimen) would show considerable efficacy in patients with advanced myelodysplastic syndromes (MDS) or MDS-transformed acute myeloid leukemia (t-AML). The efficacy of HHT has been reported in de novo and relapsed AML and in chronic myeloid leukemia, too. In a study after one course of CHG medication, 46.9% of patients achieved CR and 25.0% achieved PR, with an ORR of 71.9%. The response rate among patients aged >70 years was 78.6% (64.3% CR and 14.3% PR), whereas in patients aged <70 years the response rate was reported 66.7% (33.3% CR and 33.3% PR). Moreover, there was no severe non-hematological toxicity or myelosuppression which are usually observed in patients under conventional induction chemotherapies. Therefore, an induction therapy consisting of CHG priming regimen was recommended, since it is well tolerated and effective in patients with advanced MDS or t-AML. So, stronger and alternative subsequent chemotherapy was used for patients with CR to maintain longer CR and better OS. Importantly, in post-remission chemotherapy of patients achieving CR, combined mitoxantrone or idarubicin with cytarabine besides homoharringtonine and daunorubicin caused a CR duration of about 10 months in 64% and continuous CR in 36%, whereas those receiving only homoharringtonine and cytarabine or daunorubicin and cytarabine all suffered a relapse and had a mean CR of about 6 months.1,12
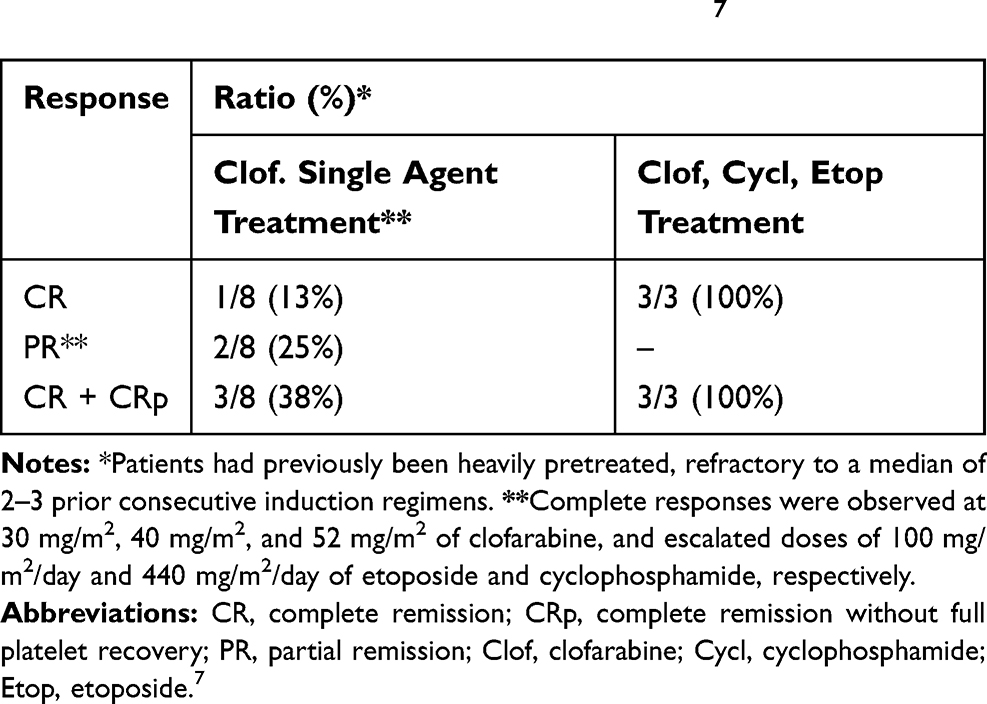 |
Table 1 Superior Response to Combination of Clofarabine/Cyclophosphamide/Etoposide in Pediatric Patients with Relapsed and Refractory AML in Phase I Trials7 |
 |
Table 2 FDA approved purine /pyrimidine anti-metabolites used in the combination therapy of AML6 |
Molecular Mechanisms in AML & Combination Therapies
Like other malignant diseases, AML is a result of somatically acquired genetic lesions (eg, numerical and structural chromosome aberrations and single nucleotide mutations), accumulated in hematopoietic stem cells (HSCs) during the lifetime of an individual which consequently lead to the formation of preleukemic stem cells. Additional mutations in these cells are required, often in genes coding for signaling proteins or transcription factors, to promote the transformation to leukemic stem cells and, finally, overt AML. Over 100 gene mutations and genetic abnormalities accumulated in a specific order have been reported in AML, reflecting the heterogeneity of the disease and the presence of multiple active pathogenetic pathways. Remarkably, there are a subset of healthy individuals exhibiting low levels of clonal hematopoietic cells that could, but did not necessarily, involve early leukemogenic driver mutations. Accordingly, these affected persons carry a substantially increased, albeit in absolute terms still low, risk to develop hematological malignancies. Furthermore, early mutations would exist in phenotypically and functionally normal HSCs in a substantial proportion of AML patients, which often persist in remission. Thus, a large proportion of stem cells whose a subset may already have attained molecular changes (sometimes very small), or may have emerged during cytostatic therapy, can regrow and lead to post-remission recurrence.2,3,10,11 Mutations that activate signal transduction pathways lead to proliferative survival advantages and may be targeted with small molecule inhibitors of the specific mutations, such as FLT3, C-Kit, and RAS, while mutations that lead to inhibition of differentiation may be targeted with differentiating agents (eg ATRA for patients with APL). Targeted agents need therefore to be used in combination with each other and with cytotoxic agents to inhibit the multiple pathways present in AML and to improve response rates (Figure 1) (Table 3). For instance, FLT3-targeted tyrosine kinase inhibitors target signal transduction pathways of cell growth, while deoxynucleoside anti-metabolites would target DNA synthesis and repair which impair DNA metabolism.5,7,11 Regarding DNA repair and its crucial roles in the development and progression of a wide array of cancers and their response to therapy, it is not surprising that there are increased efforts to validate DNA damage response and repair proteins as therapeutic targets and develop agents against these targets (Figures 1 and 2).11,13 The repair of DNA damage relies on particular pathways classified into the excision repair, nucleotide excision repair (NER), base excision repair (BER), and mismatch repair (MMR) (Figure 2). Additionally, the homologous recombination (HR) repair or homology-directed repair (HDR) is a major pathway in DNA damage in which the damage is not necessarily removed, but tolerated by using homologous template sequences to synthesize the sequence complementary to the damaged area of the genome. Importantly, the ability of cancer cells to repair therapeutically induced DNA damage impacts drug efficacy. Many cytotoxic agents including the widely prescribed anthracyclines, cyclophosphamide and cisplatin impart their clinical efficacy via the induction of DNA damage. This has led to targeting DNA repair pathways and proteins to develop anti-cancer agents that will increase sensitivity to traditional chemotherapeutics (Figure 2) (Table 3).9,13,14 Herein, cisplatin, as a cytotoxic agent in the contest of high-risk RR-AML and in combination with high-dose cytarabine and etoposide has shown effective salvage chemotherapy. Cisplatin is a DNA-damaging agent that is widely used in cancer chemotherapy of solid tumors and has successful therapeutic outcomes for head and neck, lung, ovarian, and testicular cancers. The traditionally accepted mechanism of action of cisplatin involves its cross-linking to DNA, forming intra- and interstrand adducts (Figure 2), which unwind the duplex and attract high-mobility-group domain and other proteins. The shielding effect of these proteins results in the poor repair of the cisplatin-modified DNA, thereby leading to activation of several signal transduction pathways (including those involving ATF, p53, p73, and MAPK) and ultimately cell apoptosis.14,15
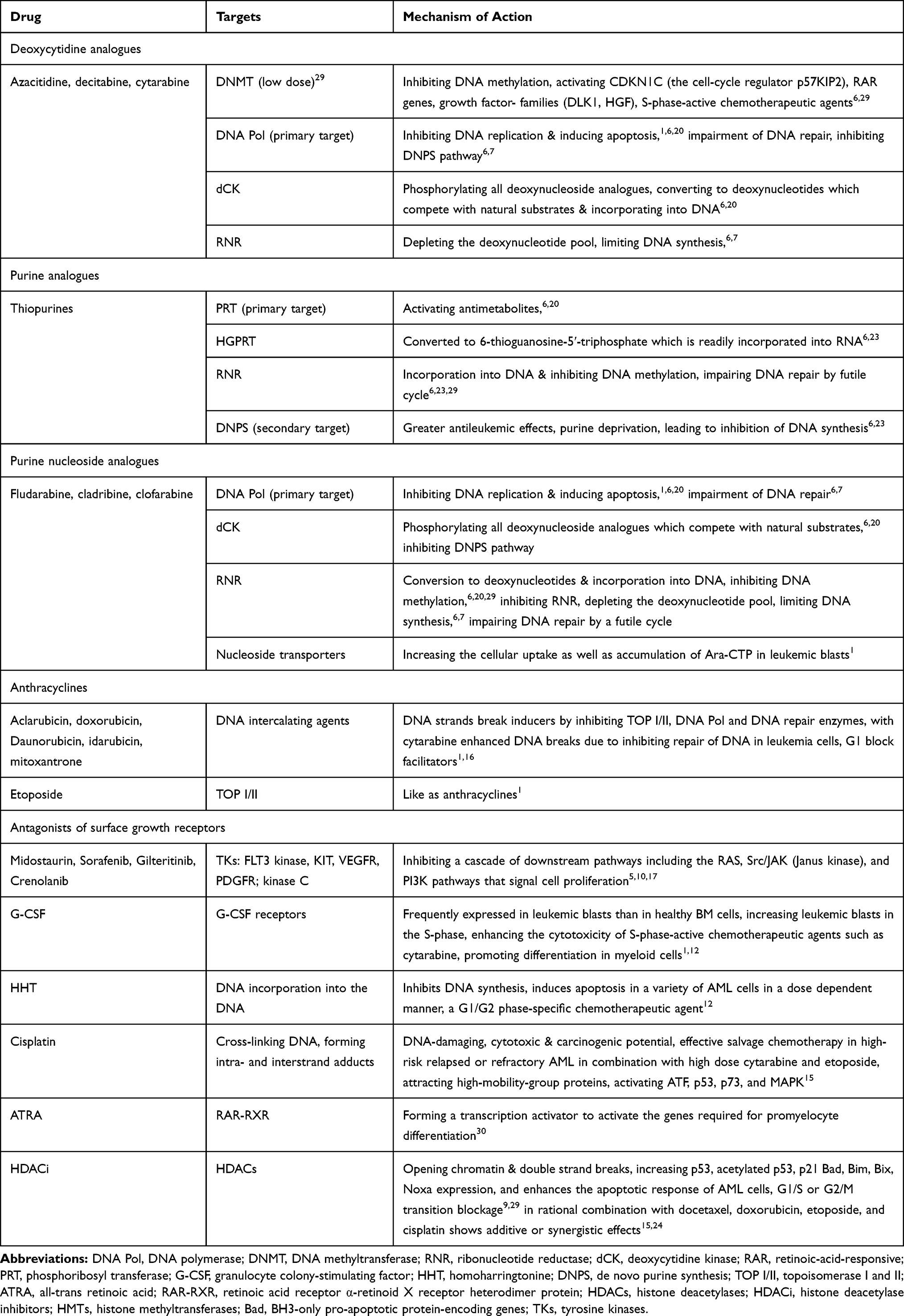 |
Table 3 A Summary of Anti-AML Drugs, Targets and Mechanisms of Action |
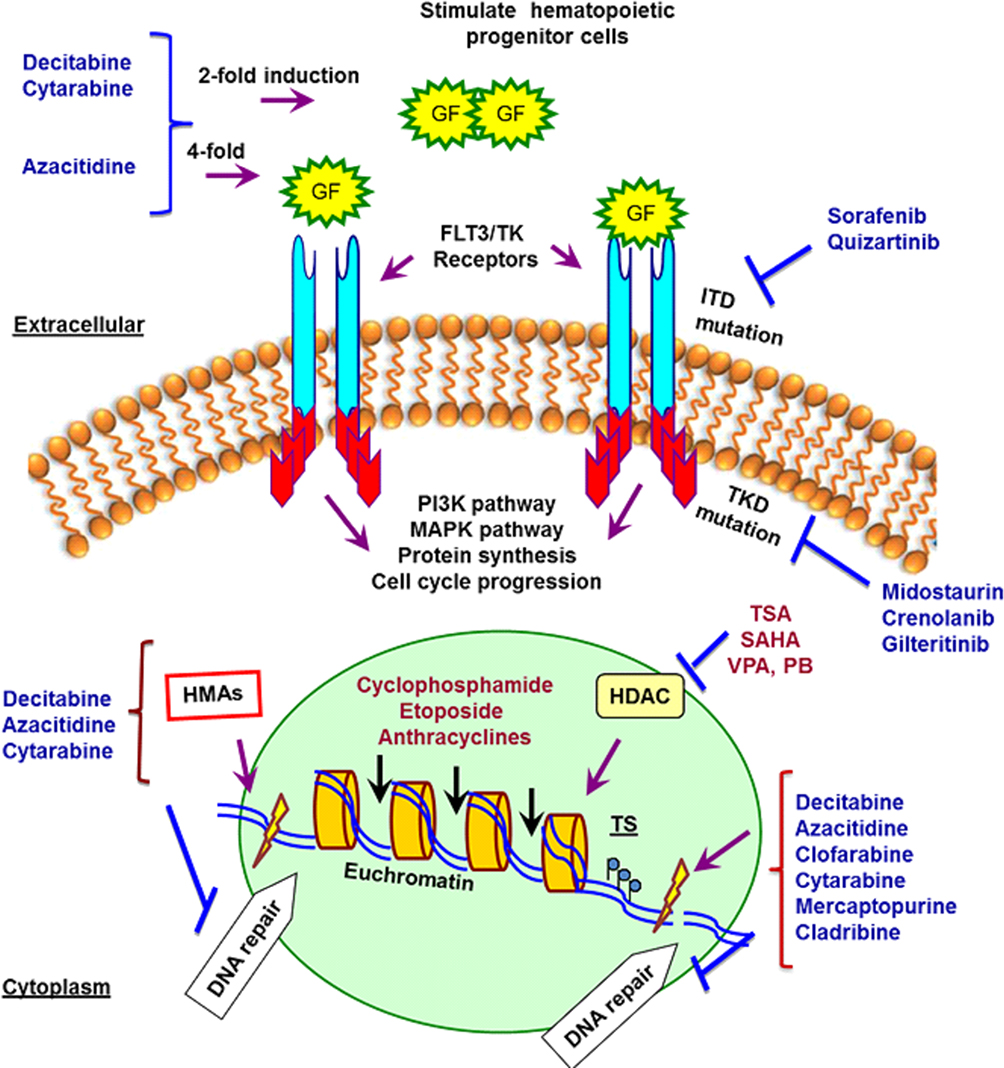 |
Figure 1 Summary of agents that target genome, epigenome, and signal transduction pathways in a cancerous hematopoietic cell. Inhibition of HDACs and TKRs in cancer cells displays anti-proliferative and pro-apoptotic activities in drug-resistant in vivo tumor models. The mechanisms of deoxynucleoside anti-metabolites are quite similar. They are converted to their respective nucleotide analogues which inhibit DNA synthesis, repair and methylation by inhibition of DNA polymerases and DNMTs besides inducing DNA damages by incorporation into DNA. The incorporation of these agents into DNA is difficult to repair and causes a lasting inhibition of DNA synthesis or disruption of DNA function. HDACi increases the chromatin accessibility for DNA-damaging agents and Top II inhibitors and down-regulation of DNA repair. Abbreviations: HDACs, histone deacetylases; TKRs, tyrosine kinase receptors; FLT3, FMS-like tyrosine kinase 3; ITD, FLT3-internal tandem duplications (FLT3-ITD mutations in or near the juxtamembrane domain); TKD, FLT3-tyrosine kinase domain mutations; DNMTs, DNA methyltransferase; SAHA, vorinostat; TSA, trichostatin A; VPA, valproic acid; PB, phenylbutyrate; HMAs, hypomethylating agents. |
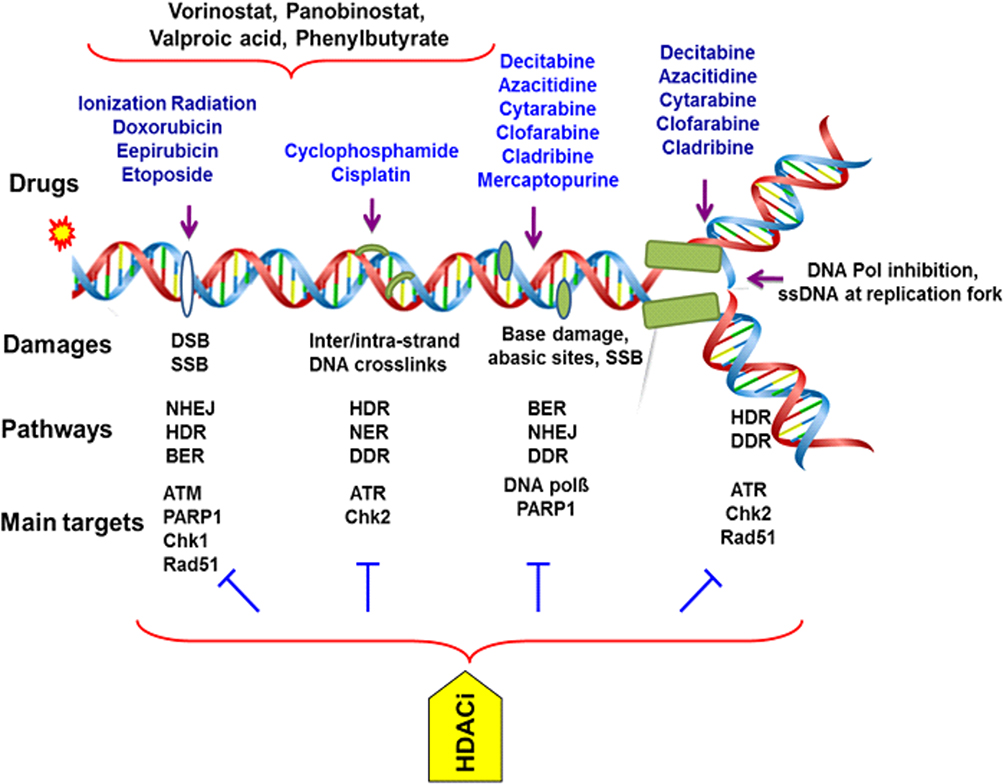 |
Figure 2 Targeting DNA and its repairing pathways by chemotherapy agents in AML. Therapeutics agents commonly impart their clinical efficacy via the induction of DNA damage or targeting DNA repair factors directly or indirectly. The repair of DNA damage relies on particular pathways to remedy specific types of damage to DNA. The range of insults to DNA includes small, modest changes in the structure including mismatched bases and simple methylation events to oxidized bases, intra- and interstrand DNA crosslinks, DNA double-strand breaks and protein–DNA adducts. HDACi causes down-regulation of dsDNA-break repair by affecting components of both NHEJ and HR repair pathways. Anthracyclines are widely used with deoxynucleoside antimetabolites where they exert their anti-leukemia effects through interacting with Top II and inhibiting replication and repair of DNA in leukemia cells, respectively. HDACi increases the chromatin accessibility for DNA-damaging agents and Top II inhibitors and down-regulation of DNA repair. Cisplatin and cyclophosphamide form intra- and interstrand adducts and leading to cross-linking of DNA. Abbreviations: AML, acute myeloid leukemia; NER, nucleotide excision repair; BER, base excision repair; HDR, homology-directed repair; FA, Fanconi anemia; HR, homologous recombination; DDR, DNA damage response; NHEJ, non-homologous end joining; SSBs, single-strand breaks; DSB, double-strand breaks; PARP, poly(ADP-ribose) polymerase; NAD+, nicotinamide adenine dinucleotide; PARPi, PARP inhibitors; AMLXP, xeroderma pigmentosum; ATM, ataxia telangiectasia mutated kinase; ATR, ATM-Rad3 related kinase; Chk, checkpoint kinase; DNA pol, DNA polymerase; DNA-PK, DNA-dependent protein kinase; Top II, topoisomerase II; HDACi, Histone deacetylase inhibitors. |
Anthracyclines, DNA-Intercalating Therapeutics of AML
Anthracyclin and cytosine arabinoside-based chemotherapy are widely used in the treatment of AML with successful CR in the majority of patients with AML; however, approximately 50% of patients receiving this chemotherapy will relapse within 1 to 2 years, and 5-year survival rates for patients <60 years old remain less than 50%.9,16 The anthracycline compounds seem to activate multiple signaling events including a sphingomyelinase-initiated sphingomyelin-ceramide pathway, mitogen-activated kinase and stress-activated protein/c-Jun N-terminal kinase signaling, Fas/Fas-ligand pathway and transcription factors activation such as nuclear factor-kB.16 In fact, the most known mechanisms of action of anthracyclines are to inhibit DNA and RNA synthesis by intercalating between base pairs of the DNA/RNA strand, thus preventing the replication of rapidly growing cancer cells. They also inhibit the activity of topoisomerase II (Top II) enzyme, which ultimately leads to broken DNA chains. The major cytotoxicity effect mediated by anthracyclines is through the drug-induced double-strand breaks to DNA. This damage is mediated by intercalation-induced distortion of the double helix, or stabilization of the cleavable complex formed between DNA and Top II (Figures 2 and 3) (Table 3).9 Studies display that anthracycline-induced apoptosis in AML cells also involves the induction of the WAF1/CIP1 p21 pathway. The WAF1/CIP1 p21 protein is a strong inhibitor of cyclin-dependent kinases involved in G1 to S transition, a mechanism that may account for G1 block after anthracycline exposure in p53 proficient cells.16 Anthracyclines are not the only antitumor agents that activate the DNA damage pathways; indeed, other genotoxic agents such as ultraviolet ray, ionizing radiation, alkylating agents, etoposide and nucleoside anti-metabolites as well as nongenotoxic agents such as microtubule poisons also activate these pathways. Observations suggest a general DNA damage pathway in mediating cell death induced by antitumor agents.6,14,16 In this regard and as mentioned above, the alkaloid ester HHT in combination with the low-dose cytarabine and G-CSF (CHG regimen) exhibits considerable efficacy in patients with advanced MDS, t-AML, de novo and relapsed AML and in chronic myeloid leukemia. Herein, HHT induces apoptosis in a variety of AML cells in a dose-dependent manner, by incorporation into the DNA of leukemic cells and inhibition of DNA synthesis. HHT is a G1/G2 phase-specific chemotherapeutic agent, whereas cytarabine is an S-phase-active chemotherapeutic agent. On the other side, G-CSF receptors are more frequently expressed in leukemic blasts than in healthy BM cells, wherein may increase the fraction of leukemic blasts in the S-phase, thereby enhancing the cytotoxicity of S-phase-active chemotherapeutic agents such as cytarabine and reducing the LC50 of cytarabine. Besides effectively inhibiting leukemic colony formation (by 50%), HHT also induces the endoplasmic reticulum stress pathway.1,12
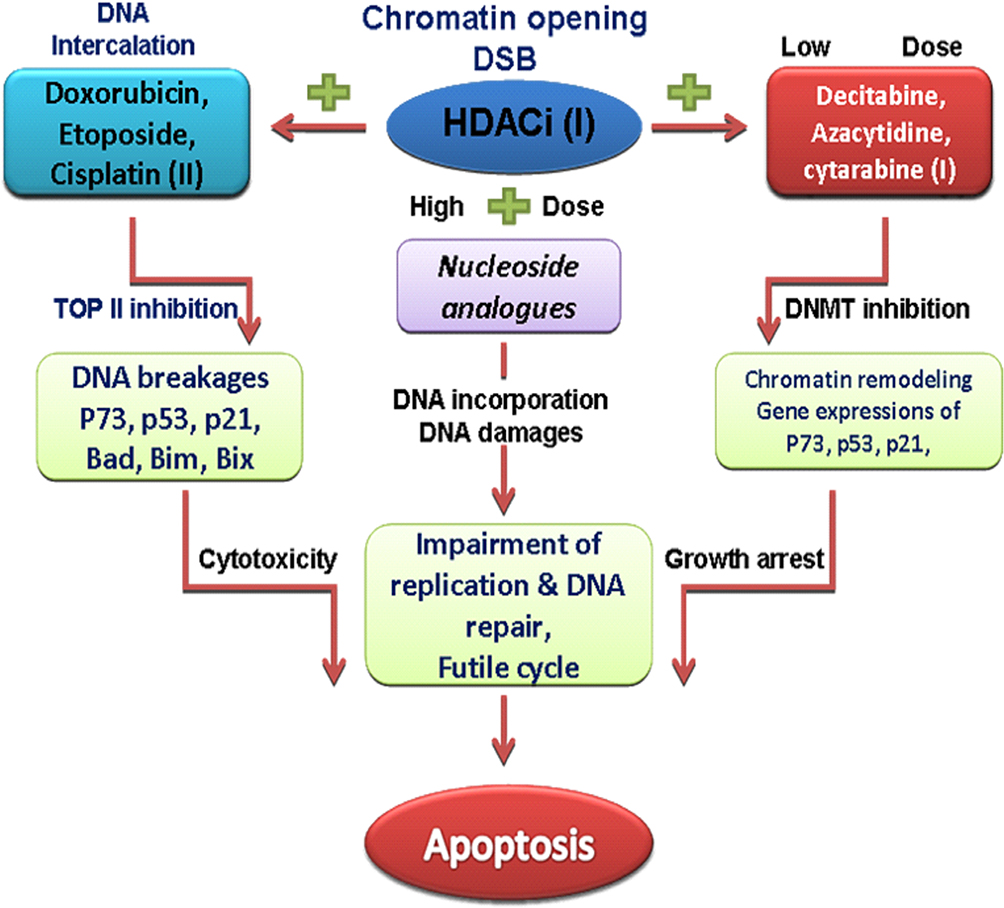 |
Figure 3 The sequence-specific administration of HDAC and DNMT inhibitors prior to topoisomerase II–inhibitor administration is of an utmost importance in the chemotherapy of AML. Concurrently and continuously administration of HDACi and DNMTi before chemotherapy sensitizes tumor cells to genotoxic agents, and synergistically increases expression of silenced tumor suppressors and promotes cell death and differentiation. Cisplatin, doxorubicin, etoposide are DNA-damaging agents and topoisomerase II (TOP II) inhibitors which induce DSB in the DNA of cancer cells and induce cytotoxic effects. Abbreviations: HDACi, histone deacetylase inhibitors; DNMTi, DNA methyltransferase inhibitors; DSB, DNA strand breaks. |
Topoisomerase Inhibitors, DNA Damage-Inducing Therapeutics of AML
Topoisomerases (Top) catalyze the cleavage of DNA backbone in order to facilitate DNA unwinding during replication. Topoisomerase targeting causes topoisomerase-mediated DNA strand breaks.14 Several cytotoxic Top II inhibitors are currently supplied in salvage chemotherapy for RR-AML.1,4,5 The Top I inhibitors; irinotecan, topotecan, karenitecin and camptothecin are used for colorectal, gastric, lung and cervical cancers. However, inhibitors of Top II like anthracyclines (eg doxorubicin), epirubicin, etoposide, and mitoxantrone are used in the treatment of lymphomas, leukemias and breast cancer.13,14 By inducing DNA damage and inhibiting repair of DNA in leukemia cells, anthracyclines daunorubicin and idarubicin are widely used in combination with cytarabine in RR-AML. Etoposide, a direct Top II inhibitor, and mitoxantrone which is a DNA-intercalating agent and acts similar to anthracyclines, are both used in salvage therapy for the treatment of RR-AML patients (Table 3) (Figures 2 and 3).1,4 TOP II inhibitors especially induce dsDNA breaks. DNA repair pathways involved in repairing dsDNA-breaks include ATM, BRCA1, CHK2 and Ku70–Ku80 heterodimers whose recruitment to the site of damage leads to their activation and initiation of DNA repair response. Downstream of these pathways, there is the DNA damage response protein p53 phosphorylated by ATM.13,14 The oligosaccharide aclarubicin is another anthracycline inhibiting DNA replication which as well repairs DNA by interacting with Top I/II. Regardless of gene status, it is effective against multidrug resistance and shows lower cardiac toxicity than doxorubicin.1,4 Additionally, the combination of low-dose cytarabine and aclarubicin with the concurrent administration of G-CSF (CAG regimen) is efficacious and recommended in the treatment of RR-AML as well as in the treatment of high-risk MDS and t-AML. In old patients with AML and t-AML and also in patients with advanced MDS, the CAG regimen has achieved a high response rate. Myelosuppression and other toxicities have been apparently lower and less frequent in patients with the CAG regimen.1,12 Importantly, in patients >60 y old, with relapse AML or refractory to 1 or two cycles of previous anthracycline-containing induction chemotherapy, a combination of vosaroxin and cytarabine has shown the greatest benefit. These patients survived after a median of 39.9 months of follow-up. This has been particularly observed among those with refractory disease or early relapse. Vosaroxin is an anti-cancer quinolone derivative that intercalates DNA and inhibits Top II, thereby inducing cell-cycle arrest and p53-independent apoptosis. The recommended dose of vosaroxin in old patients is 70 mg/m2 on days 1 and 4, in combination with standard dose decitabine as the preferred dose.5
Nucleoside Anti-Metabolites, Inhibitors of DNA Metabolism in AML
There are three primary classes of FDA-approved purine and pyrimidine antimetabolites: thiopurines, fluoropyrimidines, and the deoxynucleoside analogues (the largest class and the most efficient compounds) (Table 2). The basic mechanism of action of purine and pyrimidine antimetabolites is quite similar (Figures 1 and 2). These compounds are diffused into the cells mainly through the membrane nucleoside transporters and are converted into their respective nucleotide analogues of cellular nucleotides by enzymes of the purine or pyrimidine metabolic pathways. The nucleotide analogues then inhibit one or more enzymes that are critical for DNA synthesis, DNA repair and replication, culminating in DNA damage and induction of apoptosis. They inhibit DNA replication and repair by inhibiting DNA polymerases and ribonucleotide reductase, as well as inhibiting DNA methylation by DNMTs.6,17,18 The incorporation of these agents into DNA is the most important aspect of their mechanism of action. Drug incorporation into DNA induces an apurinic/apyrimidinic site which is recognized by the apurinic/apyrimidinic endonuclease 1 (APE 1), and causes a DNA-single strand break damage. DNA repair enzymes then recognize single-strand break, where a futile cycle of re-repair and re-synthesis is initiated (Figure 2). Mismatch repair enzymes recognize incorporated antimetabolite, which also consequently causes a futile cycle of repair and results in lethal DNA damage.6,13,19 On the other side, human DNA polymerases α, δ and Є involved in DNA replication and repair are the primary targets of the nucleoside analogues (Figure 2).6 So, the analogues’ incorporation is difficult to repair and causes a lasting inhibition of DNA synthesis or disruption of DNA function.6,13 However, in spite of some similarities, each of these agents and their metabolites imparts unique properties and results in unique clinical activity. There are differences in the interaction of these agents and their metabolites with various metabolic enzymes and intracellular targets.17–19 For example, clofarabine demonstrates more excellent efficacy in the treatment of pediatric RR-acute lymphoblastic leukemia/AML than cladribine which differs in only one fluorine atom (Table 2). Clinical results indicate that small structural modifications of nucleoside analogues can have profound effects on their chemical stability and biological activity.12,20 Higher anticancer activity of a drug is also associated with higher affinity for nucleoside transporters, deoxycytidine kinase (dCK, a constitutively expressed key cytosolic enzyme involved in the salvage pathway of DNA synthesis), and key enzymes involved in DNA synthesis, eg ribonucleotide reductase (RNR) and DNA polymerase α, δ and Є which are primarily involved in chromosomal replication. In this regard, the primary target of the deoxynucleoside analogues is the DNA polymerases involved in DNA replication.12,13,21 Although there are five kinase enzymes in human cells phosphorylating deoxynucleoside analogues (eg deoxycytidine and deoxyguanosine kinase, thymidine kinase 1/2, and 5′-nucleotidase), but among them, the primary rate-limiting enzyme for activation of most of the approved nucleoside analogues is dCK. This enzyme recognizes and phosphorylates all deoxynucleoside analogues, preferred as natural substrates.12,22 A known molecular mechanism underlying antimetabolite resistance is reduced activity of the dCK, as has previously been found to be the case with cladribine. Therefore, the primary or acquired drug resistance in AML treatment can be due to alteration in the activity of one or more actors in the metabolic pathway of these antimetabolites.13
Deoxycytidine Analogues (Cytarabine, Azacitidine, Decitabine)
Cytarabine and its analogs azacytidine (aza-Cyd) and decitabine (aza-dCyd) are primary components of numerous chemotherapy regimens in AML and play a crucial role in treating RR-AML patients (Table 2). A combination of cytarabine with other cytotoxic agents or hematopoietic growth factors is often used as an intensive chemotherapy regimen of AML or as salvage therapy for RR-AML.1,4,6,12 High-dose (HD) cytarabine has been applied as a post-remission treatment for decades; however, recent recommendations favor lower doses which incur less toxicities. It is proposed that HD cytarabine given as consolidation treatment for patients with AML has no significant impact on survival parameters in comparison to intermediate-/low-dose (ID/LD) cytarabine. There are treatment trials with cumulative HD cytarabine doses between 20,000 mg/m2 and 72,000 mg/m2 and cumulative ID/LD cytarabine doses between 700 mg/m2 and 18,000 mg/m2, while RFS and OS rates at 3–5 years show the statistically insignificant difference between them (in both patient groups, those of age ≤64 or >64 years).22 Once cytarabine and its analogs enter into leukemic cells, they form triphosphate products, which inhibit DNA polymerase and ultimately induce apoptosis by terminating the chain elongation of DNA.1,6 Cytarabine has been mostly administered in combination with DNA-damaging/Top II–inhibitor drugs daunorubicin, idarubicin, aclarubicin, azathioprine, etoposide, cyclophosphamide, and mitoxantrone.22 Cytarabine, decitabine and azacytidine at low doses (75 mg/m2/day × 5) exhibit DNMT-inhibitory activity while high doses of drug (1.5 g/m2/day × 4) act as a cytotoxic chemotherapeutic for the treatment of AML (Figure 3).6,17 Azacytidine and decitabine are two DNMT inhibitors that have been extensively used in clinic to treat AML and MDS (Tables 3 and 4).17,20 They are incorporated into newly synthesized DNA strands, and lead to a marked decrease in DNMT activity, which may be the catalyst that allows differentiation to proceed. Replacement of deoxycytidine residues by aza-dCyd inhibits DNA methylation in daughter cells, eventually resulting in the activation of epigenetically repressed genes.6 This is important in disease states such as MDS and AML, in which immature myeloid cells are often arrested in the early stages of development (Figure 2).20 Similar to thiopurines (mercaptopurine (MP)/thioguanine (TG)), aza-Cyd/aza-dCyd also readily incorporates into DNA strands and extend into internal positions in DNA by the DNA polymerase α. Once incorporated, aza-dCyd/aza-Cyd inhibits DNA methylation (Figure 1).6,20,22 Although the primary mechanism of action of aza-dCyd/aza-Cyd is supposed to be through the inhibition of DNA methylation, at high doses it can impart other effects that may contribute to its antitumor activity (Figure 3). The mechanisms of action of deoxynucleoside analogues (eg cladribine, cytarabine and aza-dCyd/aza-Cyd) can be explained as below (Table 3):
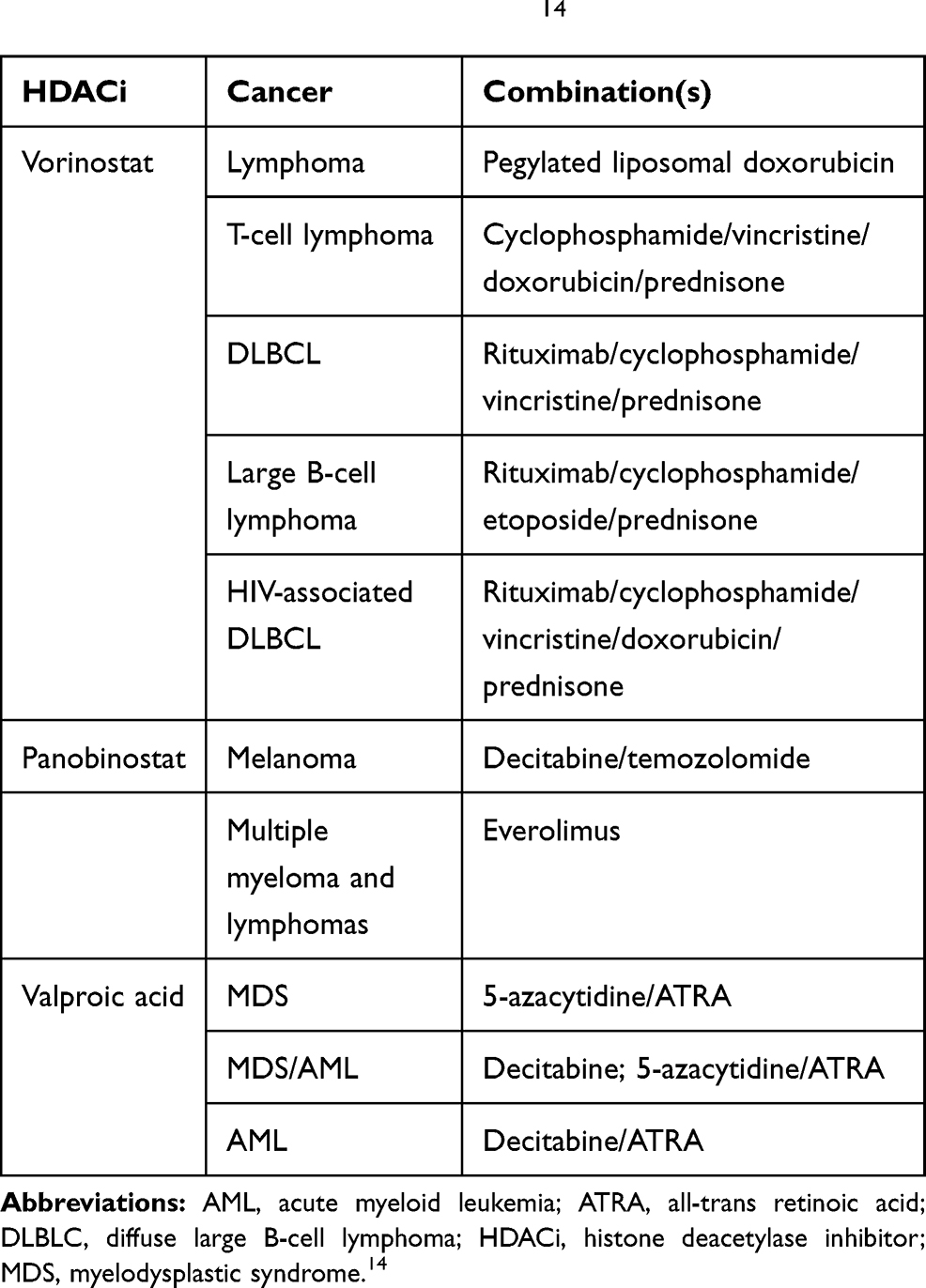 |
Table 4 Conventional Chemotherapy of Leukemia and Rational Chemotherapy Combinations with Histone Deacetylase Inhibitors in Phase II/III Clinical Trials.14 |
Purine Anti-Metabolites (Thiopurines, Clofarabine, Cladribine, Fludarabine)
Until now, five purine deoxynucleoside analogs have been approved for the treatment of cancers. Clofarabine, cladribine and fludarabine are deoxyadenosine analogs of adenosine which retain the 2-halogenated aglycone and confer resistance to cellular degradation by adenosine deaminase (Table 2). To increase the stability of adenosine analogs, the 2-hydrogen atom is replaced with a halogen atom (eg, fluorine or chlorine) which results in a molecule resistant to inactivation by adenosine deaminase. Additionally in clofarabine, the second halogen atom at the 2’ position of the arabino configuration is replaced with fluorine, which prevents cleavage of the glycosidic bond by bacterial purine nucleoside phosphorylase and stabilizes the compound in acidic environments of the gastrointestinal tract. These substitutions improve compound oral bioavailability and prevent the release of neurotoxic halogenated adenine.6–8 Likepyrimidines, purine analog triphosphates are substrates for DNA polymerases for incorporation into DNA which inhibits DNA polymerases and RNR (Table 3). Moreover, as inhibitors of DNA repair, deoxyadenosine analogues enhance the cytotoxicity of DNA-damaging agents such as cyclophosphamide and etoposide. Combinations of deoxyadenosine analogue with cyclophosphamide and etoposide are utilized during consolidation therapy for pediatric AML patients with the first relapse. Etoposide and cyclophosphamide can be escalated to their target doses of 100 mg/m2/day and 440 mg/m2/day, respectively (Figures 2 and 3).6,19,22 Herein, clofarabine, fludarabine and cladribine have been designed to integrate similar molecular mechanistic properties. They require intracellular phosphorylation by dCK to form the active dATP analogues before inhibition of DNA polymerase. The produced analogues are potent inhibitors of RNR, whereby in combination with cytarabine leads to accumulation of the cytotoxic triphosphate form of cytarabine (ara-CTP) in leukemic cells (Table 3).6–8 Ara-CTP is the active metabolite of cytarabine and its accumulation is cytotoxic to leukemic cells. Moreover, they also differ in interaction with enzymes.1,6 Fludarabine primarily inhibits DNA polymerases and cladribine mainly inhibits RNR, while clofarabine inhibits both DNA polymerases and RNR. Regarding the deoxyadenosine analog clofarabine, the inhibition of DNA polymerases leads to impairment of DNA synthesis and repair through the incorporation of clofarabine monophosphate into the DNA chain. Inhibition of RNR depletes the deoxynucleotide pool primarily of dCTP and dATP. The reduction in dCTP concentrations limits DNA synthesis, while the reduction in dATP creates a favorable environment for clofarabine triphosphate to compete with dATP for incorporation into DNA. In addition, clofarabine has a greater affinity than cladribine or fludarabine for dCK, the enzyme required for intracellular phosphorylation to its monophosphorylated form.6,19 The third mechanism of action by these compounds is by inducing DNA strand breaks (Figure 1) leading to the release of cytochrome c from mitochondria and activation of apoptotic pathways.6,7 Human studies have indicated a potent anti-leukemic nature for clofarabine. In a study, there was a consistent decline (68–99%) in the WBC count in all acute leukemia patients treated with 40 mg/m2 of clofarabine after 5 days, indicating its high potency. DNA synthesis is inhibited by 75–95% at the end of the infusion of clofarabine at doses ranging from 22.5 to 55 mg/m2. Specifically, the decrease in DNA synthesis has been shown to be associated with the accumulation of clofarabine triphosphates in the blasts of adults with refractory leukemias. There is a dose-dependent effect by clofarabine treatment.19 However, thiopurine antimetabolites (Table 2) exhibit two mechanisms of action contributing to their anticancer activity; inhibition of de novo purine synthesis and incorporation into DNA as 6-thio-2′-deoxyguanosine. Mercaptopurine (MP) is a good substrate for phosphoribosyl transferase, a primary rate-limiting enzyme in the de novo pathway. Phosphoribosyl transferases are the most important enzymes in activating thiopurine antimetabolites. MP does not inhibit DNA polymerase activity, but its incorporation into DNA results in DNA damage as its antitumor activity (Table 3).6,19,23 Full de novo purine synthesis (DNPS) inhibition would be associated with greater antileukemic effects compared with partial or no inhibition. Studies report constitutive activity of DNPS in T cell- and B cell-lineage ALL and they are more DNPS-dependent compared with other leukemias. Furthermore, the rate of DNPS in bone marrow leukemia cells is 3-fold higher in patients with T-lineage ALL compared with those with B-lineage ALL. In patients with B-lineage ALL or T-lineage ALL, just those having full inhibition of DNPS show a greater decrease in circulating leukocytes compared with those with partial or no inhibition of DNPS. The consequence of DNPS inhibition by antimetabolites is purine deprivation, leading to inhibition of DNA synthesis, decreased cell proliferation, and cytotoxicity. Methotrexate (MTX) and MP synergistically inhibit DNPS. DNPS is not consistently inhibited following MP alone but is markedly inhibited following MTX plus MP.6,23
Targeting Epigenetics in AML
Encouraging results from clinical trials using newly developed epigenetic treatments in hematological malignancies MDS, CML and AML have been obtained (Table 4) (Figure 3).24 The silencing of tumor-suppressor genes such as p16, BRCA1 and DAP kinase is observed in these malignancies, which is mediated by concert work of DNA methyltransferases (DNMTs) and histone deacetylases (HDACs).14,17 Therefore, these genes are representing rational therapeutic targets for the re-expression of silenced tumor suppressors in malignancies. A combination of both strategies (silencing epigenetics and re-expression of tumor suppressor) would improve the limited anticancer efficacy observed with either therapeutic class alone.14,25 Exposure of leukemia cell lines with extensive CpG island hypermethylation of silenced genes to a combination of epigenetic modifying drugs results in increased expression of the majority of silenced genes, whereas few genes would be down-regulated.26 Inhibition of the HDACs and DNMTs results in transcriptional activation of the corresponding genes including tumor-suppressor genes often silenced in cancers,25,27 wherein several cycles of therapeutics are required and duration of inhibitors is emphasized for the manifestations of hematologic responses. The most common class of agents to be combined with DNMT inhibitors has been pan-HDAC inhibitors which improve survival in AML patients treated with azacitidine (Table 4) (Figure 3).27 In addition, therapeutic combinations of HDAC inhibitors with DNA damage-inducing agents such as docetaxel, doxorubicin, etoposide and cisplatin have shown superior and additive or synergistic effects. HDAC inhibitors cause an open chromatin conformation that is more accessible to DNA-targeting agents and augment the activity of therapeutics (Figures 2 and 4).13,24
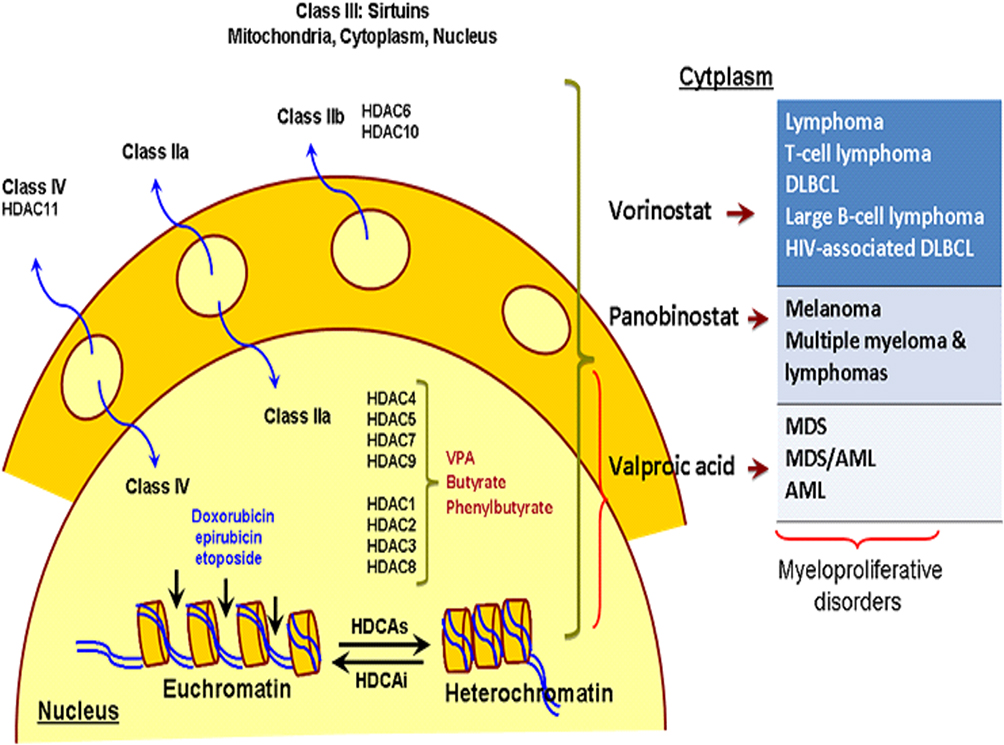 |
Figure 4 Histone deacetylase inhibitors and mechanism of action which have shown benefits in early-phase clinical trials for the treatment of myeloproliferative disorders (eg, MDS and AML). Vorinostat, panobinostat and valproic acid are pan-HDAC inhibitors. |
Demethylating Agents in AML
Deoxycytidine analogues azacitidine, decitabine and zebularine are DNA methyltransferase inhibitors acting as hypomethylating agents and are currently considered as non-intensive therapy for MDS and AML (Table 4). It was reported that the DNMT inhibitor cytarabine causes an S-phase arrest through hypomethylating activity. At low doses, the compounds reduce genomic DNA methylation as a consequence of their irreversible binding to DNMT after incorporation into newly synthesized DNA.20,26,27 Once incorporated in the genome, deoxycytidine analogues form irreversible adducts with DNMTs in their interactions with DNA, then depleting DNMTs from the cell during replication and reducing DNA methylation in subsequent rounds of cell division. In Phase III randomized trials using both azacytidine and decitabine, overall survival, hematopoietic response and time of progression to AML in patients with low- and high-risk MDS were increased. Thus, both are recommended for the treatment of low-risk MDS (Table 3). For high-risk MDS patients not eligible for intensive therapy (eg, HSCT or chemotherapy), azacytidine is the preferred treatment option, as increased survival compared with decitabine was observed.14,23 Interference with aberrant DNA methylation, leading to the reactivation of silenced tumor-suppressor genes, accounts, at least in part, for the DNMT inhibitors’ antitumoral effects. The CDKN1C gene, which encodes the cell-cycle regulator p57KIP2 and has a canonical 50-CpG island, is markedly up-regulated following treatment with azacytidine and decitabine, but not cytarabine. The hepatocyte growth factor (HGF) gene shows fourfold induction after azacytidine whereas it is upregulated only twofold by all other substances. The HGF is involved in proliferation and survival pathways in leukemia. Further genes of interest have been found up-regulated following treatment with the four cytosine nucleoside analogues including three members of the inhibitor of DNA-binding protein family (ID1, ID2 and ID3), which were previously identified as retinoic-acid-responsive genes in acute promyelocytic leukemia.20,26,27 Especially, RR-AML treatment with hypomethylating agents is time course-depended. For example in a Phase II trial, patients receiving 60 mg/m2/day DNA hypomethylating agents (HMA) for 10 days per cycle exhibited superior outcomes and better rates for CR and OS in comparison with those who received the drug for 5 days per cycle. The longer 10-day dosing induced significantly more potent and longer demethylation besides significantly more anemia and thrombocytopenia.5
DNMT inhibitors preferentially azacitidine have been recommended for old patients in whom improving overall survival is observed. In this aspect, the AZA-001 study firmly established azacitidine as the treatment of choice for patients with high-risk MDS not proceeding to allogeneic HSCT. Compared with the induction conventional chemotherapy composed of cytarabine plus anthracycline, the azacitidine-treated group demonstrated increased median survival and twice the 2-year survival compared with patients in the conventional care group.27 A proposed strategy for increasing the response rate, quality, or duration of responses has been to combine azanucleosides with other agents, either strategically or empirically (Tables 3 and 4). For example, HDAC inhibitor vorinostat in combination with idarubicin and cytarabine as induction therapy for AML patients about 65 years old or younger [57] exhibited a higher ORR of 85% and 100% in those with additional FLT3–ITD mutations.17,27 In many trials, treatment of patients with a combination of HDAC and DNMT inhibitors has decreased global DNA methylation and reversed aberrant hypermethylation of specific tumor-suppressor promoters; however, it is not the only mechanism elicited by a combination of these inhibitors (Figure 3) (Table 5).14 The administration of azacytidine (75 mg/m2 for 5 days) with escalating doses of panobinostat (3 times a week for 7 doses) on a monthly basis in AML and high-risk MDS patients resulted in high ORR in MDS (~50%) and AML patients (~31%). Panobinostat, a hydroxamic acid derivative, is a pan-HDAC inhibitor that causes a transient reduction in AML blasts (Table 5). The pan-HDAC inhibitor can transiently increase the expression levels of silenced genes including p53 and p21 tumor suppressors which may subsequently enhance the apoptotic response of AML cells, predominantly in those with higher CD34 expression (Figure 4).9,28 Data for DNMT inhibitors emphasize the importance of patience and therapy duration in the use of these drugs, and several cycles are required for the manifestations of hematologic responses. The most common class of agents to be combined with DNMT inhibitors has been found pan-HDAC inhibitors which improve survival in AML patients treated with azacitidine.9,27
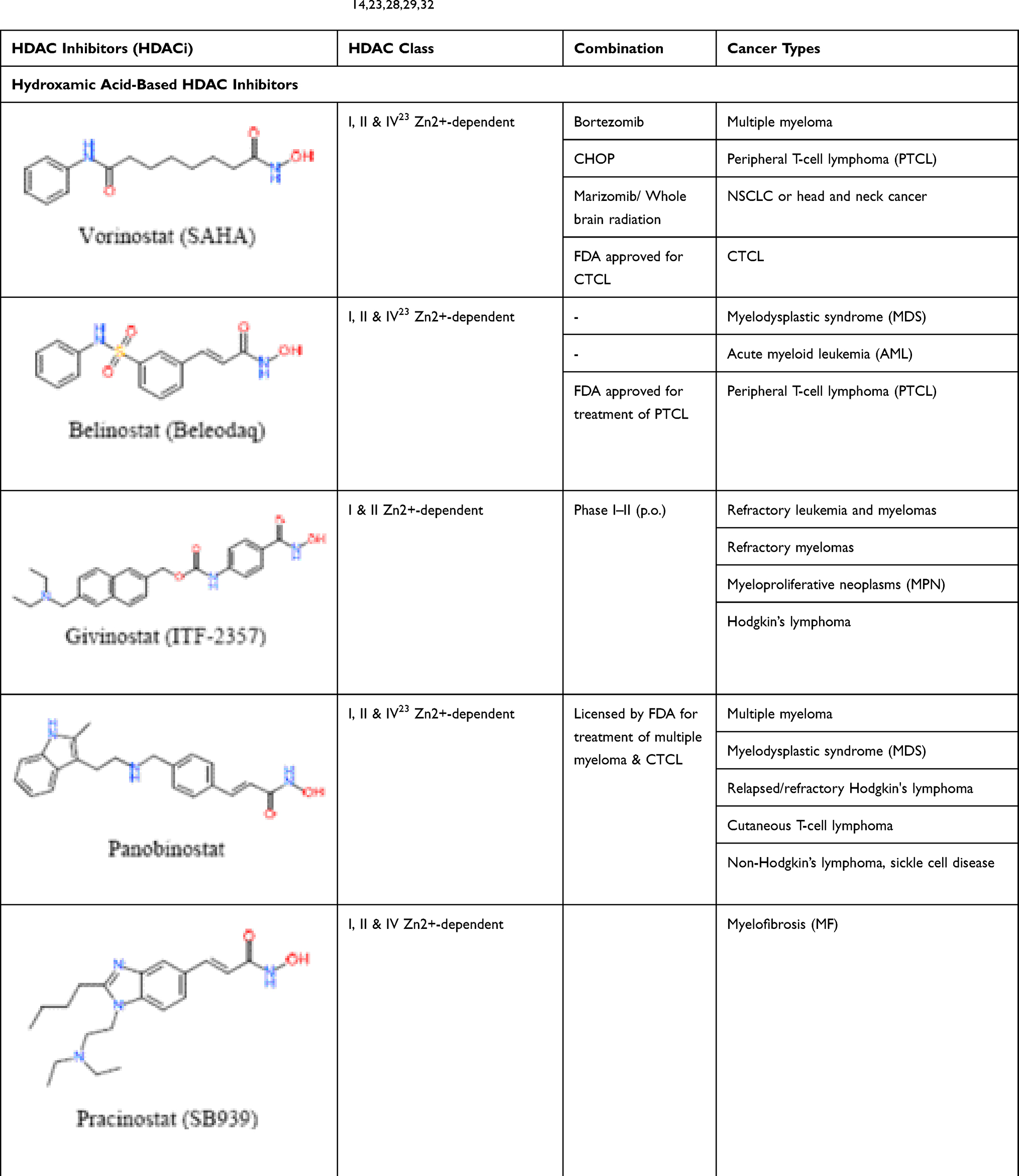 |
Table 5 Rational Combinations of Histone Deacetylase Inhibitors: Hydroxamic Acid-Based HDAC Inhibitors, The Zinc-Dependent HDAC Inhibitors, in Phase II/III Clinical Trials.14,23,28,29,32 |
HDAC Inhibitors in AML
Class I, II and IV HDACs require Zn2+ for deacetylase activity and are inhibited by zinc-chelating compounds. Class III HDACs belong to the sirtuins family and are NAD-dependent which is inhibited by nicotinamide analogues (Table 6). Class I HDACs (HDACs 1–3, 8) are generally localized in the nucleus, whereas class IIa (HDACS 4, 5, 7, 9) can shuttle between the nucleus and the cytoplasm. Class IV represents only one enzyme HDAC11, which shares some structural similarity with both classes I and II (Figure 4).14,25,28 HDAC inhibitors have come out as targets for specific epigenetic changes associated with cancer and other epigenetic-related disorders. The rationales and clinical progress of several combinations with HDAC inhibitors in the treatment of leukemia have shown clinical benefits for patients with cancer, and prompt the investigation of novel treatment combinations with other conventional therapeutics. The most rational combinations include DNA-damaging chemotherapeutic agents, DNMT inhibitors and various small-molecule TKR inhibitors (Figure 1–3).24,28 HDAC inhibitors appear to be active in AML, MDS, lymphomas and cutaneous T-cell lymphoma (CTCL) (Table 4). Vorinostat and romidepsin are two HDAC inhibitors approved by the FDA to be administered for patients with progressive, persistent or recurrent CTCL after one or more lines of chemotherapy.14,24,29 In the case of persistent or recurrent CTCL, a phase II trial of daily oral administration of vorinostat (400 mg) exhibited an objective response of approximately 30% wherein continuous daily administration of vorinostat led to the greater response of 31% vs 9% in patients treated with intermittent dosing.14 In clinical trials, HDAC-inhibitor combinations with conventional chemotherapy have demonstrated improved response rates despite uniformly disappointing results attained so far from HDAC-inhibitor monotherapy (Table 4). In clinical trials for the treatment of myeloproliferative disorders (eg, MDS and AML), HDAC inhibitors showed limited benefit when were used as single therapeutic agents, while in concurrent administration with DNMT inhibitors synergistically increased the expression of silenced tumor suppressors and promoted cell death and differentiation of HSCs (Figure 3).14,29 In a study by Gogo and colleagues treating advanced and high-risk AML patients with vorinostat at escalating doses for 7 days followed by etoposide (100 mg/m2) and cytarabine (1 to 2 g/m2) on days 11 to 14, they documented an ORR of 33% and a median remission time of 7 months. The CR was achieved in 46% of those patients treated with the MTD of 200 mg vorinostat twice daily.9 A phase Ib/II study of azacytidine (75 mg/m2/day × 5) combined with escalating doses of panobinostat (10–40 mg/day) in intensive chemotherapy, naïve AML and MDS patients produced an ORR of 31% for AML and 50% for MDS. In another phase Ib/II study, a combination of panobinostat with intensive induction chemotherapy was used for old patients with newly diagnosed AML. In this study, patients received the standard idarubicin (8 mg/m2/day × 3) and cytarabine (100 mg/m2/day × 7) plus panobinostat at escalating doses (10–40 mg/day). Patients who attained CR received a consolidation cycle with the same combination, followed by panobinostat maintenance until progression. CR was observed in 64% of the patients, with a time to relapse of 17 months.17 HDAC inhibitors are recommended to be used continuously and concurrently with intensive combination chemotherapy in newly diagnosed AML (Table 5).14,17,27 In a phase I study of vorinostat (400 mg/day) used either sequentially or concurrently with decitabine (20 mg/m2/day × 5), 2 of 13 AML patients with relapsed/refractory disease treated concurrently attained complete remission but none of the 15 patients treated on the sequential protocol responded.17 The pan-HDAC inhibitor vorinostat has been demonstrated to act synergistically with Top inhibitor, potentiate the cytotoxic effect of gemcitabine and sensitize cancer cells to cisplatin. These agents can induce the expression of pro-apoptotic genes, cellular differentiation and/or cell-cycle arrest.29 It has been established that G1 arrest is also a result of HDAC inhibitor-mediated down-regulation of cyclin proteins, mainly cyclin D and A.25 Molecularly, HDACs are participating to remodel chromatin and playing a crucial role in the epigenetic regulation of gene expression (Figure 4). HDACs through the deacetylation process regulate the expression of a variety of genes involved in cell-cycle control, proliferation, survival, DNA repair and differentiation (Figures 2 and 4). Moreover, HDACs inhibitors mediate epigenetic silencing of common translocations associated with certain hematological malignancies (eg, AML-ETO fusion protein).14,28,29 Accordingly, a combination of arsenic trioxide and ATRA without chemotherapy can yield a 100% CR and a 97% 2-year event-free survival in low-risk APL patients. Reading this, patients with APL are treated with ATRA which alters the corepressor activity of the fusion protein PML-RARα, bypasses the differentiation block of the leukemic cells, and eventually induces differentiation of the myeloid lineage with very successful results.11,21 PML-RARα acts as a transcriptional repressor of differentiation and apoptosis, by recruiting the co-repressor complexes containing HDACs. However, transcriptional repression by PML-RARα can be turned off in the pharmacological concentrations of ATRA and can induce transcription activation by dissociation of corepressor complexes from PML-RARα and subsequent degradation of PML-RARA.21,30,31 Therefore, it is expected that in combination with HMAs, HDAC inhibitors restore expression of silenced genes by remodeling the tightly coiled chromatin, leading to subsequent induction of differentiation, arrest in the progression of the cell cycle, or apoptosis.20 Importantly, the specific sequence of drug administration would significantly affect the synergy between HDAC inhibitors and topoisomerase II inhibitors (Figure 3). In preclinical studies, the greater efficacies were observed when tumor cells were first exposed to HDAC inhibitors prior to exposure to DNA damage-inducing agents. In breast cancer and other tumor cells, HDAC-inhibitors pretreatment correlates with the decondensation of chromatin observed in these tumors.14,24,29 Based on clinical results, any role expected for HDAC inhibitor in the future development of therapeutics for AML will involve sequence-specific combination with intensive conventional chemotherapy (Figure 3).17,27
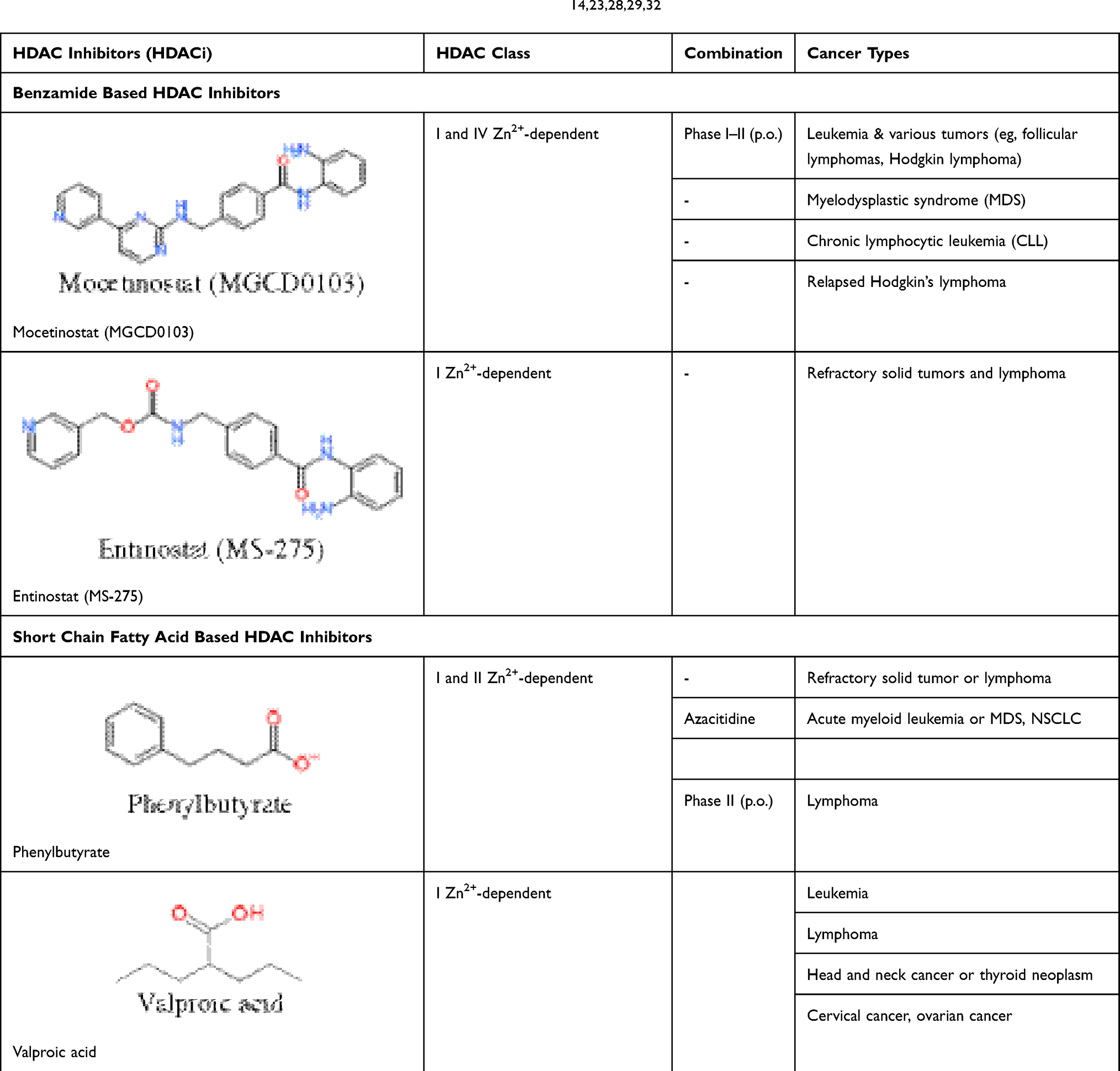 |
Table 6 Rational Combinations of Histone Deacetylase Inhibitors: Benzamide- and Short-Chain Fatty Acid-Based HDAC Inhibitors, The Zinc-Dependent HDAC Inhibitors, in Phase II/III Clinical Trials.14,23,28,29,32 |
Mechanism of Rational Combinations of Epigenetic Modifiers in AML
Rational combinations of DNMT and HDAC inhibitors with DNA damage-inducing therapies synergistically enhance the growth inhibition and apoptotic effects of DNA-damaging agents. DNMT and HDAC inhibitors increase chromatin accessibility while at the same time cause downregulation of DNA repair processes (Figures 2 and 3). Pharmacological inhibition of DNMTs and HDACs leads to local relaxation of chromatin which subsequently affects chromatin structure on a larger scale and decreases condensed heterochromatin regions within the nucleus. Herein, combined inhibitors permit greater access of intercalating agents into the nuclear dsDNA. Therefore, combinations of DNMT and HDAC inhibitors with DNA-damaging agents have a global impact on the accessibility of DNA-damaging agents to their targets as well as affecting DNA dynamics directly (Figure 4).14,25,28 Until now, five clinically relevant combinations have been defined; HDAC inhibitors, DNMT inhibitors, DNA-damaging chemotherapy, hormonal therapy and receptor tyrosine kinase pathway inhibitors (Tables 3 and 4). Prior administration of HDAC and DNMT inhibitors to topoisomerase inhibitors would enhance cleavage of the DNA backbone in order to facilitate DNA damage. Inhibitors of topoisomerase II, which include doxorubicin, epirubicin and etoposide, are widely used in lymphomas, leukemias and many other cancers.27,29,32 The sequence of drug administration in such schemes has been shown to be of an utmost importance. HDACi administration before conventional chemotherapy sensitizes tumor cells to genotoxic agents. In an in vitro assay, administration of trichostatin A and vorinostat before doxorubicin and cisplatin increased cell sensitivity more than 10-fold, whereas the inverse order of drug administration was useless (Figure 3).14,25,29 In several phase I/II clinical trials, the sequence-specific administration of HDAC inhibitors prior to topoisomerase II–inhibitor administration demonstrated a considerable high response rate to chemotherapy (~64%). The results imply that pretreatment with an HDAC inhibitor (eg valproic acid for 48 h prior to administration of the topoisomerase II inhibitor epirubicin), would be an efficient therapy.14 Additionally, HDAC inhibitors reduce HDAC interactions with a variety of vital DNA repair components including ATM, ATR, BRCA1 and p53 whereby decrease their interaction with DNA. At the same time, HDACi diminishes repair kinetics by down-regulating the activity of repairing proteins such as Ku70 and Ku80. The sequence-specific administration of HDAC inhibitors and DNA-damaging agents can act synergistically in damaging the DNA and diminishing the DNA repair system in cancerous cells (Figure 2).14,25 This complex biochemical reaction of HDACi can eventually lead to double-strand breaks (DSBs) in DNA.29 Indeed, the sensitivity of cancer cells to HDACi can be attributed to causing DNA damage where normal but not cancer cells do have time to repair.32 In transformed cells, the level of a biomarker of DBSs in DNA, phosphorylated histone variant γH2AX, increases with continued culture with vorinostat whereas normal cells repair the DSBs despite continued exposure to vorinostat.29 On the other side, the nucleotide antimetabolites 5-aza-cytidine and 5-aza-2′-deoxycytidine, which inhibit DNMT and promote an open chromatin frame conducive for gene transcription and a greater chance for DNA-damaging agents, are currently used to kill cancer cells. In one manner or another, they also inhibit the synthesis of DNA or interfere with the synthesis of other major macromolecules (protein, RNA, lipid, etc.).6,14 Therefore, combining an HDAC inhibitor with a DNMT inhibitor may lead to a greater expression of silenced genes, a greater DNA damage through the interaction of topoisomerase inhibitors in DNA and a stronger signaling to the cell death.14 In this regard, a phase I/II clinical trial with a combination of the HDAC inhibitor valproic acid and HMA decitabine reported that hypomethylation of p15 is the best indicator of response in leukemia.33 Additionally, a phase I clinical study conducted on MDS or AML patients receiving 5-azacitidine followed by sodium phenylbutyrate showed that the molecular mechanism responsible for responses to DNMTi and HDACi combination therapy might include the reversal of aberrant epigenetic gene silencing. In the trial, responding patients with pretreatment methylation of p15 or CDH-1 promoters exhibited a reversal of methylation during the first cycle of therapy, while the non-responding patients showed no demethylation. The study concludes that DNMTi and HDACi combination therapy would reverse the aberrant epigenetic silencing of genes in leukemia.34
Integration of Tyrosine Kinase Inhibitors in AML Therapeutics
While the activity of tyrosine kinase receptor (TKR) is tightly regulated in normal cells, TKR signaling is dysregulated in many cancers. Therefore, TKRs are promising therapeutic targets. Activation of TKR has been demonstrated to activate RAS-RAF-MEK-MAPK, Src/JAK (Janus kinase) and PI3K-AKT pathways that trigger a cascade of downstream events related to the activation of cell proliferation (Figure 1).17 Among them, mutations of TKR-FLT3 are predominantly encountered in approximately 30–35% of AML cases, including internal tandem duplications (ITD~25% of patients) and point mutations at the tyrosine kinase domain (TKD~5–10%).5,10,11 FLT3 mutations result in a constitutively active kinase. In patients with FLT3/ITD mutations, the duration of remission is usually short and the relapse rate is high. They have high white cell counts at disease presentation and have normal or intermediate-risk karyotypes. FLT3/TKD mutations tend to confer slightly better prognosis.11,17 Preliminary data suggest an EFS advantage for the addition of FLT3 inhibitors to conventional induction chemotherapy in patients with AML.5 To date, most of the combinations have used a targeted-FLT3 inhibitor combined with standard agents such as anthracyclines, cytarabine or hypomethylating agents. Rationally designed combinations that will more specifically and directly alter the biology of the disease and show synergism have been investigated.8,10 FLT3 inhibitors include sorafenib (type I TKi) and quizartinib (type II TKi) that inhibit FLT3/ITD mutant receptor, and midostaurin, crenolanib and gilteritinib that inhibit both FLT3/ITD and FLT3/TKD mutant receptors (Figure 1). Most of these agents are multi-kinase inhibitors. The preclinical results reveal that the combination of midostaurin with the standard “3 + 7“ induction regimen as first-line therapy would be beneficial for young AML patients with FLT3 mutations. Remarkably, midostaurin is an oral multi-kinase inhibitor, with activity against not only FLT3 kinase but also KIT, VEGFR, PDGFR, and protein kinase C, and is well tolerated.5,17 Preclinical studies have shown synergism between HMAs and midostaurin against FLT3-ITD AML (Figure 1).5 Midostaurin in combination with azacytidine in a phase I/II study on patients with RR-AML caused an ORR of approximately 26% in all patients and ~33% in patients with FLT3–ITD mutations. The combination was well tolerated. Accordingly, 75 mg midostaurin, three times daily, induced a ≥50% reduction in circulating and/or bone marrow blast count in about 79% of patients and in 70% of patients with FLT3-mutated RR-AML or high-grade MDS who could not routinely be candidates for common intensive chemotherapy with the goal of achieving a second CR (CR2).2 Another clinical trial evaluating the combination of midostaurin (50 mg twice daily), with “7 + 3” induction and high-dose cytarabine consolidation in young, newly diagnosed AML patients reported a 80% CR rate in FLT3-mutated patients vs 74% in FLT3-wild type patients.5,10,17 This study used a combination of midostaurin (50 mg twice a day), with daunorubicin (60 mg/m2/day × 3) and cytarabine (200 mg/m2/day × 7) induction therapy. This combination has been recommended for young patients newly diagnosed with AML to produce a high CR rate and OSR.17 Other studies have reported the benefits of adding sorafenib to ”7 + 3” induction chemotherapy followed by up to three cycles of high-dose cytarabine consolidation in young AML patients that also used ‘maintenance’ sorafenib for 12 months for decreasing the risk of relapse.5 Additionally, two small molecules, imatinib mesylate (a tyrosine kinase inhibitor) and ATRA (which can induce complete responses in patients with the APL; AML M3) have spawned great interests as a rational combination for the treatment of AML. TKi imatinib mesylate represses the function of BCR-ABL kinase as well as other related tyrosine kinases, which has resulted in an 80% complete cytogenetic response rate in patients with chronic phase CML.9 Moreover, combinations of TKR targeted therapies and HDACi have been proposed which are expected to represent a novel approach for targeted cancer therapy. Recent studies have revealed that the combination of vorinostat (a pan-HDACi) and sorafenib (TKI) synergistically kills tumor cells.32
Combination of Intracellular Toxins with Surface Receptor mAb in AML
Another strategy for targeted therapy of AML has been exemplified by gemtuzumab ozogamicin. It consists of monoclonal anti-CD33, a cell surface protein on tumor cells, which is chemically linked to a toxin from the class of calicheamicins. The drug initially received accelerated approval in 2000 as a stand-alone therapy for older patients with CD33-positive AML whose disease had returned after initially successful treatment. However, confirmatory clinical trials showed that it did not improve survival and was associated with serious side effects and early death. In 2010, the drug was voluntarily removed from the market by its manufacturer. Again in 2017, it re-emerged with an FDA approval for its use in adults with newly CD33-positive AML. The approval also covers the treatment of patients aged 2 and older with CD33-positive AML who have experienced a relapse or whose disease has not responded to initial treatment.35 Gemtuzumab ozogamicin also received European approval for combination therapy with daunorubicin and cytarabine for the treatment of patients age 15 years and above with previously untreated, de novo CD33-positive AML, except APL.36
Other Promising Combination Therapies for AML
On November 21, 2018, the FDA granted accelerated approval to venetoclax (a selective BCL-2 inhibitor) in combination with azacitidine or decitabine or low-dose cytarabine for the treatment of newly-diagnosed AML in patients ≥75 years, or those who have comorbidities that preclude the use of intensive induction chemotherapy. The approval was based on two open-label non-randomized trials.37 It is hoped that further randomized trials will strengthen its application for patients with AML.
Conclusion
AML is a complex disease with multiple aberrant molecular pathways involved. It is therefore unlikely that blockage of any one specific pathway will lead to prolonged remissions and cures in more than a fraction of the patient population. Thus, combination strategies are absolutely required to improve outcomes. To date, most of the combinations have used a targeted agent combined with standard chemotherapy such as anthracyclines, cytarabine, or HMAs. Rationally designed combinations and sequence-specific administration that will more specifically and directly alter the biology of the disease and show synergism are required. Preliminary data suggest complete response advantages for prior addition of HDAC inhibitors and HMAs to the conventional induction chemotherapy of AML, and an induction rate advantage for the addition of the TKIs for patients with TKR mutations.
Abbreviations
AML, Acute myeloid leukemia; RR-AML, refractory or relapsed AML; ALL, acute lymphoid leukemia; DNMT, DNA methyltransferase; HDAC, histone deacetylase; BM, bone marrow; HSCT, hematopoietic stem cell transplantation; CRRs, complete remission rates; DFS, disease-free survival; OS, overall survival; OSR, overall survival rate; CR, complete remission; ORRs, overall response rates; APL, acute promyelocytic leukemia; ATRA, all-trans retinoic acid; RARA, retinoic acid receptor alpha; G-CSF, granulocyte colony-stimulating factor; FLAG, cytarabine, G-CSF, and fludarabine; CAG/DAG, cytarabine, G-CSF and aclarubicin/daunorubicin; CLAG, cytarabine, G-CSF, and cladribine; MEC, cytarabine, etoposide, and mitoxantrone; IA/DA/MA, cytarabine, idarubicin/daunorubicin/mitoxantrone; FLAG, homoharringtonine, cytarabine, and aclarubicin/daunorubicin (HAA/HAD) and fludarabine; CLAG, cladribine, high-dose cytarabine, and G-CSF; MEC, etoposide, high-dose cytarabine, and mitoxantrone; PR-AML, primary refractory AML; ORR, overall remission rate; EFS, event-free survival; HHT, homoharringtonine; MDS, myelodysplastic syndromes; t-AML, MDS-transformed acute myeloid leukemia; FLT3, fms-like tyrosine kinase 3; stem cell factor, SCF, C-Kit; a small GTPase, Ras; NER, nucleotide excision repair; BER, base excision repair; MMR, mismatch repair; HR, homologous recombination; HDR, homology-directed repair; MAPK, mitogen-activated protein kinase; ATF, activating transcription factor; Top II, topoisomerase II; CHG, HHT, low-dose cytarabine and G-CSF; Top, topoisomerases; ATM, ataxia–telangiectasia mutated protein; BRCA1, breast cancer type 1 susceptibility protein; CHEK2, checkpoint kinase 2; Ku70-Ku80, XRCC6 and XRCC5; APE 1, apurinic/apyrimidinic endonuclease 1; dCK, deoxycytidine kinase; HD, high-dose; ID/LD, intermediate-/low-dose; aza-dCyd, decitabine; aza-Cyd, azacytidine; MP, mercaptopurine; TG, thioguanine; RNR, ribonucleotide reductase; ara-CTP, cytarabine; DNPS, de novo purine synthesis; CDKN1C, cyclin-dependent kinase inhibitor 1C; HGF, hepatocyte growth factor; p57, Kip2, cyclin-dependent kinase inhibitor 1C; FLT3-ITD, FLT3 internal tandem duplication; CTCL, cutaneous T-cell lymphoma; HMAs, DNA hypomethylation agents; HDACi, HDAcC inhibitor; DSBs, double-strand breaks; TKR, tyrosin kinase receptor; Src, proto-oncogene tyrosine-protein kinase; JAK, Janus kinase; PI3Ks, phosphoinositide 3-kinases; AKT, protein kinase B; TKD, tyrosine kinase domain; VEGFR, vascular endothelial growth factor receptor; PDGFR, platelet-derived growth factor receptors; CR2, second CR; TKi, tyrosin kinase inhibitor; mAb, monoclonal antibody.
Author Contributions
Fatemeh Pourrajab: writing the draft and literature review; Mohamad Reza Zare-Khormizi: literature survey; Azam Sadat Hashemi, supervising and consulting on data; Seyedhossein Hekmatimoghaddam: supervising and editing the final manuscript. All authors contributed to designing, data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Xu J, Lv TT, Zhou XF, Huang Y, Liu DD, Yuan GL. Efficacy of common salvage chemotherapy regimens in patients with refractory or relapsed acute myeloid leukemia: a retrospective cohort study. Medicine (Baltimore). 2018;97(39):e12102. doi:10.1097/MD.0000000000012102
2. Masoumi-Dehshiri R, Hashemi AS, Neamatzadeh H, Zare-Zardeini H, Case Report: A. Acute myeloid leukemia (FAB M7). Iran J Ped Hematol Oncol. 2014;4(4):188–190.
3. Hekmatimoghaddam S, Jebali A, Dargahi M. Folic acid-functionalized gold and silver nanoparticles: their cytotoxic effect on cancerous myeloid cells with microwave irradiation. Nano Life. 2013;3(2):
4. Price SL, Lancet JE, George TJ, et al. Salvage chemotherapy regimens for acute myeloid leukemia: is one better? Efficacy comparison between CLAG and MEC regimens. Leuk Res. 2011;35(3):301–304. doi:10.1016/j.leukres.2010.09.002
5. Bose P, Vachhani P, Cortes JE. Treatment of relapsed/refractory acute myeloid leukemia. Curr Treat Options Oncol. 2017;18(3):17. doi:10.1007/s11864-017-0456-2
6. Parker WB. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem Rev. 2009;109(7):2880–2893. doi:10.1021/cr900028p
7. Harned TM, Gaynon PS. Treating refractory leukemias in childhood, role of clofarabine. Ther Clin Risk Manag. 2008;4(2):327–336. doi:10.2147/TCRM.S2941
8. Cooper TM, Alonzo TA, Gerbing RB, et al. AAML0523: a report from the children’s oncology group on the efficacy of clofarabine in combination with cytarabine in pediatric patients with recurrent acute myeloid leukemia. Cancer. 2014;120(16):2482–2489. doi:10.1002/cncr.28674
9. Al-Hussaini M, DiPersio JF. Small molecule inhibitors in acute myeloid leukemia: from the bench to the clinic. Expert Rev Hematol. 2014;7(4):439–464. doi:10.1586/17474086.2014.932687
10. Hackl H, Astanina K, Wieser R. Molecular and genetic alterations associated with therapy resistance and relapse of acute myeloid leukemia. J Hematol Oncol. 2017;10(51):1–16. doi:10.1186/s13045-017-0416-0
11. Pourrajab F, Zare-Khormizi MR, Hashemi AS, Hekmatimoghaddam S. Genetic characterization and risk stratification of acute myeloid leukemia. Cancer Manag Res. 2020;12:2231–2253. doi:10.2147/CMAR.S242479
12. Wu L, Li X, Su J, et al. Effect of low-dose cytarabine, homoharringtonine and granulocyte colony-stimulating factor priming regimen on patients with advanced myelodysplastic syndrome or acute myeloid leukemia transformed from myelodysplastic syndrome. Leuk Lymphoma. 2009;50(9):1461–1467. doi:10.1080/10428190903096719
13. Gavande NS, VanderVere-Carozza PS, Hinshaw HD, et al. DNA repair targeted therapy: the past or future of cancer treatment? Pharmacol Ther. 2016;160:65–83. doi:10.1016/j.pharmthera.2016.02.003
14. Thurn KT, Thomas S, Moore A, Munster PN. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future Oncol. 2011;7(2):263–283. doi:10.2217/fon.11.2
15. Mody MD, Gill HS, Higgins KA, Saba NF, Kota VK. Complete remission of acute myeloid leukemia following cisplatin based concurrent therapy with radiation for squamous cell laryngeal cancer. Case Rep Hematol. 2016;2016:8581421.
16. Laurent G, Jaffrézou JP. Signaling pathways activated by daunorubicin. Blood. 2001;98(4):913–924. doi:10.1182/blood.V98.4.913
17. Lim SH, Dubielecka PM, Raghunathan VM. Molecular targeting in acute myeloid leukemia. J Transl Med. 2017;15(1):183. doi:10.1186/s12967-017-1281-x
18. Iacobucci I, Mullighan CG. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol. 2017;35(9):975–983. doi:10.1200/JCO.2016.70.7836
19. Zhenchuk A, Lotfi K, Juliusson G, Albertioni F. Mechanisms of anti-cancer action and pharmacology of clofarabine. Biochem Pharmacol. 2009;78(11):1351–1359. doi:10.1016/j.bcp.2009.06.094
20. Navada SC, Steinmann J, Lübbert M, Silverman LR. Clinical development of demethylating agents in hematology. J Clin Invest. 2014;124(1):40–46. doi:10.1172/JCI69739
21. Braoudaki M, Tzortzatou-Stathopoulou F. Clinical cytogenetics in pediatric acute leukemia: an update. Clin Lymphoma Myeloma Leuk. 2012;12(4):230–237. doi:10.1016/j.clml.2012.04.004
22. Magina KN, Pregartner G, Zebisch A, et al. Cytarabine dose in the consolidation treatment of AML: a systematic review and meta-analysis. Blood. 2017;130(7):946–948. doi:10.1182/blood-2017-04-777722
23. Dervieux T, Brenner TL, Hon YY, et al. De novo purine synthesis inhibition and antileukemic effects of mercaptopurine alone or in combination with methotrexate in vivo. Blood. 2002;100(4):1240–1247. doi:10.1182/blood-2002-02-0495
24. Zagni C, Floresta G, Monciino G, Rescifina A. The search for potent, small-molecule HDACIs in cancer treatment: a decade after vorinostat. Med Res Rev. 2017;37(6):1373–1428. doi:10.1002/med.21437
25. Wanczyk M, Roszczenko K, Marcinkiewicz K, Bojarczuk K, Kowara M, Winiarska M. HDACi–going through the mechanisms. Front Biosci. 2011;16(1):340–359. doi:10.2741/3691
26. Flotho C, Claus R, Batz C, et al. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia. 2009;23(6):1019–1028. doi:10.1038/leu.2008.397
27. Gore SD. New ways to use DNA methyltransferase inhibitors for the treatment of myelodysplastic syndrome. Hematology Am Soc Hematol Educ Program. 2011;2011(1):550–555. doi:10.1182/asheducation-2011.1.550
28. Hekmatimoghaddam S, Zare-Khormizi MR, Pourrajab F. Underlying mechanisms and chemical/biochemical therapeutic approaches to ameliorate protein misfolding neurodegenerative diseases. Biofactors. 2017;43(6):737–759. doi:10.1002/biof.1264
29. Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. 2015;20(3):3898–3941. doi:10.3390/molecules20033898
30. Prada-Arismendy J, Arroyave JC, Röthlisberger S. Molecular biomarkers in acute myeloid leukemia. Blood Rev. 2017;31(1):63–76. doi:10.1016/j.blre.2016.08.005
31. Zaidi SZ, Owaidah T, Al Sharif F, Ahmed SY, Chaudhri N, Aljurf M. The challenge of risk stratification in acute myeloid leukemia with normal karyotype. Hematol Oncol Stem Cell Ther. 2008;1(3):141–158. doi:10.1016/S1658-3876(08)50023-9
32. Suraweera A, O’Byrne KJ, Richard DJ. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: achieving the full therapeutic potential of HDACi. Front Oncol. 2018;8:92. doi:10.3389/fonc.2018.00092
33. Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, et al. Phase 1/2 study of the combination of 5-aza-2’-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006;108(10):3271–3279. doi:10.1182/blood-2006-03-009142
34. Gore SD, Baylin S, Sugar E, et al. Combined DNA methyltransferase and histone deacetylase inhibitors in the treatment of myeloid neoplasms. Cancer Res. 2006;66(12):6361–6369. doi:10.1158/0008-5472.CAN-06-0080
35. U.S. Food and Drug Administration. Gemtuzumab ozogamicin: FDA-Approved Drugs. Retrieved from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=761060.
36. European Medicines Agency. Mylotarg EPAR. Retrieved from: https://www.ema.europa.eu/en/medicines/human/EPAR/mylotarg-0.
37. FDA approves venetoclax in combination for AML in adults; 2020. Retrieved from: https://www.fda.gov/drugs/fda-approves-venetoclax-combination-aml-adults.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.