")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Molecular pharmacodynamics of new oral drugs used in the treatment of multiple sclerosis
Authors di Nuzzo L, Orlando R, Nasca C, Nicoletti F
Received 2 August 2013
Accepted for publication 12 December 2013
Published 19 May 2014 Volume 2014:8 Pages 555—568
DOI https://doi.org/10.2147/DDDT.S52428
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Luigi di Nuzzo,1 Rosamaria Orlando,2 Carla Nasca,1 Ferdinando Nicoletti1,3
1Department of Physiology and Pharmacology, Sapienza University of Rome, 2IRCCS Associazione Oasi Maria S.S., Institute for Research on Mental Retardation and Brain Aging, Troina, Enna, 3IRCCS Neuromed, Pozzilli, Italy
Abstract: New oral drugs have considerably enriched the therapeutic armamentarium for the treatment of multiple sclerosis. This review focuses on the molecular pharmacodynamics of fingolimod, dimethyl fumarate (BG-12), laquinimod, and teriflunomide. We specifically comment on the action of these drugs at three levels: 1) the regulation of the immune system; 2) the permeability of the blood–brain barrier; and 3) the central nervous system. Fingolimod phosphate (the active metabolite of fingolimod) has a unique mechanism of action and represents the first ligand of G-protein-coupled receptors (sphingosine-1-phosphate receptors) active in the treatment of multiple sclerosis. Dimethyl fumarate activates the nuclear factor (erythroid-derived 2)-related factor 2 pathway of cell defense as a result of an initial depletion of reduced glutathione. We discuss how this mechanism lies on the border between cell protection and toxicity. Laquinimod has multiple (but less defined) mechanisms of action, which make the drug slightly more effective on disability progression than on annualized relapse rate in clinical studies. Teriflunomide acts as a specific inhibitor of the de novo pyrimidine biosynthesis. We also discuss new unexpected mechanisms of these drugs, such as the induction of brain-derived neurotrophic factor by fingolimod and the possibility that laquinimod and teriflunomide regulate the kynurenine pathway of tryptophan metabolism.
Keywords: demyelinating diseases, pharmacotherapy, fingolimod, dimethyl fumarate, laquinimod, teriflunomide
Introduction
The advent of new oral drugs opens a new era in the treatment of multiple sclerosis (MS). These drugs, some of which are already marketed or in the final stages of clinical development, are highly heterogeneous in terms of mechanism of action and clinical efficacy. Here, we discuss the pharmacodynamics of fingolimod, dimethyl fumarate, laquinimod, and teriflunomide in an attempt to offer a head-to-head comparison of their mechanisms of action at three levels: the immune system, the blood–brain barrier, and the central nervous system (CNS). We searched PubMed-MEDLINE using the headings fingolimod, sphingosine-1-phosphate, dimethyl fumarate, fumarate, nuclear factor (erythroid-derived 2)-related factor 2 (Nrf2), laquinimod, and teriflunomide linked to MS, experimental autoimmune encephalomyelitis (EAE), immune system, immune function, vascular permeability, blood–brain barrier, neurons, astrocytes, oligodendrocytes, and microglia. Primary objectives of the review are to 1) provide a molecular correlate of the beneficial effect of the four drugs in MS and EAE; 2) comment on some ambiguous aspects inherent to their mechanism of action; and 3) to lay the groundwork for the choice of one of these drugs in the treatment of MS. Other issues that may critically influence the choice of one of the four drugs, such as pharmacokinetics, safety and tolerability, and costs, are not discussed here.
General mechanisms of action
Fingolimod
Fingolimod (Gilenya™, Novartis, Basel, Switzerland) was the first oral drug to receive US Food and Drug Administration (FDA) and European Medicines Agency (EMA) approval for the treatment of relapsing-remitting MS (RRMS). In two phase III clinical studies (TRANSFORMS [Trial assessing Injectable Interferon Versus FTY720 Oral in relapsing-Remitting Multiple Sclerosis] versus interferon [IFN]-β and FREEDOMS [FTY720 Research Evaluating Effects of Daily Oral therapy in Multiple Sclerosis] versus placebo) fingolimod (0.5 mg/day) demonstrated high efficacy in reducing the annual relapse rate (by about 55%) and progression of neurological disability.1–3 Fingolimod also had a remarkable effect on brain atrophy associated with MS, reducing the rate of atrophy to the same levels as those observed in healthy subjects.
Fingolimod has a unique mechanism of action among the drugs that are currently used in the treatment of MS.4 Fingolimod is an analog of sphingosine, which is a component of glycosphingolipids, and is more lipophilic than sphingosine as a result of the presence of an aromatic ring in its structure (Figure 1). Sphingosine is phosphorylated inside the cells into sphingosine-1-phosphate (S1P) by two protein kinases, named type-1 and type-2 sphingosine kinase (SphK1 and -2). S1P is then transported outside the cells.5–7
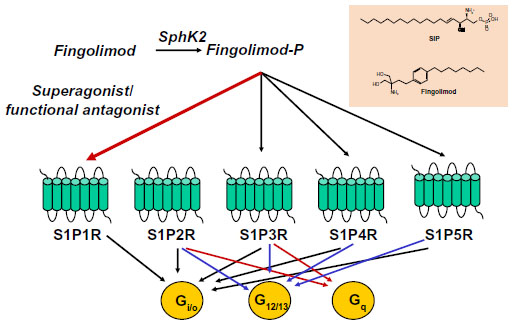 | Figure 1 Fingolimod phosphate activates four sphingosine-1-phosphate receptors: S1P1R, S1P3R, S1P4R, and S1P5R. Superactivation of S1P1R results in functional antagonism. Coupling of S1PRs to G proteins is also shown. |
S1P is present in relatively high concentrations in the blood and lymph, but in lower concentrations inside the lymph nodes and other organs.8 The concentration gradient between circulatory fluids and tissue interstitium is critical for the regulation of cell migration by S1P. S1P acts as an ‘inside-out’ signaling molecule by activating five types of G-protein-coupled receptors (S1P1R to S1P5R), which are linked to multiple transduction pathways. Fingolimod shares the same metabolic fate of sphingosine, and is phosphorylated by SphK2 into fingolimod phosphate. Because of the high distribution volume of fingolimod, high levels of fingolimod phosphate are found in the interstitium of lymph nodes and other organs.9 Fingolimod phosphate activates S1P1Rs with high potency (KD=0.3 nM) and higher efficacy than S1P. Over-activation of S1P1Rs results in rapid receptor desensitization and internalization.10 Thus, fingolimod behaves as a functional antagonist of S1P1Rs, although binding of arrestins to S1P1Rs may redirect signaling to alternative G-protein-independent pathways. Fingolimod phosphate can also activate S1P3Rs, S1P4Rs, and S1P5Rs, but not S1P2Rs (Figure 1). The following aspects must be taken into account: 1) functional antagonism is preceded by a transient activation of S1P1Rs, a mechanism that underlies the first-dose bradycardia associated with fingolimod in MS patients; and 2) only receptors that are actively coupled to G proteins can be desensitized in response to agonist activation. Thus, the balance between S1P1R activation and functional antagonism by fingolimod critically depends on the receptor reserve (ie, the proportion of S1P1Rs that are not functionally recruited to the biological response in a particular context and cell type).
Dimethyl fumarate
Dimethyl fumarate (also known as BG-12) is an oral prodrug that is converted into monomethyl fumarate and fumarate inside cells. The extent to which dimethyl fumarate, monomethyl fumarate, and fumarate contribute to the immune suppressant and cytoprotective activity of dimethyl fumarate is unknown. Fumaric acid esters, under the trade name of Fumaderm® (Fumapharm AG, Lucerne, Switzerland), have been used in Germany since 1994 in the treatment of psoriasis.11,12 The efficacy of oral dimethyl fumarate in the treatment of MS has been proven in two phase III studies named DEFINE (Placebo-controlled Phase 3 Study of Oral BG-12 for Relapsing Multiple Sclerosis) and CONFIRM (Placebo-controlled Phase 3 Study of Oral BG-12 or Glatiramer in Multiple Sclerosis).13,14 The latter study included an arm with glatiramer acetate as an active comparator. In both studies, dimethyl fumarate reduced relapse rates by about 50% compared with placebo. Reduction of disability progression was significant in the DEFINE study,13 but not in the CONFIRM study.14 Dimethyl fumarate is administered at a dose of 240 mg, two or three times per day.
Dimethyl fumarate enhances the activity of Nrf2, a basic leucine zipper transcription factor that controls the expression of a wide range of antioxidant proteins and detoxification enzymes.15,16 Under physiological conditions, Nrf2 is retained in the cytoplasm by a dimer of the Kelch-like ECH-associated protein (Keap1), which serves as an adapter for the ring box protein (Rbx1)-bound Cullin3 (Cul3)-based E3 ligase. Cul3 primes Nrf2 to degradation by the ubiquitin-proteasome system. Keap1, which is a Zn-finger protein, acts as the redox sensor of the system. Its highly reactive cysteine residues can be oxidized to sulfenic acid, form disulfides, or be alkylated by electrophiles. This leads to destabilization of the complex and release of Nrf2, which is no longer degraded and translocates into the nucleus. Nuclear import of Nrf2 is facilitated by protein kinase C-δ/ι-mediated Nrf2 phosphorylation. Nrf2 binds in a complex with a small Maf protein or a member of the activator protein-1 (AP-1) family to the antioxidant response element sequence in the promoter of its target genes.17,18 Activation of Nrf2 results in the induction of genes encoding enzymes that detoxify reactive oxygen species (ROS) or enhance glutathione (GSH) levels, eg, GSH-synthesizing enzymes, NAD(P)H: quinone oxidoreductase, thioredoxin, and heme oxygenase-1 (HO-1).19 Some of the proteins encoded by these genes are vital for cells’ defense against oxidative damage. Nuclear export of Nrf2 is promoted by phosphorylation of Tyr568 mediated by the tyrosine kinase, Fyn. Activation of glycogen synthase kinase-3β promotes nuclear translocation of Fyn, thus limiting the nuclear availability of glycogen-synthase kinase-3β (Figure 2).20,21
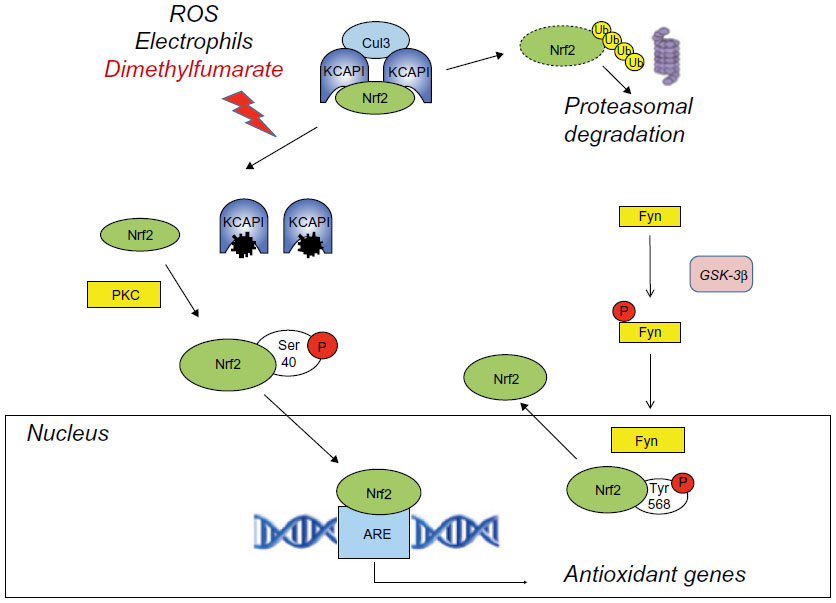 | Figure 2 Activation of the Nrf2 pathway by dimethyl fumarate. |
Activators of Nrf2 include ROS, electrophiles, and other molecules that are potentially hazardous to cells and can be neutralized by Nrf2-regulated genes. Subtoxic concentrations of oxidants or electrophiles can afford cell protection against toxic concentrations of the same or other molecules via the activation of the Keap1-Nrf2 pathway. This toxicological phenomenon, called “hormesis”, is reminiscent of the phenomenon of ischemic preconditioning, in which a sub-lethal ischemic insult protects cells against a subsequent ischemic challenge. Activation of Nrf2 by dimethyl fumarate fits into this scenario. Dimethyl fumarate forms conjugates with GSH, thereby causing depletion of intracellular free GSH.12,19–21 This potentially harmful event may, in principle, activate the Nrf2 pathway in response to dimethyl fumarate, although other mechanisms of activation are also possible. There are some caveats: 1) one cannot exclude that the initial GSH depletion may reach the threshold for cell toxicity if combined with additional toxic insults; 2) GSH conjugation is considered to be one of the major defensive mechanisms against oxidative damage, but can paradoxically generate harmful species (eg, episulfonium ions) under particular circumstances; 3) activation of the Nrf2 system may inhibit tumor initiation but may also facilitate tumor growth by inhibiting apoptosis and autophagy and by enhancing the resistance to chemotherapeutic drugs via the multidrug-resistant proteins;20,22 and 4) agents that activate the Nrf2 pathway may cause toxic effects due to their interaction with nucleophiles such as GSH.23 This may explain the high frequency of gastrointestinal adverse effects found in patients treated with dimethyl fumarate in the DEFINE and CONFIRM studies.13,14
Laquinimod
Laquinimod is structurally similar to roquinimex (linomide), a quinoline-3-carboxamide analog. The efficacy of laquinimod in MS was evaluated in a placebo-controlled phase III study (ALLEGRO [Placebo-controlled Trial of Oral Laquinimod for Multiple Sclerosis]), in which the drug reduced disability progression and showed a modest effect on annualized relapse rate.24
The mechanism of action of laquinimod is only partially known. Interestingly, laquinimod is also structurally similar to kynurenic acid and xanthurenic acid, which are two metabolites of the kynurenine pathway of tryptophan metabolism (Figure 3). Kynurenic acid acts as an antagonist at the glycine site of N-methyl-D-aspartate (NMDA) receptors, quinolinic acid is an orthosteric NMDA receptor agonist, and xanthurenic acid is a putative type-2/3 metabotropic glutamate receptor agonist.25 Interestingly, indoleamine 2,3-dioxygenase (IDO), an enzyme involved in the kynurenine pathway is induced by pro-inflammatory cytokines, and its expression in antigen-presenting cells (APC) supports T-regulatory (TReg) cell generation and immune tolerance.26,27 Some of the metabolites of the kynurenine pathway have been considered as potential effectors of IDO in the regulation of immune tolerance.28,29 Thus, the kynurenine pathway is highly relevant to the pathophysiology of neuroinflammation and EAE, being at the interface between immune regulation and mechanisms of neurodegeneration/neuroprotection. In one report, laquinimod was indicated as an inhibitor of the kynurenine pathway.30 However, no information was provided on the enzymatic reaction of the kynurenine pathway that is targeted by laquinimod.
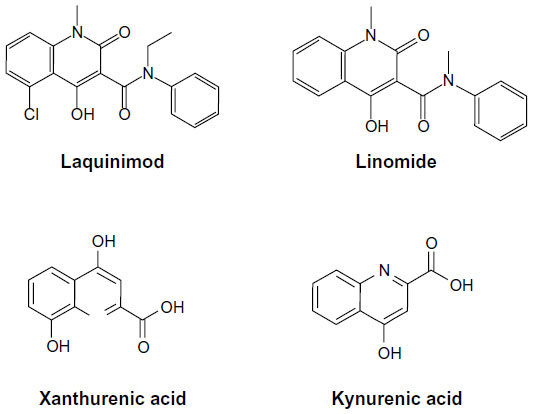 | Figure 3 Molecular structures of laquinimod, linomide, xanthurenic acid, and kynurenic acid. |
Teriflunomide
Teriflunomide is the active metabolite of leflunomide, an immunosuppressive drug approved as a disease-modifying agent in the treatment of rheumatoid arthritis.31 The efficacy of teriflunomide in MS was shown in a placebo-controlled phase III study (Teriflunomide Multiple Sclerosis Oral [TEMSO]), in which the drug reduced both the annualized relapse rate (by about 30% versus placebo) and the disability progression.32 In contrast with laquinimod, the mechanism of action of teriflunomide is well established.
Teriflunomide behaves as a high-affinity inhibitor of dihydroorotate dehydrogenase (DHODH), the enzyme that converts dihydroorotate into orotate in the de novo synthesis of pyrimidines.33,34 De novo synthesis of pyrimidines is pivotal for the metabolism of highly proliferative cells such as activated lymphocytes, and, therefore, inhibition of DHODH accounts for the immunosuppressive effect of teriflunomide (Figure 4). Teriflunomide inhibits DHODH activity in a competitive manner, with a half maximal inhibitory concentration (IC50) of 1.25 μM.35 All DHODH-related effects of teriflunomide can be overcome by increases in intracellular levels of pyrimidines.
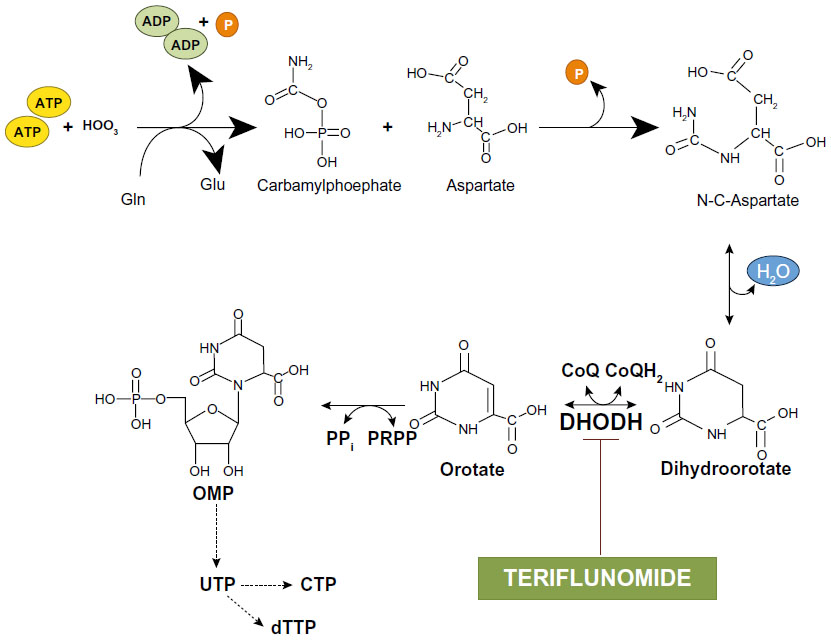 | Figure 4 De novo synthesis of pyrimidines: inhibition of dihydroorotate dehydrogenase by teriflunomide. |
Pyrimidine nucleotides originate either from the de novo synthesis described above or from a ‘salvage pathway’ that is independent of DHODH. Thus, cells might become resistant to teriflunomide by enhancing the activity of the salvage pathway. However, this mechanism is effective in resting cells, but not in highly proliferating cells, such as blasting lymphocytes, which strictly depend on de novo synthesis of pyrimidines. Teriflunomide can be considered as an ‘atypical’ antimetabolite because it inhibits proliferation of blasting lymphocytes without affecting resting and homeostatically expanding lymphocytes.36
Interestingly, DHODH-independent effects of teriflunomide have been described in vitro. For example, teriflunomide inhibits Janus kinases 1 and 3 (JAK1/3), two particular tyrosine kinases involved in cytokine signaling.37,38 Src family tyrosine kinases and receptor tyrosine kinases can also be targeted by teriflunomide.39 Two of the most important intracellular signaling pathways, the mitogen-activated protein kinase (MAPK) and nuclear factor-kappa B (NF-κB) pathways, are inhibited by teriflunomide in cultured cells. Finally, teriflunomide inhibits type-2 cyclooxygenase (COX-2) activity, with an IC50 of 0.5–20 μM,40 and it has also been suggested that teriflunomide acts as an inhibitor of the kynurenine pathway.30
Effects on the immune system
Fingolimod
The main action of fingolimod in the immune system is to regulate the trafficking of lymphocytes between primary and secondary lymphoid organs and circulating fluids (blood or lymph).41 Fingolimod causes transient and reversible lymphopenia without affecting lymphocyte proliferation or survival. Lymphopenia results from the retention of T-naïve and central memory T-cells (Tn and TCM) in secondary lymphoid organs. The retention of TCM cells strongly contributes to the therapeutic efficacy of fingolimod because TCM cells account for the vast majority of T-cells invading the CNS in MS.42,43 Fingolimod has no effect on circulating granulocytes, monocytes, eosinophils, erythrocytes, and platelets, and does not affect T-cell activation, proliferation, differentiation, or cytokine and antibody production.
The S1P1R, which is the main target of fingolimod, is one of the G-protein-coupled receptors that regulate migration of immune cells from primary and secondary lymphoid organs to the blood or lymph and vice versa. This particular function is also shared by S1P3Rs, S1P5Rs, the CXCR4, CXCR5, and CCR7 chemokine receptors, and the CB2 cannabinoid receptors.44 What is critical for the chemoattractive or chemorepulsive action of S1PRs is the concentration gradient of the ligand, S1P, between the blood or lymph and the interstitial fluid. S1P is present in relatively high concentrations in the blood or lymph, but only in low amounts in the interstitial fluid of lymphoid organs. Thus, S1P1Rs are maintained in a desensitized state by the high amount of S1P in the blood and lymph, whereas they are fully active in the interstitium of lymph nodes or other lymphoid organs. Circulating lymphocytes enter the lymph nodes across high endothelial venules, and T lymphocytes are attracted to the T zone in response to CCR7 activation by their cognate chemokine ligands (CCL19/CCL21). In contrast, B lymphocytes are attracted to the follicles by the CXCR5 ligand, CXCL13. Activation of S1P1Rs by endogenous S1P allows the egress of CCR7+ cluster of differentiation (CD)-45RA+ Tn cells and CCR7+CD45RA− TCM cells from the lymph nodes by over-riding the retention signal of CCR7. It is curious that the two ‘antagonistic’ receptors, S1P1R and CCR7, are both coupled to Gi proteins. Because of its high lipophilicity, fingolimod penetrates in large amounts inside the lymph nodes, and over-stimulates S1P1Rs after its transformation into fingolimod phosphate. This results in rapid desensitization and functional antagonism of S1P1Rs in spite of the low levels of S1P. Thus, fingolimod tips the balance between CCR7 and S1P1R in favor of CCR7 by simulating an environment rich in extracellular S1P. Cells lacking the chemokine retention signal, ie, CCR7−CD45RA+ and CCR7−CD45RA− effector memory T-cells (TEM) exit the lymph nodes independently of S1P1Rs and are therefore insensitive to fingolimod. TEM cells play an important role in mechanisms of immune surveillance at the site of entry of pathogens (eg, the intestinal mucosa), and represent a major defense against viral infections. Thus, fingolimod acts as an immune suppressant in MS with minimal or no effect on peripheral mechanisms of immune surveillance (Figure 5). Using a metaphor, fingolimod retains the army in the barracks, leaving the sentinels outside. S1P1Rs also regulate T-cell egress from the thymus as well as the cyclical shuttling of B-cells between the marginal and follicular zones of the spleen.45–49
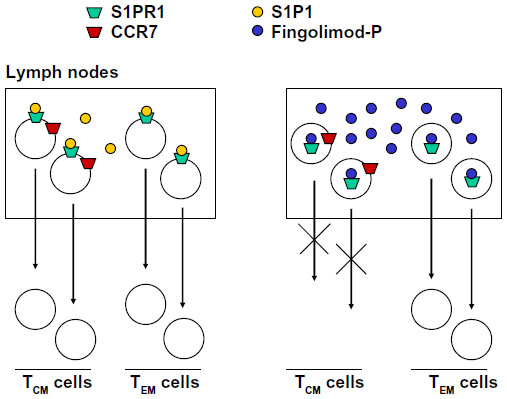 | Figure 5 Fingolimod inhibits the egress of TCM cells from lymph nodes without affecting the egress of TEM cells. TEM cells do not express CCR7 receptors. |
Differentiation of CD4+ Tn cells into diverse effectors and regulatory lineages is a central event in the regulation of immune function, with T-helper (TH)-1 and TH17 cells supporting autoimmunity in EAE and MS, and TReg cells mediating mechanisms of immune tolerance. Liu et al50 found that activation of S1P1Rs restrains the thymic generation, peripheral maintenance, and suppressive activity of TReg cells. S1P1Rs signal via the phosphatidylinositol-3-kinase/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) kinase pathway to over-ride the TReg-promoting function of transforming growth factor-β (TGF-β). More specifically, S1PRs restrain the action of the TGF-β-related signaling molecule, Smad-350,51 (Figure 6). One expects that fingolimod, acting as a functional antagonist of S1P1Rs, enhances TReg differentiation and activity at the expense of TH1 and TH17. Accordingly, fingolimod enhances the conversion of Foxp3−CD4+ T-cells into Foxp3+ TReg cells in lymphoid organs of non-obese diabetic mice,52 and improves TH1-mediated colitis and acute kidney injury induced by ischemia/reperfusion by enhancing the formation and activity of TReg cells.53,54 There are no published studies on the effect of fingolimod on TReg cells in MS patients. However, it is known that fingolimod reduces blood levels of TH17 cells by >90% in MS patients, perhaps because these cells are found primarily within the pool of TCM cells that are retained in the lymph nodes.55
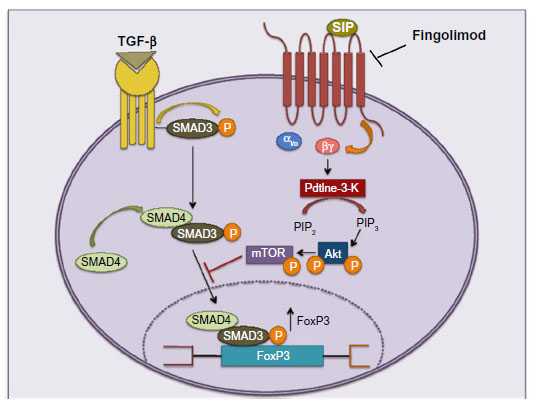 | Figure 6 Fingolimod enhances the production of TReg cells by acting as a functional antagonist of S1P1Rs. |
Dimethyl fumarate
Treumer et al56 showed that dimethyl fumarate causes apoptotic death of purified human T-lymphocytes stimulated with anti-CD3 antibodies and/or cytokines, a mechanism that might explain the beneficial effect of Fumaderm® in the treatment of psoriasis.11,12,57,58 How does dimethyl fumarate cause T-cell death when the Nrf2 system represents one of the cell’s main defenses against oxidative damage? One possible explanation is that T-cell death results from the initial trigger of Nrf2 activation, represented by the GSH depletion caused by dimethyl fumarate. If dimethyl fumarate concentrations at the target site are high, then the resulting GSH depletion may be sufficient to reach the threshold for the induction of cell death. Accordingly, dimethyl fumarate was found to cause death of activated splenocytes only at high micromolar concentrations, but did not affect cell viability at concentrations of 1 or 10 μM. However, low micromolar concentrations of dimethyl fumarate were sufficient to inhibit cytokine production from splenocytes,59 suggesting that the real mechanism responsible for the immune-suppressive action of dimethyl fumarate involves changes in the cytokine network rather than a direct effect on T-cell viability.
Recent evidence indicates that dimethyl fumarate acts at the core of the immunological synapse by inhibiting dendritic cell maturation and differentiation of Tn cells into autoreactive TH1 and TH17 cells. Dimethyl fumarate suppresses NF-κB signaling in dendritic cells by reducing p65 nuclear translocation and phosphorylation by the ERK (extracellular signal-regulated kinase)1/2 kinase target, mitogen stress-activated kinase 1.60 Dimethyl fumarate treatment in humans generates type-II dendritic cells that produce interleukin (IL)-10 instead of IL-12 and IL-23, and drive the differentiation of Tn cells into IL-4-producing TH2 cells instead of TH1 and TH17. The underlying mechanism has been clarified in mice, where dimethyl fumarate produced an initial GSH depletion followed by Nrf2 activation, induction of HO-1, and impaired signal transducers and activators of transcription 1 (STAT1) phosphorylation. HO-1 translocates to the nucleus, where it prevents transcription of the p19 subunit of IL-23, whereas STAT1 inactivation inhibits transcription of the p35 subunit of IL-12.61 This epigenetic mechanism may underlie the ability of dimethyl fumarate to inhibit the TH1 and TH17 branches of the adaptive immune response, which are critically involved in the pathogenesis of MS and psoriasis. No data are available on the effect of dimethyl fumarate on differentiation and activity of TReg cells. Thus, it is not known whether dimethyl fumarate affects mechanisms of immune tolerance in autoimmune disorders.
Laquinimod
Laquinimod reduces the expression of pro-inflammatory cytokines and enhances the expression of regulatory cytokines, thus hindering over-reactive immune responses.62,63 Central to the anti-inflammatory action of laquinimod is the inhibition of NF-κB, a signaling pathway that is activated by pro-inflammatory cytokines and, in turn, drives the expression of pro-inflammatory cytokines. Gurevich et al64 found that laquinimod inhibits the NF-κB pathway by suppressing the beta-transducin repeat containing encoding gene that increases ubiquitination and degradation of the NF-κB inhibitor protein, IκB. This was followed by the suppression of NF-κB downstream signaling genes.64 In vitro studies have shown that laquinimod does not affect viability or proliferation of immune cells and does not influence humoral immune responses.62
Laquinimod acts at the core of the immunological synapse by reducing the expression of genes related to the antigen presentation pathway in APCs.64 In addition, laquinimod down-regulates the expression of genes that are involved in the activation of APCs and T-lymphocyte chemotaxis.64 As a result of a primary action on APCs, laquinimod polarizes the immune response towards TH2 and TReg cells at the expense of TH1 and TH17 cells, thus protecting mice against EAE or experimental autoimmune neuritis (animal model of the Guillain–Barrè syndrome).65–68 This effect on antigen presentation is similar to that produced by glatiramer acetate, a first-line drug in the treatment of MS. The precise molecular mechanism by which laquinimod influences antigen presentation in dendritic cells and other APCs is unknown. Laquinimod might influence the kynurenine pathway, thus driving kynurenine metabolism towards the formation of molecules that promote immune tolerance at the immunological synapse. This interesting hypothesis warrants in-depth investigation.
Laquinimod can also modulate B-cell phenotypes by increasing the percentage of CD25+ cells in B lymphocytes isolated from healthy donors. However, this effect was not seen when lymphocytes were cultured from MS patients.63
Finally, laquinimod affects innate immunity by targeting monocytes and APC subpopulations. In macrophages, laquinimod down-regulates genes related to cell proliferation, phagocytosis, and T-cell stimulation; in addition, laquinimod treatment reduces monocyte infiltration and promotes the proliferation of anti-inflammatory subsets of monocytes and dendritic cells in mouse models of EAE and in MS patients.64,68
Teriflunomide
Teriflunomide restrains T-cell activation by inhibiting the formation of the immunological synapse, and by altering intracellular signaling.69,70 Activated T lymphocytes expand the intracellular pyrimidine pool by up to 8-fold, which not only serves as a substrate for DNA synthesis but is also involved in lipid biosynthesis, protein glycosylation, and other metabolic processes.71 Thus, teriflunomide behaves as a cytostatic agent. T-cells exposed to teriflunomide are still able to proliferate when activated ex vivo by mitogens,36 and teriflunomide administration does not reduce CD4+ and CD8+ blood counts in patients.
In addition to its activity on cell proliferation, teriflunomide directly affects lymphocyte function, affects B-cell proliferation and activation,36 and polarizes T-cell differentiation towards the TH2 phenotype at the expense of TH1 cells.36,72 The inhibition of JAK/STAT signaling, COX-2 activity, and NF-κB may contribute to the activity of teriflunomide against autoreactive immune responses.
Effects on the permeability of the blood–brain barrier
Fingolimod
S1P regulates the permeability of the endothelial barrier by interacting with different S1PRs expressed by endothelial cells. Activation of S1P1Rs reinforces the endothelial barrier by a Rac-dependent mechanism, which leads to membrane translocation of claudin (CLN)-5, occludins, or zonula occludens proteins (ZO-1 and -2).73 In contrast, activation of S1P2Rs and S1P3Rs enhances the permeability of the endothelial barrier by stimulating Rho activity, with ensuing activation of Rho-associated protein kinase (ROCK) and phosphate and tensin homologue (PTEN).74,75 Fingolimod reinforces the endothelial barrier in models of neuroinflammation,76,77 as well as in other models of vascular leakage.43 The endothelium of human brain microvessels expresses type-2 SphK, which phosphorylates fingolimod into fingolimod phosphate.78 It is possible that fingolimod phosphate acts as an agonist (and not as a functional antagonist) of endothelial S1P1Rs, which might be resistant to receptor desensitization because of a high receptor reserve or other mechanisms. However, this remains to be demonstrated. An alternative hypothesis is that fingolimod acts on reactive astrocytes associated with MS lesions by reducing the activity of acidic sphingomyelinase and the resulting formation of ceramide. Ceramide acts as a pro-inflammatory lipid molecule in EAE and MS, enhancing the permeability of the blood–brain barrier.79 The two hypotheses on the mechanism of action of fingolimod are not mutually exclusive. Interestingly, both fingolimod and S1P reduce the activity of P-glycoprotein at the blood–brain barrier.80,81 P-glycoprotein is the main drug efflux pump, mediating mechanisms of drug resistance in the CNS and other organs. By inhibiting P-glycoprotein activity, fingolimod might enhance drug delivery in the CNS and boost the activity of multiple classes of drugs acting inside the brain parenchyma.
There are exceptions to the reinforcing action of fingolimod on the endothelial barrier. Of 2,564 MS patients treated with fingolimod in phase III clinical studies, 13 developed reversible macular edema, suggesting that fingolimod may enhance permeability of the blood–retinal barrier. However, it should be highlighted that this adverse effect was dose-dependent, and macular edema was observed only in two patients treated with the approved dose of fingolimod (0.5 mg/day) for the treatment of RRMS.82 The exact mechanism by which fingolimod affects permeability at the blood–retinal barrier is unknown.
Dimethyl fumarate
To our knowledge, studies on the action of dimethyl fumarate on blood–brain barrier permeability have not been published. Dimethyl fumarate inhibits nuclear translocation and activation of NF-κB in human endothelial cells, thereby suppressing the induction of adhesion molecules in response to pro-inflammatory cytokines.83 More recent data demonstrate that dimethyl fumarate suppresses the expression of type-2 vascular endothelial growth factor (VEGF) receptors (VEGFR2) in human endothelial cells by inhibiting the interaction of the Sp1 transcription factor with specific responsive elements at the VEGFR2 gene promoter.84 This particular mechanism might restrain the action of VEGF at the blood–brain barrier, and, therefore, be relevant to the treatment of MS. Whether, and in which particular context, dimethyl fumarate inhibits the action of VEGF in the endothelium of brain microvessels remains to be established.
Laquinimod
The effect of laquinimod on the permeability of the blood–brain barrier remains to be investigated. Wegner et al66 demonstrated a reduced migratory capacity of immune cells into the CNS in EAE-challenged mice treated with laquinimod. However, when the number of T-cells and macrophages was analyzed within CNS lesions, there was no significant difference between control mice and mice treated with laquinimod.66
Teriflunomide
To the best of our knowledge, there are no published studies on the effect of teriflunomide on the blood–brain barrier.
Direct effects on CNS cells
Fingolimod
Fingolimod, which is highly lipophilic, easily crosses the blood–brain barrier. In addition, S1P1Rs, which are the main targets of fingolimod phosphate, are present in neurons, astrocytes, oligodendrocytes, and microglia, and are more abundant in the CNS than in any other organ. S1P3Rs are less abundant in the CNS, but they are found in neurons, astrocytes, and microglia. S1P5Rs are predominantly expressed in oligodendrocytes.43,85 S1P1Rs and S1P3Rs are strongly up-regulated in astrocytes present in MS lesions,86 suggesting that these receptors have a role in the process of reactive gliosis and contribute to the overall mechanism of inflammation-induced cell death or repair. The specific role of astrocytic S1P1Rs in the action of fingolimod has been examined in an elegant study using conditional null mouse mutants lacking S1P1Rs either in astrocytes or neurons and challenged with EAE. Treatment with fingolimod, administered at the peak of the neurological symptoms, caused the expected peripheral lymphopenia in all mutants, but showed efficacy in relieving neurological signs of EAE only in wild-type mice or in mice lacking S1P1Rs in neurons. Mice lacking S1P1Rs in astrocytes developed less severe EAE, which was completely resistant to fingolimod.87 These data suggest that S1P1Rs present in astrocytes support the development of EAE and are specifically targeted by fingolimod. However, the study is not sufficient to exclude the involvement of neuronal S1P1Rs in the overall effect of fingolimod in MS.87 We and others have found that fingolimod protects cultured neurons against excitotoxic death. Neuroprotection was mediated by S1P1R-mediated activation of the MAPK and the phosphatidylinositol-3-kinase pathways, which are known to support cell survival.88,89 This mechanism might contribute to the protective activity of fingolimod in models of brain ischemia.90–92 Interestingly, fingolimod enhances the production of brain-derived neurotrophic factor (BDNF) in cultured neurons and in the mouse brain.88 This effect has been shown to mediate the protective effect of fingolimod phosphate against β-amyloid neurotoxicity in cultured neurons.93 Carriers of the low-BDNF expressing Met-66 allele show memory deficits and low hippocampal volume.94–97 In addition, BDNF is tightly linked to the pathophysiology of major depression, and serum BDNF levels predict response to antidepressant medication.98–100 We expect that fingolimod improves cognitive function and depressive mood in patients with MS by enhancing brain BDNF levels. This interesting hypothesis warrants further investigation.
The presence of S1PRs in oligodendrocytes suggests that fingolimod may have direct effects on mechanisms of demyelination and remyelination in MS. In vitro studies show that fingolimod phosphate promotes the differentiation of neural stem cells towards the oligodendroglial lineage and supports the proliferation and survival of oligodendrocyte progenitors.78,101–104 In non-immunological in vivo models of demyelination, fingolimod protects against myelin and axonal damage, but does not promote remyelination.54,105 In contrast, fingolimod has been found to enhance remyelination markers in in vitro models of demyelination.106,107 Thus, the possibility that fingolimod facilitates mechanisms of myelin repair via a direct action on oligodendrocytes is still open.108
Dimethyl fumarate
Dimethyl fumarate activates the Nrf2 system, which represents one of the major intracellular protective pathways against oxidative damage. ROS are critically involved in the pathophysiology of MS by promoting transendothelial leukocyte migration and causing myelin and axonal damage.109–111 Thus, in principle, dimethyl fumarate has the potential to protect neurons and oligodendrocytes in EAE and MS.
Dimethyl fumarate activates the Nrf2 pathway by causing an initial GSH depletion, which does not reach the threshold for the induction of cell death. This mechanism is reminiscent of ‘ischemic tolerance’, whereby short episodes of ischemia protect the brain or the heart against a subsequent ischemic insult. In vitro studies have shown that dimethyl fumarate protects neurons against oxidative damage.15,59 However, neuroprotection is critically time- and context-dependent. In one study, cultured neurons were challenged with high concentrations of glutamate, which reduce GSH levels by limiting the intracellular transport of the GSH precursor, cystine.59 Under these conditions, pretreatment with dimethyl fumarate was highly neuroprotective. In contrast, dimethyl fumarate became neurotoxic when co-applied with sub-maximal concentrations of glutamate. Once again, the initial GSH depletion caused by dimethyl fumarate might be permissive in causing neuronal death if combined with proinflammatory cytokines, excitotoxins, or other harmful stimuli.59 This raises the question of whether the drug exerts neuroprotective or neurotoxic effects under conditions in which neurons are repeatedly exposed to toxic insults, as occurs during neuroinflammatory flare-ups in MS. Studies in models of relapsing-remitting EAE in which the drug is applied at time intervals that coincide with the onset or peak of relapses may help to clarify this issue.
Astrocytes and microglia, which are critically involved in the neuroinflammatory cascade in EAE and MS, are also targeted by dimethyl fumarate via the activation of Nrf2 and the inhibition of NF-κB. This is associated with induction of HO-1, activation of GSH synthesis, and inhibition of proinflammatory cytokine production and inducible nitric oxide synthase.19,112 However, again, dimethyl fumarate applied to cultured astrocytes was shown to acutely deplete GSH levels, which was not associated with cell death.113 The precise consequences of acute GSH depletion in astrocytes under conditions of neuroinflammation remain to be determined. Only a few studies have examined the effects of dimethyl fumarate or other fumarate esters on oligodendrocytes. Dimethyl fumarate causes GSH depletion and Mrf2 activation in an OLN-93 oligodendroglial cell line, similar to what is observed in other cell types.114 However, in a non-immune-dependent in vivo model of demyelination (the cuprizone model in mice), dimethyl fumarate showed no protective activity on oligodendrocytes.115
Laquinimod
Laquinimod can cross the blood–brain barrier,62 and a series of indirect observations suggest that the drug affects the viability of neurons and oligodendrocytes independently of its anti-inflammatory effects. In the ALLEGRO study, laquinimod showed a greater efficacy on disability progression than on annualized relapse rate, in contrast with the majority of drugs used in the treatment of MS. The evidence that laquinimod reduces demyelination, axonal damage, and lesion size in the EAE model65–68 is also consistent with direct action of the drug in the CNS. Regulation of BDNF levels may represent a point of convergence between the central actions of fingolimod and laquinimod. Accordingly, laquinimod treatment enhances BDNF expression and reduces CNS injury in EAE mice.67 Laquinimod can also modulate inhibitory gamma-aminobutyric acid (GABA)-ergic transmission in the CNS, under both normal and inflammatory conditions.116 However, it should be noted that the enhancing effect of laquinimod on GABA-mediated inhibitory postsynaptic currents requires concentrations of the drug that are several-fold higher than those reached inside the CNS during treatment.116
Laquinimod reduces astrogliosis and impairs astrocyte activation in vitro by negatively regulating the NF-κB pathway. Interestingly, in contrast with dimethyl fumarate, laquinimod protects oligodendrocytes in the cuprizone model of demyelination, an effect that has been ascribed to its action on astrocytes.117
As highlighted above, laquinimod is a quinoline carboxamide derivative, and, as such, has the potential to modulate the activity of the kynurenine pathway.30 This pathway has been implicated in the pathophysiology of multiple disorders of the CNS, including acute and chronic neurodegenerative disorders.25 Two questions must be addressed: 1) which specific enzyme of the kynurenine pathway is inhibited by laquinimod; and 2) whether, and to what extent, activity of laquinimod on this pathway contributes to the central effects of the drug in EAE and MS.
Teriflunomide
There is no evidence suggesting a direct effect of teriflunomide on neurons and other cells resident in the CNS. However, this is a possibility if the modulation of the kynurenine pathway by teriflunomide30 is confirmed and better characterized.
Conclusion
We have discussed the molecular pharmacodynamics of fingolimod, dimethyl fumarate, laquinimod, and teriflunomide in an attempt to compare their mechanism of action in MS.
In brief, fingolimod has a unique mechanism of action among current MS therapies. It prevents the egress of particular subpopulations of T lymphocytes (ie, Tn and TCM cells) from secondary lymphoid organs, thereby restraining autoimmunity in patients with MS. This particular mechanism results from desensitization and internalization of S1P1Rs on T-cells. Interestingly, fingolimod does not cause peripheral depletion of TEM cells, which mediate immune surveillance at the site of entry of pathogens. Fingolimod also decreases the permeability of the blood–brain barrier and acts inside the CNS parenchyma by supporting neuronal survival. The latter effect may be mediated by an increased BDNF formation.
Dimethyl fumarate is thought to work in MS in two ways: via immune-suppressive activity and via protective effects in the CNS. Dimethyl fumarate alters the balance in the blood of immune reactive and immune tolerant T-cells, thereby causing immune suppression. Dimethyl fumarate activates the Nrf2 pathway of cell defense as a result of an initial depletion of GSH. In principle, this mechanism may result in cell protection; however, the initial GSH depletion may synergize with environmental insults, thereby promoting cell death. Thus, the impact of dimethyl fumarate on mechanisms of cell death and survival may be context-dependent.
Laquinimod has multiple (but less defined) mechanisms of action in MS. It is thought to act by modulating the immune system towards an anti-inflammatory rather than a pro-inflammatory response. Laquinimod may also prevent autoreactive T-cells from entering the CNS. Teriflunomide acts as a specific inhibitor of the de novo pyrimidine synthesis, thereby inhibiting lymphocyte proliferation. In addition, teriflunomide polarizes T-cell differentiation towards a TH2 phenotype at the expense of autoreactive TH1 cells.
While there are no available data on a direct effect of laquinimod and teriflunomide on CNS cells, it is possible that both drugs influence mechanisms of neuronal death and survival by interfering with the kynurenine pathway of tryptophan metabolism.
The pharmacodynamic profile of new oral drugs at the level of the immune system, blood–brain barrier, and CNS cells should be taken into account when choosing one of these drugs in the treatment of MS.
Acknowledgments
We thank Carmen Innes, an independent medical writer, who provided writing assistance on behalf of Springer Healthcare Communications. This assistance was sponsored by Novartis Farma, Origgio, Italy.
Disclosure
The authors report no conflicts of interest in this work.
References
Aktas O, Küry P, Kieseier B, Hartung HP. Fingolimod is a potential novel therapy for multiple sclerosis. Nat Rev Neurol. 2010;6(7):373–382. | |
Cohen JA, Barkhof F, Comi G, et al; TRANSFORMS Study Group. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–415. | |
Kappos L, Radue EW, O’Connor P, et al; FREEDOMS Study Group. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. | |
Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 2010;33(2):91–101. | |
Boujaoude LC, Bradshaw-Wilder C, Mao C, et al. Cystic fibrosis transmembrane regulator regulates uptake of sphingoid base phosphates and lysophosphatidic acid: modulation of cellular activity of sphingosine 1-phosphate. J Biol Chem. 2001;276(38):35258–35264. | |
Kobayashi N, Nishi T, Hirata T, et al. Sphingosine 1-phosphate is released from the cytosol of rat platelets in a carrier-mediated manner. J Lipid Res. 2006;47(3):614–621. | |
Mitra P, Oskeritzian CA, Payne SG, Beaven MA, Milstien S, Spiegel S. Role of ABCC1 in export of sphingosine-1-phosphate from mast cells. Proc Natl Acad Sci U S A. 2006;103(44):16394–16399. | |
Rivera J, Proia RL, Olivera A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol. 2008;8(10):753–763. | |
David OJ, Kovarik JM, Schmouder RL. Clinical pharmacokinetics of fingolimod. Clin Pharmacokinet. 2012;51(1):15–28. | |
Arnon TI, Xu Y, Lo C, et al. GRK2-dependent S1PR1 desensitization is required for lymphocytes to overcome their attraction to blood. Science. 2011;333(6051):1898–1903. | |
Mrowietz U, Asadullah K. Dimethylfumarate for psoriasis: more than a dietary curiosity. Trends Mol Med. 2005;11(1):43–48. | |
Rostami-Yazdi M, Mrowietz U. Fumaric acid esters. Clin Dermatol. 2008;26(5):522–526. | |
Gold R, Kappos L, Arnold DL, et al; DEFINE Study Investigators. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–1107. Erratum in: N Engl J Med. 2012;367(24):2362. | |
Fox RJ, Miller DH, Phillips JT, et al; CONFIRM Study Investigators. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087–1097. Erratum in: N Engl J Med. 2012;367(17):1673. | |
Scannevin RH, Chollate S, Jung MY, et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J Pharmacol Exp Ther. 2012;341(1):274–284. | |
Ashrafian H, Czibik G, Bellahcene M, et al. Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metab. 2012;15(3):361–371. | |
Itoh K, Chiba T, Takahashi S, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236(2):313–322. | |
Venugopal R, Jaiswal AK. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene. 1998;17(24):3145–3156. | |
Lin SX, Lisi L, Dello Russo C, et al. The anti-inflammatory effects of dimethyl fumarate in astrocytes involve glutathione and haem oxygenase-1. ASN Neuro. 2011;3(2):75–84. | |
Wakabayashi N, Slocum SL, Skoko JJ, Shin S, Kensler TW. When NRF2 talks, who’s listening? Antioxid Redox Signal. 2010;13(11):1649–1663. | |
Brigelius-Flohé R, Flohé L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid Redox Signal. 2011;15(8):2335–2381. | |
Ji L, Li H, Gao P, et al. Nrf2 pathway regulates multidrug-resistance-associated protein 1 in small cell lung cancer. PLoS One. 2013;8(5):e63404. | |
Kensler TW, Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis. 2010;31(1):90–99. | |
Comi G, Jeffery D, Kappos L, et al; ALLEGRO Study Group. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med. 2012;366(11):1000–1009. | |
Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ. Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci. 2012;13(7):465–777. | |
Hönig A, Rieger L, Kapp M, Sütterlin M, Dietl J, Kämmerer U. Indoleamine 2,3-dioxygenase (IDO) expression in invasive extravillous trophoblast supports role of the enzyme for materno-fetal tolerance. J Reprod Immunol. 2004;61(2):79–86. | |
Fallarino F, Vacca C, Orabona C, et al. Functional expression of indoleamine 2,3-dioxygenase by murine CD8 alpha(+) dendritic cells. Int Immunol. 2002;14(1):65–68. | |
Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. 2002;196(4):459–468. | |
Belladonna ML, Grohmann U, Guidetti P, et al. Kynurenine pathway enzymes in dendritic cells initiate tolerogenesis in the absence of functional IDO. J Immunol. 2006;177(1):130–137. | |
Chen Y, Meininger V, Guillemin GJ. Recent advances in the treatment of amyotrophic lateral sclerosis. Emphasis on kynurenine pathway inhibitors. Cent Nerv Syst Agents Med Chem. 2009;9(1):32–39. | |
Killestein J, Rudick RA, Polman CH. Oral treatment for multiple sclerosis. Lancet Neurol. 2011;10(11):1026–1034. | |
O’Connor P, Wolinsky JS, Confavreux C, et al; TEMSO Trial Group. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365(14):1293–1303. | |
Greene S, Watanabe K, Braatz-Trulson J, Lou L. Inhibition of dihydroorotate dehydrogenase by the immunosuppressive agent leflunomide. Biochem Pharmacol. 1995;50(6):861–867. | |
Williamson RA, Yea CM, Robson PA, et al. Dihydroorotate dehydrogenase is a high affinity binding protein for A77 1726 and mediator of a range of biological effects of the immunomodulatory compound. J Biol Chem. 1995;270(38):22467–22472. | |
Fox RI, Herrmann ML, Frangou CG, et al. Mechanism of action for leflunomide in rheumatoid arthritis. Clin Immunol. 1999;93(3):198–208. | |
Claussen MC, Korn T. Immune mechanisms of new therapeutic strategies in MS: teriflunomide. Clin Immunol. 2012;142(1):49–56. | |
Siemasko K, Chong AS, Jäck HM, Gong H, Williams JW, Finnegan A. Inhibition of JAK3 and STAT6 tyrosine phosphorylation by the immunosuppressive drug leflunomide leads to a block in IgG1 production. J Immunol. 1998;160(4):1581–1588. | |
González-Alvaro I, Ortiz AM, Domínguez-Jiménez C, Aragón-Bodi A, Díaz Sánchez B, Sánchez-Madrid R. Inhibition of tumour necrosis factor and IL-17 production by leflunomide involves the JAK/STAT pathway. Ann Rheum Dis. 2009;68(10):1644–1650. | |
Mattar T, Kochhar K, Bartlett R, Bremer EG, Finnegan A. Inhibition of the epidermal growth factor receptor tyrosine kinase activity by leflunomide. FEBS Lett. 1993;334(2):161–164. | |
Hamilton LC, Vojnovic I, Warner TD. A771726, the active metabolite of leflunomide, directly inhibits the activity of cyclo-oxygenase-2 in vitro and in vivo in a substrate-sensitive manner. Br J Pharmacol. 1999;127(7):1589–1596. | |
Cohen JA, Chun J. Mechanisms of fingolimod’s efficacy and adverse effects in multiple sclerosis. Ann Neurol. 2011;69(5):759–777. | |
Kivisäkk P, Mahad DJ, Callahan MK, et al. Expression of CCR7 in multiple sclerosis: implications for CNS immunity. Ann Neurol. 2004;55(5):627–638. | |
Brinkmann V, Billich A, Baumruker T, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9(11):883–897. | |
Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol. 2012;30:69–94. | |
Hla T, Brinkmann V. Sphingosine 1-phosphate (S1P): physiology and the effects of S1P receptor modulation. Neurology. 2011;76(8 Suppl 3):S3–S8. | |
Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355–360. | |
Sallusto F, Mackay CR. Chemoattractants and their receptors in homeostasis and inflammation. Curr Opin Immunol. 2004;16(6):724–731. | |
Brinkmann V. FTY720 (fingolimod) in multiple sclerosis: therapeutic effects in the immune and the central nervous system. Br J Pharmacol. 2009;158(5):1173–1182. | |
Oo ML, Thangada S, Wu MT, et al. Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. J Biol Chem. 2007;282(12):9082–9089. | |
Liu G, Burns S, Huang G, et al. The receptor S1P1 overrides regulatory T cell-mediated immune suppression through Akt-mTOR. Nat Immunol. 2009;10(7):769–777. | |
Liu G, Yang K, Burns S, Shrestha S, Chi H. The S1P(1)-mTOR axis directs the reciprocal differentiation of T(H)1 and T(reg) cells. Nat Immunol. 2010;11(11):1047–1056. | |
Sun Y, Wang W, Shan B, et al. FTY720-induced conversion of conventional Foxp3- CD4+ T cells to Foxp3+ regulatory T cells in NOD mice. Am J Reprod Immunol. 2011;66(5):349–362. | |
Daniel C, Sartory N, Zahn N, Geisslinger G, Radeke HH, Stein JM. FTY720 ameliorates Th1-mediated colitis in mice by directly affecting the functional activity of CD4+CD25+ regulatory T cells. J Immunol. 2007;178(4):2458–2468. | |
Kim HJ, Miron VE, Dukala D, et al. Neurobiological effects of sphingosine 1-phosphate receptor modulation in the cuprizone model. FASEB J. 2011;25(5):1509–1518. | |
Mehling M, Lindberg R, Raulf F, et al. Th17 central memory T cells are reduced by FTY720 in patients with multiple sclerosis. Neurology. 2010;75(5):403–410. | |
Treumer F, Zhu K, Gläser R, Mrowietz U. Dimethyl fumarate is a potent inducer of apoptosis in human T cells. J Invest Dermatol. 2003;121(6):1383–1388. | |
Mrowietz U, Asadullah K. Dimethyl fumarate for psoriasis: more than a dietary curiosity. Trends Mol Med. 2005;11(1):43–48. | |
Ormerod AD, Mrowietz U. Fumaric acid esters, their place in the treatment of psoriasis. Br J Dermatol. 2004;150(4):630–632. | |
Albrecht P, Bouchachia I, Goebels N, et al. Effects of dimethyl fumarate on neuroprotection and immunomodulation. J Neuroinflammation. 2012;9:163–172. | |
Peng H, Guerau-de-Arellano M, Mehta VB, et al. Dimethyl fumarate inhibits dendritic cell maturation via nuclear factor κB (NF-κB) and extracellular signal-regulated kinase 1 and 2 (ERK1/2) and mitogen stress-activated kinase 1 (MSK1) signaling. J Biol Chem. 2012;287(33):28017–28026. | |
Ghoreschi K, Brück J, Kellerer C, et al. Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. J Exp Med. 2011;208(11):2291–2303. | |
Brück W, Wegner C. Insight into the mechanism of laquinimod action. J Neurol Sci. 2011;306(1–2):173–179. | |
Toubi E, Nussbaum S, Staun-Ram E, et al. Laquinimod modulates B cells and their regulatory effects on T cells in multiple sclerosis. J Neuroimmunol. 2012;251(1–2):45–54. | |
Gurevich M, Gritzman T, Orbach R, Tuller T, Feldman A, Achiron A. Laquinimod suppress antigen presentation in relapsing-remitting multiple sclerosis: in-vitro high-throughput gene expression study. J Neuroimmunol. 2010;221(1–2):87–94. | |
Zou LP, Abbas N, Volkmann I, et al. Suppression of experimental autoimmune neuritis by ABR-215062 is associated with altered Th1/Th2 balance and inhibited migration of inflammatory cells into the peripheral nerve tissue. Neuropharmacol. 2002;42(5):731–739. | |
Wegner C, Stadelmann C, Pförtner R, et al. Laquinimod interferes with migratory capacity of T cells and reduces IL-17 levels, inflammatory demyelination and acute axonal damage in mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. 2010;227(1–2):133–143. | |
Aharoni R, Saada R, Eilam R, Hayardeny L, Sela M, Arnon R. Oral treatment with laquinimod augments regulatory T-cells and brain-derived neurotrophic factor expression and reduces injury in the CNS of mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. 2012;251(1–2):14–24. | |
Schulze-Topphoff U, Shetty A, Varrin-Doyer M, et al. Laquinimod, a quinoline-3-carboxamide, induces type II myeloid cells that modulate central nervous system autoimmunity. PLoS One. 2012;7(3):e33797. | |
Layseca-Espinosa E, Pedraza-Alva G, Montiel JL, et al. T cell aggregation induced through CD43: intracellular signals and inhibition by the immunomodulatory drug leflunomide. J Leukoc Biol. 2003;74(6):1083–1093. | |
Zeyda M, Poglitsch M, Geyeregger R, et al. Disruption of the interaction of T cells with antigen-presenting cells by the active leflunomide metabolite teriflunomide: involvement of impaired integrin activation and immunologic synapse formation. Arthritis Rheum. 2005;52(9):2730–2739. | |
Quéméneur L, Gerland LM, Flacher M, Ffrench M, Revillard JP, Genestier L. Differential control of cell cycle, proliferation, and survival of primary T lymphocytes by purine and pyrimidine nucleotides. J Immunol. 2003;170(10):4986–4995. | |
Dimitrova P, Skapenko A, Herrmann ML, Schleyerbach R, Kalden JR, Schulze-Koops H. Restriction of de novo pyrimidine biosynthesis inhibits Th1 cell activation and promotes Th2 cell differentiation. J Immunol. 2002;169(6):3392–3399. | |
Wang L, Chiang ET, Simmons JT, Garcia JG, Dudek SM. FTY720-induced human pulmonary endothelial barrier enhancement is mediated by c-Abl. Eur Respir J. 2011;38(1):78–88. | |
Sanchez T, Skoura A, Wu MT, Casserly B, Harrington EO, Hla T. Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arterioscler Thromb Vasc Biol. 2007;27(6):1312–1318. | |
Niessen F, Furlan-Freguia C, Fernández JA, et al. Endogenous EPCR/aPC-PAR1 signaling prevents inflammation-induced vascular leakage and lethality. Blood. 2009;113(12):2859–2866. | |
Balatoni B, Storch MK, Swoboda EM, et al. FTY720 sustains and restores neuronal function in the DA rat model of MOG-induced experimental autoimmune encephalomyelitis. Brain Res Bull. 2007;74(5):307–316. | |
Foster CA, Mechtcheriakova D, Storch MK, et al. FTY720 rescue therapy in the dark agouti rat model of experimental autoimmune encephalomyelitis: expression of central nervous system genes and reversal of blood-brain-barrier damage. Brain Pathol. 2009;19(2):254–266. | |
Miron VE, Hall JA, Kennedy TE, Soliven B, Antel JP. Cyclical and dose-dependent responses of adult human mature oligodendrocytes to fingolimod. Am J Pathol. 2008;173(4):1143–1152. | |
van Doorn R, Nijland PG, Dekker N, et al. Fingolimod attenuates ceramide-induced blood-brain barrier dysfunction in multiple sclerosis by targeting reactive astrocytes. Acta Neuropathol. 2012;124(3):397–410. | |
Cannon RE, Peart JC, Hawkins BT, Campos CR, Miller DS. Targeting blood-brain barrier sphingolipid signaling reduces basal P-glycoprotein activity and improves drug delivery to the brain. Proc Natl Acad Sci U S A. 2012;109(39):15930–15935. | |
Cartwright TA, Campos CR, Cannon RE, Miller DS. Mrp1 is essential for sphingolipid signaling to p-glycoprotein in mouse blood-brain and blood-spinal cord barriers. J Cereb Blood Flow Metab. 2013;33(3):381–388. | |
Jain N, Bhatti MT. Fingolimod-associated macular edema: incidence, detection, and management. Neurology. 2012;78(9):672–680. | |
Loewe R, Holnthoner W, Gröger M, et al. Dimethyl fumarate inhibits TNF-induced nuclear entry of NF-kappa B/p65 in human endothelial cells. J Immunol. 2002;168(9):4781–4787. | |
Meissner M, Doll M, Hrgovic I, et al. Suppression of VEGFR2 expression in human endothelial cells by dimethyl fumarate treatment: evidence for anti-angiogenic action. J Invest Dermatol. 2011;131(6):1356–1364. | |
Soliven B, Miron V, Chun J. The neurobiology of sphingosine 1-phosphate signaling and sphingosine 1-phosphate receptor modulators. Neurology. 2011;76(8 Suppl 3):S9–S14. | |
Van Doorn R, Van Horssen J, Verzijl D, et al. Sphingosine 1-phosphate receptor 1 and 3 are upregulated in multiple sclerosis lesions. Glia. 2010;58(12):1465–1476. | |
Choi JW, Gardell SE, Herr DR, et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci U S A. 2011;108(2):751–756. | |
Deogracias R, Yazdani M, Dekkers MP, et al. Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2012;109(35):14230–14235. | |
Di Menna L, Molinaro G, Di Nuzzo L, et al. Fingolimod protects cultured cortical neurons against excitotoxic death. Pharmacol Res. 2013;67(1):1–9. | |
Czech B, Pfeilschifter W, Mazaheri-Omrani N, et al. The immunomodulatory sphingosine 1-phosphate analog FTY720 reduces lesion size and improves neurological outcome in a mouse model of cerebral ischemia. Biochem Biophys Res Commun. 2009;389(2):251–256. | |
Hasegawa Y, Suzuki H, Sozen T, Rolland W, Zhang JH. Activation of sphingosine 1-phosphate receptor-1 by FTY720 is neuroprotective after ischemic stroke in rats. Stroke. 2010;41(2):368–374. | |
Wei Y, Yemisci M, Kim HH, et al. Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann Neurol. 2011;69(1):119–129. | |
Doi Y, Takeuchi H, Horiuchi H, et al.85. Fingolimod phosphate attenuates oligomeric amyloid β-induced neurotoxicity via increased brain-derived neurotrophic factor expression in neurons. PLoS One. 2013;8(4):e61988. | |
Molendijk ML, Bus BA, Spinhoven P, et al. A systematic review and meta-analysis on the association between BDNF val(66)met and hippocampal volume: a genuine effect or a winners curse? Am J Med Genet B Neuropsychiatr Genet. 2012;159B(6):731–740. | |
Kanellopoulos D, Gunning FM, Morimoto SS, et al. Hippocampal volumes and the brain-derived neurotrophic factor val66met polymorphism in geriatric major depression. Am J Geriatr Psychiatry. 2011;19(1):13–22. | |
Erickson KI, Prakash RS, Voss MW, et al. Brain-derived neurotrophic factor is associated with age-related decline in hippocampal volume. J Neurosci. 2010;30(15):5368–5375. | |
Benjamin S, McQuoid DR, Potter GG, et al. The brain-derived neurotrophic factor Val66Met polymorphism, hippocampal volume, and cognitive function in geriatric depression. Am J Geriatr Psychiatry. 2010;18(4):323–331. | |
Neto FL, Borges G, Torres-Sanchez S, Mico JA, Berrocoso E. Neurotrophins role in depression neurobiology: a review of basic and clinical evidence. Curr Neuropharmacol. 2011;9(4):530–552. | |
Wolkowitz OM, Wolf J, Shelly W, et al. Serum BDNF levels before treatment predict SSRI response in depression. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(7):1623–1630. | |
Yu H, Wang DD, Wang Y, Liu T, Lee FS, Chen ZY. Variant brain-derived neurotrophic factor Val66Met polymorphism alters vulnerability to stress and response to antidepressants. J Neurosci. 2012;32(12):4092–4101. | |
Coelho RP, Payne SG, Bittman R, Spiegel S, Sato-Bigbee C. The immunomodulator FTY720 has a direct cytoprotective effect in oligodendrocyte progenitors. J Pharmacol Exp Ther. 2007;323(2):626–635. | |
Jung CG, Kim HJ, Miron VE, et al. Functional consequences of S1P receptor modulation in rat oligodendroglial lineage cells. Glia. 2007;55(16):1656–1667. | |
Miron VE, Jung CG, Kim HJ, Kennedy TE, Soliven B, Antel JP. FTY720 modulates human oligodendrocyte progenitor process extension and survival. Ann Neurol. 2008;63(1):61–71. | |
Bieberich E. There is more to a lipid than just being a fat: sphingolipid-guided differentiation of oligodendroglial lineage from embryonic stem cells. Neurochem Res. 2011;36(9):1601–1611. | |
Hu Y, Lee X, Ji B, et al. Sphingosine 1-phosphate receptor modulator fingolimod (FTY720) does not promote remyelination in vivo. Mol Cell Neurosci. 2011;48(1):72–81. | |
Miron VE, Ludwin SK, Darlington PJ, et al. Fingolimod (FTY720) enhances remyelination following demyelination of organotypic cerebellar slices. Am J Pathol. 2010;176(6):2682–2694. | |
Jackson SJ, Giovannoni G, Baker D. Fingolimod modulates microglial activation to augment markers of remyelination. J Neuroinflammation. 2011;8:76. | |
Kipp M, Amor S. FTY720 on the way from the base camp to the summit of the mountain: relevance for remyelination. Mult Scler. 2012;18(3):258–263. | |
Vladimirova O, O’Connor J, Cahill A, Alder H, Butunoi C, Kalman B. Oxidative damage to DNA in plaques of MS brains. Mult Scler. 1998;4(5):413–418. | |
Smith KJ, McDonald WI. The pathophysiology of multiple sclerosis: the mechanisms underlying the production of symptoms and the natural history of the disease. Philos Trans R Soc Lond B Biol Sci. 1999;354(1390):1649–1473. | |
Lee DH, Gold R, Linker RA. Mechanisms of oxidative damage in multiple sclerosis and neurodegenerative diseases: therapeutic modulation via fumaric acid esters. Int J Mol Sci. 2012;13(9):11783–11803. | |
Wilms H, Sievers J, Rickert U, Rostami-Yazdi M, Mrowietz U, Lucius R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model of brain inflammation. J Neuroinflammation. 2010;7:30. | |
Schmidt MM, Dringen R. Fumaric acid diesters deprive cultured primary astrocytes rapidly of glutathione. Neurochem Int. 2010;57(4):460–467. | |
Thiessen A, Schmidt MM, Dringen R. Fumaric acid dialkyl esters deprive cultured rat oligodendroglial cells of glutathione and upregulate the expression of heme oxygenase 1. Neurosci Lett. 2010;475(1):56–60. | |
Moharregh-Khiabani D, Blank A, Skripuletz T, et al. Effects of fumaric acids on cuprizone induced central nervous system de- and remyelination in the mouse. PLoS One. 2010;5(7):e11769. | |
Ruffini F, Rossi S, Bergamaschi A, et al. Laquinimod prevents inflammation-induced synaptic alterations occurring in experimental autoimmune encephalomyelitis. Mult Scler. 2012;19(8):1084–1094. | |
Raasch J, Zeller N, van Loo G, et al. IkappaB kinase 2 determines oligodendrocyte loss by non-cell-autonomous activation of NF-kappaB in the central nervous system. Brain. 2011;134(Pt 4):1184–1198. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.