")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Molecular mechanisms of cisplatin resistance in cervical cancer
Authors Zhu H, Luo H, Zhang W , Shen Z, Hu X, Zhu X
Received 14 February 2016
Accepted for publication 10 March 2016
Published 7 June 2016 Volume 2016:10 Pages 1885—1895
DOI https://doi.org/10.2147/DDDT.S106412
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Haiyan Zhu, Hui Luo, Wenwen Zhang, Zhaojun Shen, Xiaoli Hu, Xueqiong Zhu
Department of Obstetrics and Gynecology, The Second Affiliated Hospital of Wenzhou Medical University, Wenzhou, People’s Republic of China
Abstract: Patients with advanced or recurrent cervical cancer have poor prognosis, and their 1-year survival is only 10%–20%. Chemotherapy is considered as the standard treatment for patients with advanced or recurrent cervical cancer, and cisplatin appears to treat the disease effectively. However, resistance to cisplatin may develop, thus substantially compromising the efficacy of cisplatin to treat advanced or recurrent cervical cancer. In this article, we systematically review the recent literature and summarize the recent advances in our understanding of the molecular mechanisms underlying cisplatin resistance in cervical cancer.
Keywords: cisplatin, epithelial–mesenchymal transition, microRNA, molecular mechanism, resistance
Introduction
Cervical cancer remains to be one of the leading causes of cancer-related death in women despite advances in screening, diagnosis, prevention, and treatment. It accounts for ~4% of the total newly diagnosed cancer cases and 4% of the total cancer deaths according to the GLOBOCAN 2012 estimates.1 The prognosis of patients with advanced/recurrent cervical cancer is particularly poor, and their chance of 1-year survival is only 10%–20%.2 Chemotherapy is currently the standard treatment for those patients. The chemotherapeutic agent cisplatin, which is a small-molecule platinum compound and was originally found to inhibit bacterial growth and later identified as an anticancer agent, appears to most effectively treat advanced/recurrent cervical cancer.3 The molecular mechanism underlying cisplatin-mediated anticancer effect is associated with multiple intertwined signaling pathways.4 When the concentration of cytoplasmic chloride ion reduces, the chloride ligands of cisplatin are gradually replaced by water. The resulting aquated cisplatin is highly reactive and covalently binds to DNA to form DNA–cisplatin adducts, which in turn induce DNA damage. When the cisplatin-induced DNA damage is beyond repair, the cells undergo apoptosis and die.
Combination therapy of cisplatin and paclitaxel is a standard chemotherapeutic regimen to treat recurrent or metastatic cervical cancer. The overall response rate is 29.1%–67%, and the median overall survival is 12.87 months in patients with recurrent or advanced cervical cancer receiving the combination chemotherapy.3,5 However, resistance to cisplatin, either intrinsic or acquired resistance, may develop, seriously compromising the efficacy of cisplatin. In this article, we summarize recent advances in our understanding of the mechanisms underlying cisplatin resistance (CPR) in cervical cancer and propose strategies to overcome CPR in cervical cancer.
Mechanisms underlying CPR
The molecular mechanisms underlying CPR are complex and usually associated with the following features: 1) reduction in the intracellular accumulation of the platinum compounds; 2) increase in DNA damage repair; 3) inactivation of apoptosis; 4) activation of epithelial–mesenchymal transition (EMT); 5) alteration in DNA methylation, microRNA profile, cancer stem cell characteristics, and expression of stress-response chaperones (Figure 1).
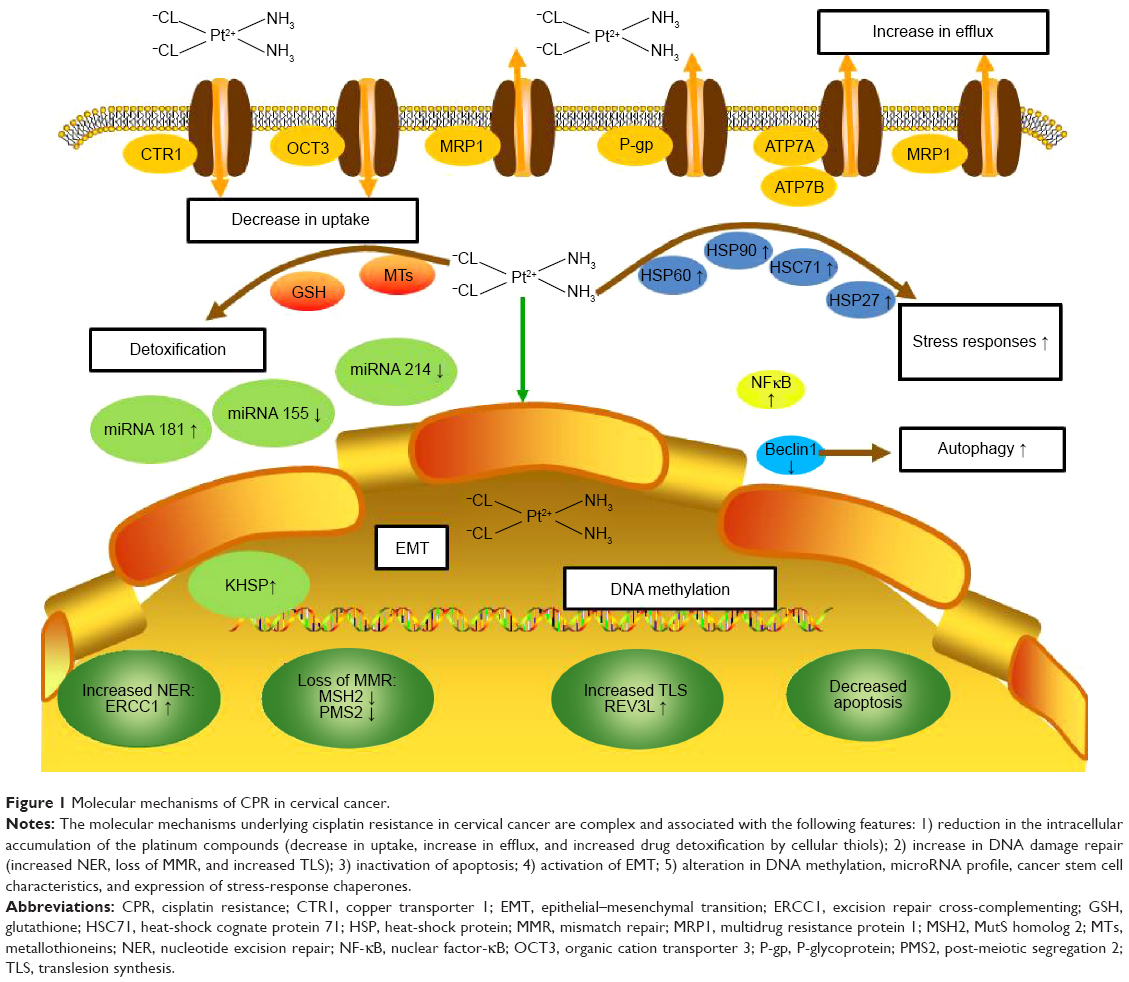 | Figure 1 Molecular mechanisms of CPR in cervical cancer. |
Reduced intracellular accumulation and CPR
Reduced intracellular accumulation of cisplatin may be the predominant cause for CPR. Decrease in uptake, increase in efflux, and inactivation by thiol-containing proteins contribute to reduction in intracellular cisplatin accumulation, which results in reduction in cisplatin–DNA adduct formation and ultimately leads to resistance to cisplatin.4
Reduced uptake
Reduced cisplatin uptake has been observed in cervical cancer cells with acquired CPR. The cisplatin-resistant HeLa cells (HeLa-CPR)6 and A431 (A431/Pt)7 cells show 50% and 77% reduction in cisplatin uptake, respectively, compared with the parental cell lines. In HeLa-CPR cells, the human cervical adenocarcinoma cells with CPR, the amount of cisplatin–DNA adducts is two- to threefold less than that in the parental cells,6 but the HeLa-CPR cells have a similar rate of cisplatin–DNA adduct removal as the parental HeLa cells. Similarly, in A431/Pt cells, which are CPR cervix squamous carcinoma cells, platinum accumulation, DNA-bound platinum, and interstrand cross-link frequency are also reduced compared with the parental cells after a short-term drug exposure.8 Thus, impaired uptake may contribute to the development of CPR in cervical cancer cells.
Up to now, the complex molecular mechanism by which cisplatin enters cells remains poorly understood. Cisplatin is generally believed to pass the cell membrane via passive diffusion, and the diffusion rate is associated with cisplatin lipophilicity.9,10 Recently, copper transporter 1 (CTR1), which is a transmembrane protein and involved in the maintenance of copper homeostasis, has been recognized to regulate the influx of cisplatin and its analogs into the cells. CTR1 is downregulated in various CPR cell lines including HeLa-CPR cells.11 HeLa cells overexpressing CTR1 show 2.2-fold increase in cisplatin accumulation compared with the mock-transfected cells.12 Du et al12 found that the C-terminus of CTR1 protein was required for cisplatin uptake in HeLa cells. In a mouse model of cervical cancer, Ishida et al13 demonstrated that the level of DNA–cisplatin adducts correlated with CTR1 mRNA level in various organs, suggesting that CTR1 may regulate cisplatin uptake in vivo. In contrast, overexpression of CTR1 in the parental cells A431 and the CPR cells A431/Pt does not affect cisplatin uptake and the sensitivity of the cells to cisplatin.7,13 These results suggest that the role of CTR1 in cisplatin transmembrane transport may vary in different types of cervical cancer cells.
Increased efflux
Previous studies suggested that adenosine 5′-triphosphate (ATP)-binding cassette (ABC) transporters, including multidrug resistance proteins (MRPs), MRP1, MRP2, MRP3, and MRP5, might mediate CPR by increasing cisplatin export.14,15 MRP1 overexpression has been found to be associated with CPR in some cervical cancer cells.16 MRP2 contributes to an increased cisplatin efflux in CPR human hepatic cancer cells, embryonic kidney cells, and melanoma cells.17–19 Contrarily, MRP2 expression is reduced significantly in CPR cervical cancer KB-CP20 cells,20 but increased in the cisplatin-sensitive KB-8-5-11 cells,21 indicating that MRP2 is inversely associated with CPR in cervical cancer cells.
In addition to MRPs, P-glycoprotein (P-gp, ABCB1), which is also an ABC transporter, mediates the efflux of cisplatin conjugates and consequently promotes CPR. P-gp is overexpressed in CPR cervical cancer cell line SiHaR.16 P-gp expression increases rapidly when HeLa cells are exposed to cisplatin,22 and overexpression of P-gp attenuates cisplatin-induced apoptosis in HeLa cells. However, Takara et al23 found that P-gp activity and expression were reduced in CPR HeLa subline. Similarly, Okada et al24 showed that neither the non-P-gp-specific inhibitor probenecid (an inhibitor of multiple ABCs) nor the P-gp-specific inhibitor verapamil affected the sensitivity to cisplatin in CPR HeLa cells. These results suggest that P-gp may not substantially contribute to CPR in HeLa cells.
ATP7A and ATP7B, which are copper-transporting ATPases, are also involved in cisplatin efflux. Beretta et al7 found that CPR A431 cells had higher ATP7A expression than the parental cells. In addition, upregulation of ATP7A and ATP7B was associated with acquired platinum resistance in CPR HeLa subline.11 Contrarily, the cisplatin-sensitive KB-8-5-11 cells had reduced ATP7A protein expression and ATP7B gene expression.21
Organic cation transporter 3 (OCT3), a widely expressed transporter for endogenous and exogenous organic cations, is also found to be associated with cisplatin transport. The CPR cervical adenocarcinoma KB-CP20 cells express extremely less OCT3 than the parental cell lines. OCT3 overexpression significantly increases intracellular cisplatin accumulation and cytotoxicity, while downregulation of OCT3 by small interfering RNA or chemical inhibitors increases the resistance of cervical cancer cells to cisplatin.20
Lysosome-associated protein transmembrane 4β-35 (LAPTM4B-35) is a member of the mammalian 4-tetratransmembrane spanning protein superfamily. Previous studies have shown that LAPTM4B-35 was overexpressed in malignant tissue specimens and significantly correlates with poor prognosis of cervical cancer.25 Recently, Li et al26 found that LAPTM4B-35 interacted with P-gp to inhibit apoptosis by activating PI3K/AKT signaling and induced multidrug resistance, such as resistance to doxorubicin, paclitaxel, and cisplatin, in cervical cancer cells by promoting drug efflux.
Thiol-containing protein-mediated inactivation
Cisplatin–DNA adducts induce DNA damage, which in turn leads to cytotoxicity. After cisplatin enters the cells, some cisplatin molecules bind to DNA and thus activate the DNA damage-induced apoptosis cascade,15 while others can avidly bind to cytoplasmic nucleophilic species, such as glutathione (GSH), methionine, metallothioneins (MTs), and thiol-containing proteins. The binding of cisplatin to the thiol-containing nucleophilic species or thiol-containing proteins not only depletes intracellular antioxidant reserves to promote oxidative stress but also reduces the availability of reactive cisplatin.15
GSH, which is a thiol-containing tripeptide (Glu-Cys-Gly), can bind to cisplatin to prevent cisplatin from binding to DNA and other targets, quench proapoptotic reactive oxygen species that are often generated by cisplatin, and reduce the sensitivity of the cells to cell death signals.15 A large body of evidence suggests that an increased expression of enzymes that promote GSH synthesis and conjugation, such as GSH-S-transferase (GST), gamma-glutamyl cysteine synthase, and gamma-glutamyl transferase, may contribute to the development of CPR. The association between the expression of GSH conjugates and CPR in cervical cancer appears controversial. GSH has been shown to positively correlate with CPR in several cervical cancer cell lines.24,27,28 KB-8-5-11 cells, which carry platinum-sensitive phenotype, have reduced expression of GSTA4, GSTK1, and GSTP1. Correspondingly, increased gene expression of GSTP1, GSTA4, and GSTK1 is associated with the development of CPR in the cervical cancer cell line.21 In contrast, Konishi et al29 found that in patients with locally advanced or bulky cervical carcinoma, poor response to cisplatin significantly correlated with the expression of P-gp but not with GST-pi. Similarly, Chao et al30 showed that neither intracellular GSH level nor GST activity was elevated in CPR HeLa cells. Roy and Mukherjee16 also found that GSH level remained unaltered in the CPR clone SiHaR, which derived from SiHa. Thus, the role of GSH and its metabolism in CPR in cervical cancer remains to be determined.
MTs are low-molecular-weight thiol-containing proteins and regulate metal homeostasis and detoxification. MTs can bind to cisplatin, thus leading to the development of CPR phenotype.31 Mellish et al27 investigated the role of MTs in CPR in five human cervical squamous carcinoma cell lines and found a significant correlation between an increased expression of MTs and CPR.
Increased DNA repair and CPR
In CPR cancer cells, inter- and intra-strand DNA adducts often fail to trigger apoptotic cascade for many reasons.15 Tumor cells with acquired CPR show an enhanced capability to repair cisplatin-induced DNA lesions or to tolerate high level of unrepaired DNA lesions compared with their parental cisplatin-sensitive counterparts.15 A high level of repair-associated DNA strand breaks and an enhanced activity of DNA excision repair have been found in CPR HeLa cells.6,32 Cisplatin-induced DNA lesions, which often cause DNA distortion, can be identified by multiple DNA repair pathways, among which nucleotide excision repair (NER) and mismatch repair (MMR) are the predominant DNA repair mechanisms.
Nucleotide excision repair
NER, which is a highly conserved DNA repair pathway and a major pathway for the repair of DNA–cisplatin adducts,33 usually targets on the DNA damages that change the DNA helical structure and interfere in DNA replication and transcription.34 More than 20 proteins participate in NER, including excision repair cross-complementation group 1 (ERCC1). ERCC1 is a single-strand DNA endonuclease and forms a tight heterodimer with ERCC4 to incise DNA on the 5′ side of bulky lesions such as DNA–cisplatin adducts.15 ERCC1 expression is upregulated in CPR cervical cancer cells HCA-1R,35 and in patients with locally advanced cervical squamous cell carcinoma. ERCC1 expression negatively correlates with responsiveness to cisplatin.36 Thus, the level of ERCC1 expression may predict responsiveness to cisplatin in patients with cervical cancer. Furthermore, low ERCC1 expression is an independent prognostic factor and associated with a survival benefit in patients receiving adjuvant cisplatin chemotherapy or chemoradiotherapy with cisplatin.37,38
DNA mismatch repair
DNA MMR, an evolutionarily conserved process, corrects mismatches that are generated during DNA replication and escape from DNA proofreading.34 A properly functioned MMR system is required to detect cisplatin-induced DNA lesions. Thus, MMR deficiency may lead to the development of DNA damage tolerance and CPR. Among MMR proteins, MutS homolog 2 (MSH2) protein has been shown to contribute to the development of CPR in cervical cancer cells.39 Lanzi et al8 found that MSH2 protein expression in CPR A431 cells was significantly less than that in the parental cells. Post-meiotic segregation 2 (PMS2) is also a major component of the MMR system. Zhang et al40 found a marked downregulation of PMS2 in human cervical carcinoma tissue, and over-expression of PMS2 in HeLa cells dramatically increased cisplatin-induced apoptosis and caspase-3 activity. Thus, upregulation of PMS2 appears to enhance the sensitivity of HeLa cells to cisplatin. Additionally, REV3L, the catalytic subunit of DNA polymerase ζ (Polζ), plays a key role in skipping DNA damage during translesion synthesis. Yang et al41 demonstrated that REV3L conferred resistance to cisplatin in cervical cancer cells by regulating apoptosis rate and the expression of antiapoptotic proteins, such as B-cell lymphoma 2 (Bcl-2), myeloid cell leukemia sequence 1 (Mcl-1), Bcl-extra large (Bcl-xL), and proapoptotic Bcl-2-associated x protein (Bax).
Inactivation of apoptosis pathway and CPR
Cisplatin-induced apoptosis is essential for the anticancer effect of cisplatin. Cisplatin stimulates apoptosis by triggering the extrinsic death receptor pathway or the intrinsic mitochondrial pathway. Multiple proteins such as the Bcl-2 family proteins and p53 and several signaling pathways including mitogen-activated protein kinase (MAPK) pathway and nuclear factor-κB (NF-κB) pathway are involved in the extrinsic and intrinsic apoptosis pathways. Dysfunction of these proteins and signaling pathways may lead to the development of CPR (Figure 2). Brozovic et al42 found that HeLa cells with acquired CPR showed lower level of cisplatin-induced apoptosis and lower levels of Bcl-2 and p-Bad than the parental cells. In addition, SiHa cells, which are more resistant to cisplatin than HeLa cells, showed reduced activity of caspase-3, caspase-8, and caspase-9 and cisplatin-induced cleavage of poly(adenosine diphosphate [ADP]-ribose) polymerase compared with HeLa cells.43
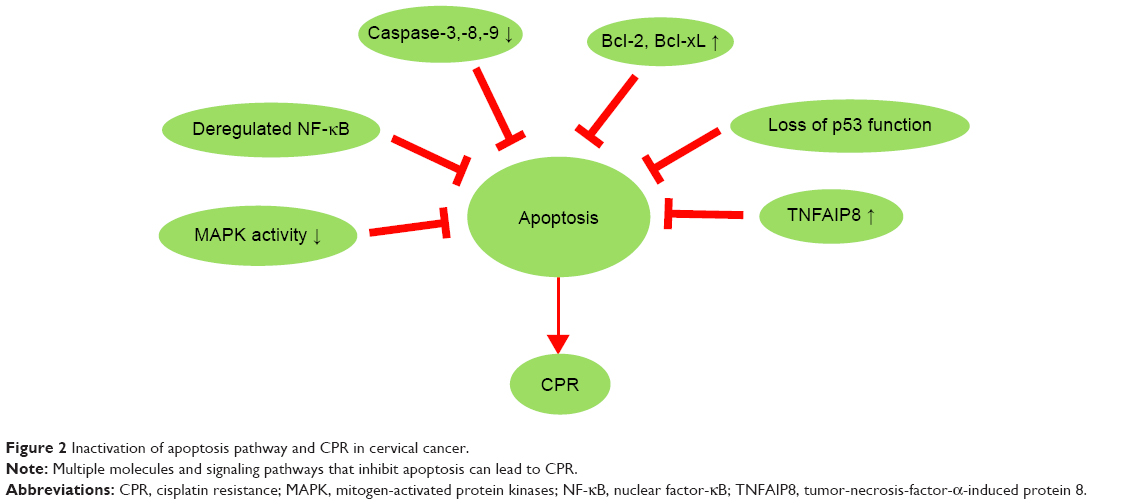 | Figure 2 Inactivation of apoptosis pathway and CPR in cervical cancer. |
Caspases
Caspases play critical roles in apoptosis. The activity of caspase-3, caspase-8, and caspase-9 is decreased in CPR cells.44 Compared with drug-sensitive parental cells, the multidrug-resistant endocervical HEN-16-2/CDDP cells are more resistant to apoptosis and exhibit reduced caspase-3 activation.45
Bcl-2 protein family
Exposure to cisplatin initiates programmed cell death. Bcl-2 protein family contains three functionally and structurally distinct subfamilies: 1) anti-apoptotic proteins, including Bcl-2, Bcl-xL, Mcl-1, Bcl-w, Bag-1, and A1; 2) proapoptotic effector proteins Bax and Bak; 3) proapoptotic BH3-only proteins. These proteins tightly control apoptotic process by regulating the permeability of the mitochondrial outer membrane and the release of cytochrome c and other proapoptotic factors. The release of proapoptotic factors activates caspases.46
CPR cervical cancer cells frequently overexpress anti-apoptotic proteins.47 For example, Brozovic et al42 found that Bcl-2 was upregulated in CPR HeLa cells compared with the parental cells. The overexpression of Bcl-2 was related to poorer clinical outcome in patients with cervical cancer receiving CDDP-based chemoradiotherapy.48 The multidrug-resistant endocervical HEN-16-2/cisplatin cells show significantly higher level of Bcl-xL and Bag-1 than the drug-sensitive parental cells.45 Similarly, overexpression of Bag-1L prevents cisplatin-induced apoptosis by affecting Raf/Ras signaling and the expression of Mcl-1 and heat-shock proteins (HSPs) in HeLa cells.49 Furthermore, stable overexpression of Bag-1 promotes resistance to cisplatin-induced apoptosis in C33A cells.47 In addition to the upregulation of anti-apoptotic proteins, suppression of proapoptotic effector proteins also contributes to the development of CPR.44
MAPK pathway
MAPKs play critical roles in the complex intracellular signaling network, which regulates gene expression in response to various extracellular stimuli.50 In mammalian cells, there are mainly three types of MAPKs: stress-activated protein kinase/c-Jun-N-terminal kinase (SAPK/JNK), p38 kinase, and extracellular signal-regulated kinase (ERK). The association of MAPK activation and CPR has recently been recognized. CPR cancer cells often have reduced MAPK activity. Cisplatin activates SAPK/JNK, p38 kinase, and ERK dose dependently in cisplatin-sensitive HeLa cells, whereas cisplatin-mediated activation of SAPK/JNK is significantly reduced in the CPR subline.42,51,52 In addition, inhibition of JNK, p38 kinase, or ERK attenuates cisplatin-induced apoptosis and cell death.42,51,52 Similarly, blockade of MEK–ERK by MEK inhibitor PD98059 induces CPR in human cervical carcinoma SiHa cells.53 Thus, sufficient MAPK activation appears to be required for cisplatin-induced apoptosis.
P53 signaling pathway
Stabilization and activation of wild-type p53 are critical for cisplatin-induced apoptosis. Thus, loss of p53 can impair apoptosis process and lead to tolerance of DNA damage, consequently promoting drug resistance.44 Sultana et al54 demonstrated that chemosensitivity was associated with p53-Bax-mediated apoptosis in cervical cancer. Cisplatin-based chemotherapy for cervical cancer is more beneficial to patients harboring wild-type p53 than to patients with mutated p53.55 Additionally, patients who respond to cisplatin well show a higher proportion of p53-positive cells than nonresponders.56 Overexpression of p53 is a predictive factor for chemoresistance in adenocarcinoma of the uterine cervix.57
NF-κB pathway
NF-κB, usually in the form of heterodimer or homodimer formed by the NF-κB family members, is ubiquitously expressed and regulates >500 genes that are involved in immunoregulation, inflammation, growth regulation, apoptosis, and carcinogenesis.58 Numerous in vitro and in vivo studies, including ours, have demonstrated that constitutive activation of NF-κB inhibits chemotherapy-induced apoptosis in different types of cancer, including cervical cancer.43,59 We found that NF-κB contributed to CPR in human cervical cancer SiHa cells and inhibition of NF-κB sensitized SiHa cells to cisplatin-induced apoptosis.59 This study suggests that combination of cisplatin and NF-κB inhibitors may have therapeutic potential.
Others
The tumor-necrosis-factor-α-induced protein 8 (TNFAIP8) family, which is a newly identified and poorly characterized group of proteins, is found to play a role in the maintenance of immune homeostasis and inhibition of apoptosis. Shi et al60 reported that high protein level of TNFAIP8 was significantly associated with CPR and poor clinical outcomes in patients with cervical cancer.
Activation of EMT and CPR
In recent years, the role of EMT in acquired CPR has been increasingly recognized. Our previous study found that 7-day low-concentration cisplatin (1 μM) treatment induced EMT in cervical cancer HeLa and C4-1 cell lines by activating transforming growth factor-β pathway. TWIST1, which is a highly conserved transcription factor and belongs to the basic helix-loop-helix protein family, plays a key role in EMT. In cervical cancer, TWIST1 expression appears to correlate with MDR1/P-gp expression positively. In HeLa cells, silencing TWIST1 expression by RNAi downregulates MDR1/P-gp expression, suppresses cell proliferation, inhibits rhodamine 123 efflux, and sensitizes the cells to cisplatin.61 In addition to TWIST1, other proteins that are involved in EMT have also been found to be associated with CPR. Shen et al4 found that human cervical cancer cells with high CPR overexpressed SNAIL1 and E-cadherin, whereas the cells with mild CPR did not. These results indicate that overexpression of SNAIL1 and E-cadherin may occur at late stage of CPR development and thus facilitate cell survival under high-dose platinum.4 Astrocyte elevated gene-1 (AEG-1), which can be induced by human immunodeficiency virus-1 (HIV-1), was initially identified from human fetal astrocytes.62 Knockdown of AEG-1 blocks EMT and reduces CPR in cervical cancer cells.63 These findings indicate that EMT may contribute to the development of CPR in cervical cancer.
MicroRNA and CPR
MicroRNAs have been found to regulate multiple pathways that are involved in the cellular response to cisplatin.64 The effects of microRNAs on the development of CPR in cervical cancer have been investigated. Pouliot et al65 found that the miR-181 family members were overexpressed in CPR cells KB-CP5 and KB-CP20 compared with the parental KB-3-1 cells, and silencing the proteins that are essential for microRNA synthesis, such as DICER and TRBP2, reversed CPR in the cells. Chen et al66 found that in cervical squamous cell carcinoma, upregulation of miR-181 appeared to correlate with CPR significantly and miR-181a inhibited apoptosis and enhanced CPR by targeting protein kinase C-δ (PRKCD). In contrast to miR-181, other microRNAs can increase drug sensitivity. Lei et al67 found that miR155 suppressed epithelial-growth-factor-induced EMT, decreased migration/invasion, inhibited cell proliferation, and increased the sensitivity to cisplatin in human CaSki cervical cancer cells. Wang et al68 showed that miR-214 increased the sensitivity of HeLa cells to cisplatin by directly inhibiting Bcl2l2 expression and increasing the expression of Bax, caspase-9, caspase-8, and caspase-3. In addition, KH-type splicing regulatory protein, which interacts with Drosha and increases the binding and subsequent processing of specific pri-microRNAs such as pri-let7a-1 and pri-miR-21,64 is upregulated in the nucleus of HeLa cells that are exposed to cisplatin.69 These results indicate that the levels of some microRNAs may predict patient response to cisplatin.
Cancer stem cells and CPR
Cancer stem cells are highly resistant to various chemotherapeutic agents, because they express abundant levels of multiple drug resistance transporters such as MDR1/P-gp and mitoxantrone resistance protein (MXR)/breast cancer resistance protein-1 (BCRP-1).50 Liu and Zheng70 demonstrated that aldehyde dehydrogenase (ALDH) was a marker for cervical cancer stem cells and high ALDH activity was associated with an increased resistant to cisplatin in cervical cancer cells. In addition, the CPR clone R-ME-180 can form three-dimensional multicellular spheroids, which show positive expression of the cancer stem cell marker ALDH, but the parental ME-180 cells do not express the marker.71 Wang et al72 enriched cervical cancer stem cells by using a nonadhesive culture system and found that the cervical cancer stem cells had high resistance to cisplatin.
DNA methylation and CPR
DNA methylation plays a critical role in the development of CPR in several cell culture models. Aberrant DNA methylation may alter the expression of genes that are critical to drug response and thus affect the sensitivity of cancer cells to cisplatin.4 Epigenetic drugs such as 5-azacytidine have been shown to block aberrant DNA methylation and thus reverse CPR.64 Combination of cisplatin and 5-aza-2′-deoxycytidine, a demethylating agent, significantly enhances the sensitivity to cisplatin in ME180 parent cells and the CPR subclones, and the removal of 5-aza-2′-deoxycytidine restores the CPR of the subclones rapidly.73
Stress response chaperones and CPR
Molecular chaperones that are involved in the general stress responses, such as autophagy and HSPs, can promote CPR via multiple, most often indirect, mechanisms.10,15 Autophagy, a process by which cells digest their own damaged organelles, has been recently shown to contribute to CPR in some cell types and inhibition of autophagy can enhance the cytotoxicity of chemotherapeutic agents.74 Cisplatin induces autophagy in HeLa cells, and inhibition of autophagy induces endoplasmic reticulum (ER) stress and thus enhances cisplatin cytotoxicity.75 Beclin-1, an autophagy-related molecule, plays a critical role in the regulation of response to chemotherapeutic agents. In CaSki cells, overexpression of Beclin-1 promotes apoptosis signaling and thus sensitizes the cells to cisplatin.76 NH4Cl, an autophagy inhibitor, blocks the activation of lysosomal enzymes and the degradation of autolysosome components. NH4Cl has been found to increase DNA damage and consequently enhance cisplatin cytotoxicity in HeLa cells.77 Similarly, pretreatment with the autophagy inhibitor bafilomycin sensitizes both cervical adenocarcinoma HeLa cells and squamous cell carcinoma CaSki cells effectively to cisplatin.78
The function of HSPs is to adapt the cells to high temperature or other stressful conditions, and HSPs promote conformational change in target proteins and protect the cells from oxidant-induced DNA damage and apoptosis.4 Because cisplatin induces DNA damage and consequently results in apoptosis, HSPs are probably affected during the development of CPR.4 Shen et al79 used two-dimensional gel electrophoreses and amino acid micro-sequencing and found HSP 60 overexpression in human cervical CPR cells. Castagna et al80 further confirmed that the expression of heat shock cognate protein 71 (HSC71) and HSP 60 was significantly increased in the CPR cells, A431/Pt cells, compared with the parental A431 cells. Furthermore, Zhang and Shen31 demonstrated that HSP 27 suppressed cisplatin-induced ASK1/p38 and Akt activation, which consequently impaired the cytotoxic response of HeLa cells to cisplatin. The HSP 90 inhibitor PU-H71 (8-[(6-iodo-1,3-benzodioxol-5-yl)sulfanyl]-9-[3-(propan-2-ylamino)propyl]purin-6-amine) induces ER stress and thus promotes apoptosis in HeLa cells, and HeLa cells treated with PU-H71 overcome the CPR conferred by Bcl-2.81 These results indicate that HSPs may contribute to CPR.
System biology of CPR
System biological approach has been used to investigate CPR.10 In our previous studies on proteomics profiling, we found that protein levels of S100A8 and S100A9 were increased and annexin A2 level was decreased in patients with cervical cancer who failed to response to cisplatin-based neoadjuvant chemotherapy treatment.82,83 S100A8, S100A9, and annexin A2 are Ca2+-binding proteins and play roles in immune response, vesicle trafficking, cell division, apoptosis, and calcium signaling. Chavez et al84 conducted quantitative proteomics, analyzed protein interaction network, and found that 374 proteins were differentially expressed in CPR versus cisplatin-sensitive HeLa cells. Those differentially expressed proteins are involved in DNA binding, DNA damage repair, energy-producing metabolic pathways, and stress response.84 Castagna et al80 used a similar approach to discover that calmodulin, microtubule-associated protein, and stathmin were downregulated in CPR A431 subline. Wu et al85 performed gene expression profiling on CPR HeLa cells and identified nine genes (NAPA, CITED2, CABIN1, ADM, HIST1H1A, EHD1, MARK2, PTPN21, and MVD), which were consistently upregulated in two CPR HeLa cell lines.
Conclusion and future direction
Cisplatin has been used to treat cervical cancer since the early 1980s86 and remains to be the most effective anticancer agent for advanced/recurrent cervical cancer. However, chemoresistance may develop, and thus seriously hinders the use of cisplatin in clinical practice. Over the past 3 decades, great efforts have been devoted to characterize the molecular mechanisms underlying CPR in cervical cancer cells. Starting with the early 1990s, GSH87 and NER88 were identified as causative of resistance to cisplatin. In the early 2000s, CTR1 turned out to play a critical role in the uptake of cisplatin.89 In the mid-2000s, EMT and microRNA have been implicated in the development of drug resistance. In the last decade, the advent of high-content and high-throughput screening technologies has accelerated the discovery of the molecular mechanisms underlying CPR at cell-intrinsic level.10 Ca2+-binding proteins such as S100A8, S100A9, and annexin A2 and energy-producing metabolic pathways have been identified to be related to CPR in cervical cancer.82 NAPA, a protein found in the ER, and CITED2, a transcriptional modulator, have been identified to confer resistance to cisplatin in HeLa cells.85 Further investigation is required to obtain additional insights into CPR at protein, DNA, RNA, mRNA, and microRNA levels.
Based on the mechanisms underlying CPR, the following strategies have been proposed to overcome the resistance in cervical cancer: 1) develop new platinum drugs;90,91 2) improve cisplatin delivery to tumor;71 3) specifically target CPR mechanisms; and 4) combine cisplatin with other drugs.92 The greatest challenge is that CPR often exhibits a multifactorial nature. Strategies that target one mechanism at a time may not sufficiently enhance the sensitivity to cisplatin. Thus, combination therapies targeting multiple mechanisms underlying CPR should be exploited. In our previous study, we found that a combination of pyrrolidine dithiocarbamate and cisplatin synergistically increased apoptosis and inhibited cell growth by suppressing NF-κB in human cervical cancer cells. Additionally, agents including genistein,93 curcumin,16 tea polyphenols,94 melatonin,95 mifepristone,96–98 epigallocatechin gallate,99,100 ursolic acid,101 Morinda citrifolia,102,103 and wogonin104 have been shown to be beneficial to overcome CPR via targeting the molecular pathways that are involved in CPR (Table 1). Understanding the molecular mechanisms underlying CPR may be helpful to identify patients with a potential to develop CPR, and thus oncologists may be able to provide an effective therapy for those patients.
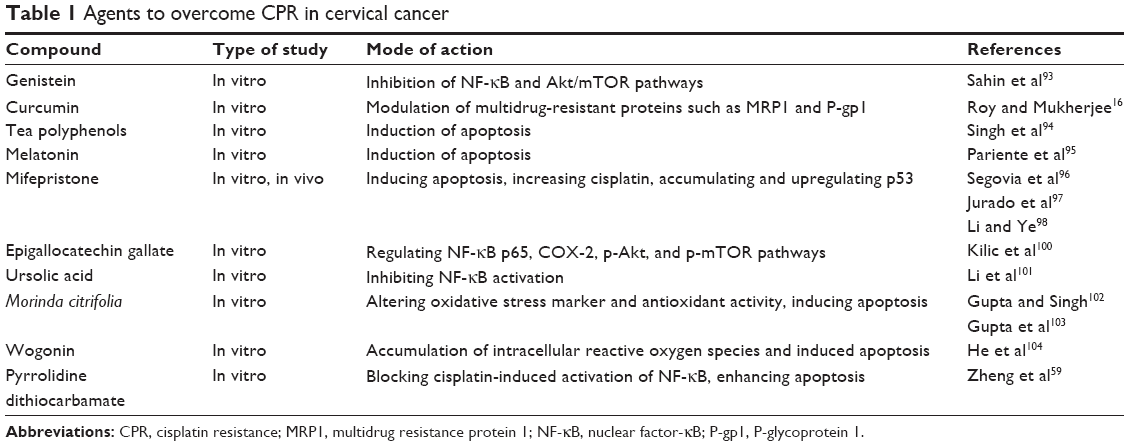 | Table 1 Agents to overcome CPR in cervical cancer |
Acknowledgments
This work was supported by a grant from the National Natural Science Foundation of China (No 81372381) and sponsored by medical and technology project of Zhejiang Province (No 2016KYA141).
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Diaz-Padilla I, Monk BJ, Mackay HJ, Oaknin A. Treatment of metastatic cervical cancer: future directions involving targeted agents. Crit Rev Oncol Hematol. 2013;85(3):303–314. | ||
Lorusso D, Petrelli F, Coinu A, Raspagliesi F, Barni S. A systematic review comparing cisplatin and carboplatin plus paclitaxel-based chemotherapy for recurrent or metastatic cervical cancer. Gynecol Oncol. 2014;133(1):117–123. | ||
Shen DW, Pouliot LM, Hall MD, Gottesman MM. Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol Rev. 2012;64(3):706–721. | ||
Seol HJ, Ulak R, Ki KD, Lee JM. Cytotoxic and targeted systemic therapy in advanced and recurrent cervical cancer: experience from clinical trials. Tohoku J Exp Med. 2014;232(4):269–276. | ||
Chao CC. Decreased accumulation as a mechanism of resistance to cis-diamminedichloroplatinum(II) in cervix carcinoma HeLa cells: relation to DNA repair. Mol Pharmacol. 1994;45(6):1137–1144. | ||
Beretta GL, Gatti L, Tinelli S, et al. Cellular pharmacology of cisplatin in relation to the expression of human copper transporter CTR1 in different pairs of cisplatin-sensitive and -resistant cells. Biochem Pharmacol. 2004;68(2):283–291. | ||
Lanzi C, Perego P, Supino R, et al. Decreased drug accumulation and increased tolerance to DNA damage in tumor cells with a low level of cisplatin resistance. Biochem Pharmacol. 1998;55(8):1247–1254. | ||
Ciarimboli G. Membrane transporters as mediators of cisplatin effects and side effects. Scientifica (Cairo). 2012;2012:473829. | ||
Galluzzi L, Vitale I, Michels J, et al. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014;5:e1257. | ||
Zisowsky J, Koegel S, Leyers S, et al. Relevance of drug uptake and efflux for cisplatin sensitivity of tumor cells. Biochem Pharmacol. 2007;73(2):298–307. | ||
Du X, Wang X, Li H, Sun H. Comparison between copper and cisplatin transport mediated by human copper transporter 1 (hCTR1). Metallomics. 2012;4(7):679–685. | ||
Ishida S, McCormick F, Smith-McCune K, Hanahan D. Enhancing tumor-specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer Cell. 2010;17(6):574–583. | ||
Borst P, Evers R, Kool M, Wijnholds J. A family of drug transporters: the multidrug resistance-associated proteins. J Natl Cancer Inst. 2000;92(16):1295–1302. | ||
Galluzzi L, Senovilla L, Vitale I, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31(15):1869–1883. | ||
Roy M, Mukherjee S. Reversal of resistance towards cisplatin by curcumin in cervical cancer cells. Asian Pac J Cancer Prev. 2014;15(3):1403–1410. | ||
Cui Y, Konig J, Buchholz JK, Spring H, Leier I, Keppler D. Drug resistance and ATP-dependent conjugate transport mediated by the apical multidrug resistance protein, MRP2, permanently expressed in human and canine cells. Mol Pharmacol. 1999;55(5):929–937. | ||
Koike K, Kawabe T, Tanaka T, et al. A canalicular multispecific organic anion transporter (cMOAT) antisense cDNA enhances drug sensitivity in human hepatic cancer cells. Cancer Res. 1997;57(24):5475–5479. | ||
Liedert B, Materna V, Schadendorf D, Thomale J, Lage H. Overexpression of cMOAT (MRP2/ABCC2) is associated with decreased formation of platinum-DNA adducts and decreased G2-arrest in melanoma cells resistant to cisplatin. J Invest Dermatol. 2003;121(1):172–176. | ||
Li Q, Peng X, Yang H, Rodriguez JA, Shu Y. Contribution of organic cation transporter 3 to cisplatin cytotoxicity in human cervical cancer cells. J Pharm Sci. 2012;101(1):394–404. | ||
Doherty B, Lawlor D, Gillet JP, Gottesman M, O’Leary JJ, Stordal B. Collateral sensitivity to cisplatin in KB-8-5-11 drug-resistant cancer cells. Anticancer Res. 2014;34(1):503–507. | ||
Sakaeda T, Nakamura T, Hirai M, et al. MDR1 up-regulated by apoptotic stimuli suppresses apoptotic signaling. Pharm Res. 2002;19(9):1323–1329. | ||
Takara K, Obata Y, Yoshikawa E, et al. Molecular changes to HeLa cells on continuous exposure to cisplatin or paclitaxel. Cancer Chemother Pharmacol. 2006;58(6):785–793. | ||
Okada T, Murata K, Hirose R, et al. Upregulated expression of FGF13/FHF2 mediates resistance to platinum drugs in cervical cancer cells. Sci Rep. 2013;3:2899. | ||
Meng F, Tan S, Liu T, Song H, Lou G. Predictive significance of combined LAPTM4B and VEGF expression in patients with cervical cancer. Tumour Biol. Epub 2015 Nov 2. | ||
Li L, Wei XH, Pan YP, et al. LAPTM4B: a novel cancer-associated gene motivates multidrug resistance through efflux and activating PI3K/AKT signaling. Oncogene. 2010;29(43):5785–5795. | ||
Mellish KJ, Kelland LR, Harrap KR. In vitro platinum drug chemosensitivity of human cervical squamous cell carcinoma cell lines with intrinsic and acquired resistance to cisplatin. Br J Cancer. 1993;68(2):240–250. | ||
Nishikawa K, Rosenblum MG, Newman RA, Pandita TK, Hittelman WN, Donato NJ. Resistance of human cervical carcinoma cells to tumor necrosis factor correlates with their increased sensitivity to cisplatin: evidence of a role for DNA repair and epidermal growth factor receptor. Cancer Res. 1992;52(17):4758–4765. | ||
Konishi I, Nanbu K, Mandai M, et al. Tumor response to neoadjuvant chemotherapy correlates with the expression of P-glycoprotein and PCNA but not GST-pi in the tumor cells of cervical carcinoma. Gynecol Oncol. 1998;70(3):365–371. | ||
Chao CC, Huang YT, Ma CM, Chou WY, Lin-Chao S. Overexpression of glutathione S-transferase and elevation of thiol pools in a multidrug-resistant human colon cancer cell line. Mol Pharmacol. 1992;41(1):69–75. | ||
Zhang Y, Shen X. Heat shock protein 27 protects L929 cells from cisplatin-induced apoptosis by enhancing Akt activation and abating suppression of thioredoxin reductase activity. Clin Cancer Res. 2007;13(10):2855–2864. | ||
Chao CC. Enhanced excision repair of DNA damage due to cis-diamminedichloroplatinum(II) in resistant cervix carcinoma HeLa cells. Eur J Pharmacol. 1994;268(3):347–355. | ||
Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–378. | ||
Martin LP, Hamilton TC, Schilder RJ. Platinum resistance: the role of DNA repair pathways. Clin Cancer Res. 2008;14(5):1291–1295. | ||
Torii Y, Kato R, Minami Y, Hasegawa K, Fujii T, Udagawa Y. ERCC1 expression and chemosensitivity in uterine cervical adenocarcinoma cells. Anticancer Res. 2014;34(1):107–115. | ||
Bai ZL, Wang YY, Zhe H, He JL, Hai P. ERCC1 mRNA levels can predict the response to cisplatin-based concurrent chemoradiotherapy of locally advanced cervical squamous cell carcinoma. Radiat Oncol. 2012;7:221. | ||
Park JS, Jeon EK, Chun SH, et al. ERCC1 (excision repair cross-complementation group 1) expression as a predictor for response of neoadjuvant chemotherapy for FIGO stage 2B uterine cervix cancer. Gynecol Oncol. 2011;120(2):275–279. | ||
Hasegawa K, Kato R, Torii Y, Ichikawa R, Oe S, Udagawa Y. The relationship between ERCC1 expression and clinical outcome in patients with FIGO stage I to stage II uterine cervical adenocarcinoma. Int J Gynecol Cancer. 2011;21(8):1479–1485. | ||
Martelli L, Ragazzi E, Di Mario F, Basato M, Martelli M. Cisplatin and oxaliplatin cytotoxic effects in sensitive and cisplatin-resistant human cervical tumor cells: time and mode of application dependency. Anticancer Res. 2009;29(10):3931–3937. | ||
Zhang Y, Shu YM, Wang SF, Da BH, Wang ZH, Li HB. Stabilization of mismatch repair gene PMS2 by glycogen synthase kinase 3beta is implicated in the treatment of cervical carcinoma. BMC Cancer. 2010;10:58. | ||
Yang L, Shi T, Liu F, et al. REV3L, a promising target in regulating the chemosensitivity of cervical cancer cells. PLoS One. 2015;10(3):e0120334. | ||
Brozovic A, Fritz G, Christmann M, et al. Long-term activation of SAPK/JNK, p38 kinase and fas-L expression by cisplatin is attenuated in human carcinoma cells that acquired drug resistance. Int J Cancer. 2004;112(6):974–985. | ||
Venkatraman M, Anto RJ, Nair A, Varghese M, Karunagaran D. Biological and chemical inhibitors of NF-kappaB sensitize SiHa cells to cisplatin-induced apoptosis. Mol Carcinog. 2005;44(1):51–59. | ||
Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22(47):7265–7279. | ||
Ding Z, Yang X, Pater A, Tang SC. Resistance to apoptosis is correlated with the reduced caspase-3 activation and enhanced expression of antiapoptotic proteins in human cervical multidrug-resistant cells. Biochem Biophys Res Commun. 2000;270(2):415–420. | ||
Brozovic A, Osmak M. Activation of mitogen-activated protein kinases by cisplatin and their role in cisplatin-resistance. Cancer Lett. 2007;251(1):1–16. | ||
Chen J, Xiong J, Liu H, Chernenko G, Tang SC. Distinct BAG-1 isoforms have different anti-apoptotic functions in BAG-1-transfected C33A human cervical carcinoma cell line. Oncogene. 2002;21(46):7050–7059. | ||
Henriquez-Hernandez LA, Lloret M, Pinar B, et al. BCL-2, in combination with MVP and IGF-1R expression, improves prediction of clinical outcome in complete response cervical carcinoma patients treated by radiochemotherapy. Gynecol Oncol. 2011;122(3):585–589. | ||
Ozfiliz P, Arisan ED, Coker-Gurkan A, et al. Bag-1L is a stress-withstand molecule prevents the downregulation of Mcl-1 and c-Raf under control of heat shock proteins in cisplatin treated HeLa cervix cancer cells. Asian Pac J Cancer Prev. 2014;15(11):4475–4482. | ||
Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–284. | ||
Wang X, Martindale JL, Holbrook NJ. Requirement for ERK activation in cisplatin-induced apoptosis. J Biol Chem. 2000;275(50):39435–39443. | ||
Basu A, Tu H. Activation of ERK during DNA damage-induced apoptosis involves protein kinase Cdelta. Biochem Biophys Res Commun. 2005;334(4):1068–1073. | ||
Yeh PY, Chuang SE, Yeh KH, Song YC, Ea CK, Cheng AL. Increase of the resistance of human cervical carcinoma cells to cisplatin by inhibition of the MEK to ERK signaling pathway partly via enhancement of anticancer drug-induced NF kappa B activation. Biochem Pharmacol. 2002;63(8):1423–1430. | ||
Sultana H, Kigawa J, Kanamori Y, et al. Chemosensitivity and p53-Bax pathway-mediated apoptosis in patients with uterine cervical cancer. Ann Oncol. 2003;14(2):214–219. | ||
Garzetti GG, Ciavattini A, Provinciali M, et al. Expression of p53 and apoptosis of tumor cells in locally advanced cervical carcinoma after cisplatin based neoadjuvant chemotherapy. Anticancer Res. 1996;16(5B):3229–3234. | ||
Garzetti GG, Ciavattini A, Lucarini G, et al. Modulation of expression of p53 and cell proliferation in locally advanced cervical carcinoma after neoadjuvant combination chemotherapy. Eur J Obstet Gynecol Reprod Biol. 1995;63(1):31–36. | ||
Saito T, Takehara M, Tanaka R, et al. Correlation between responsiveness of neoadjuvant chemotherapy and apoptosis-associated proteins for cervical adenocarcinoma. Gynecol Oncol. 2004;92(1):284–292. | ||
Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092):431–436. | ||
Zheng X, Lv J, Shen Q, et al. Synergistic effect of pyrrolidine dithiocarbamate and cisplatin in human cervical carcinoma. Reprod Sci. 2014;21(10):1319–1325. | ||
Shi TY, Cheng X, Yu KD, et al. Functional variants in TNFAIP8 associated with cervical cancer susceptibility and clinical outcomes. Carcinogenesis. 2013;34(4):770–778. | ||
Zhu K, Chen L, Han X, Wang J, Wang J. Short hairpin RNA targeting Twist1 suppresses cell proliferation and improves chemosensitivity to cisplatin in HeLa human cervical cancer cells. Oncol Rep. 2012;27(4):1027–1034. | ||
Yoo BK, Emdad L, Lee SG, et al. Astrocyte elevated gene-1 (AEG-1): a multifunctional regulator of normal and abnormal physiology. Pharmacol Ther. 2011;130(1):1–8. | ||
Liu X, Wang D, Liu H, et al. Knockdown of astrocyte elevated gene-1 (AEG-1) in cervical cancer cells decreases their invasiveness, epithelial to mesenchymal transition, and chemoresistance. Cell Cycle. 2014;13(11):1702–1707. | ||
Drayton RM. The role of microRNA in the response to cisplatin treatment. Biochem Soc Trans. 2012;40(4):821–825. | ||
Pouliot LM, Shen DW, Suzuki T, Hall MD, Gottesman MM. Contributions of microRNA dysregulation to cisplatin resistance in adenocarcinoma cells. Exp Cell Res. 2013;319(4):566–574. | ||
Chen Y, Ke G, Han D, Liang S, Yang G, Wu X. MicroRNA-181a enhances the chemoresistance of human cervical squamous cell carcinoma to cisplatin by targeting PRKCD. Exp Cell Res. 2014;320(1):12–20. | ||
Lei C, Wang Y, Huang Y, et al. Up-regulated miR155 reverses the epithelial-mesenchymal transition induced by EGF and increases chemo-sensitivity to cisplatin in human Caski cervical cancer cells. PLoS One. 2012;7(12):e52310. | ||
Wang F, Liu M, Li X, Tang H. MiR-214 reduces cell survival and enhances cisplatin-induced cytotoxicity via down-regulation of Bcl2l2 in cervical cancer cells. FEBS Lett. 2013;587(5):488–495. | ||
Zhang G, Sun L, Lu X, et al. Cisplatin treatment leads to changes in nuclear protein and microRNA expression. Mutat Res. 2012;746(1):66–77. | ||
Liu SY, Zheng PS. High aldehyde dehydrogenase activity identifies cancer stem cells in human cervical cancer. Oncotarget. 2013;4(12):2462–2475. | ||
Casagrande N, De Paoli M, Celegato M, et al. Preclinical evaluation of a new liposomal formulation of cisplatin, lipoplatin, to treat cisplatin-resistant cervical cancer. Gynecol Oncol. 2013;131(3):744–752. | ||
Wang L, Guo H, Lin C, Yang L, Wang X. Enrichment and characterization of cancer stem-like cells from a cervical cancer cell line. Mol Med Rep. 2014;9(6):2117–2123. | ||
Bai T, Tanaka T, Yukawa K, Umesaki N. A novel mechanism for acquired cisplatin-resistance: suppressed translation of death-associated protein kinase mRNA is insensitive to 5-aza-2′-deoxycitidine and trichostatin in cisplatin-resistant cervical squamous cancer cells. Int J Oncol. 2006;28(2):497–508. | ||
Liu B, Cheng Y, Liu Q, Bao JK, Yang JM. Autophagic pathways as new targets for cancer drug development. Acta Pharmacol Sin. 2010;31(9):1154–1164. | ||
Xu Y, Yu H, Qin H, et al. Inhibition of autophagy enhances cisplatin cytotoxicity through endoplasmic reticulum stress in human cervical cancer cells. Cancer Lett. 2012;314(2):232–243. | ||
Sun Y, Liu JH, Jin L, et al. Over-expression of the Beclin1 gene upregulates chemosensitivity to anti-cancer drugs by enhancing therapy-induced apoptosis in cervix squamous carcinoma CaSki cells. Cancer Lett. 2010;294(2):204–210. | ||
Xu Y, Wang N, Ding Y, et al. Ammonium chloride enhances cisplatin cytotoxicity through DNA double-strand breaks in human cervical cancer cells. Oncol Rep. 2013;30(3):1195–1200. | ||
Leisching GR, Loos B, Botha MH, Engelbrecht AM. The role of mTOR during cisplatin treatment in an in vitro and ex vivo model of cervical cancer. Toxicology. 2015;335:72–78. | ||
Shen DW, Akiyama S, Schoenlein P, Pastan I, Gottesman MM. Characterisation of high-level cisplatin-resistant cell lines established from a human hepatoma cell line and human KB adenocarcinoma cells: cross-resistance and protein changes. Br J Cancer. 1995;71(4):676–683. | ||
Castagna A, Antonioli P, Astner H, et al. A proteomic approach to cisplatin resistance in the cervix squamous cell carcinoma cell line A431. Proteomics. 2004;4(10):3246–3267. | ||
Gallerne C, Prola A, Lemaire C. Hsp90 inhibition by PU-H71 induces apoptosis through endoplasmic reticulum stress and mitochondrial pathway in cancer cells and overcomes the resistance conferred by Bcl-2. Biochim Biophys Acta. 2013;1833(6):1356–1366. | ||
Jin L, Shen Q, Ding S, Jiang W, Jiang L, Zhu X. Immunohistochemical expression of annexin A2 and S100A proteins in patients with bulky stage IB-IIA cervical cancer treated with neoadjuvant chemotherapy. Gynecol Oncol. 2012;126(1):140–146. | ||
Zou S, Shen Q, Hua Y, Jiang W, Zhang W, Zhu X. Proteomic identification of neoadjuvant chemotherapy-related proteins in bulky stage IB-IIA squamous cervical cancer. Reprod Sci. 2013;20(11):1356–1364. | ||
Chavez JD, Hoopmann MR, Weisbrod CR, Takara K, Bruce JE. Quantitative proteomic and interaction network analysis of cisplatin resistance in HeLa cells. PLoS One. 2011;6(5):e19892. | ||
Wu ZZ, Lu HP, Chao CC. Identification and functional analysis of genes which confer resistance to cisplatin in tumor cells. Biochem Pharmacol. 2010;80(2):262–276. | ||
Vogl SE, Seltzer V, Camacho F, Calanog A. Dianhydrogalactitol and cisplatin in combination for advanced cancer of the uterine cervix. Cancer Treat Rep. 1982;66(10):1809–1812. | ||
Mistry P, Kelland LR, Abel G, Sidhar S, Harrap KR. The relationships between glutathione, glutathione-S-transferase and cytotoxicity of platinum drugs and melphalan in eight human ovarian carcinoma cell lines. Br J Cancer. 1991;64(2):215–220. | ||
Koberle B, Masters JR, Hartley JA, Wood RD. Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumours. Curr Biol. 1999;9(5):273–276. | ||
Ishida S, Lee J, Thiele DJ, Herskowitz I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc Natl Acad Sci U S A. 2002;99(22):14298–14302. | ||
Martelli L, Di Mario F, Ragazzi E, et al. Different accumulation of cisplatin, oxaliplatin and JM216 in sensitive and cisplatin-resistant human cervical tumour cells. Biochem Pharmacol. 2006;72(6):693–700. | ||
Kuo DY, Blank SV, Christos PJ, et al. Paclitaxel plus oxaliplatin for recurrent or metastatic cervical cancer: a New York Cancer Consortium Study. Gynecol Oncol. 2010;116(3):442–446. | ||
Tewari KS, Sill MW, Long HJ 3rd, et al. Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med. 2014;370(8):734–743. | ||
Sahin K, Tuzcu M, Basak N, et al. Sensitization of cervical cancer cells to cisplatin by genistein: the role of NFkappaB and Akt/mTOR signaling pathways. J Oncol. 2012;2012:461562. | ||
Singh M, Bhui K, Singh R, Shukla Y. Tea polyphenols enhance cisplatin chemosensitivity in cervical cancer cells via induction of apoptosis. Life Sci. 2013;93(1):7–16. | ||
Pariente R, Pariente JA, Rodriguez AB, Espino J. Melatonin sensitizes human cervical cancer HeLa cells to cisplatin-induced cytotoxicity and apoptosis: effects on oxidative stress and DNA fragmentation. J Pineal Res. 2016;60(1):55–64. | ||
Segovia-Mendoza M, Jurado R, Mir R, Medina LA, Prado-Garcia H, Garcia-Lopez P. Antihormonal agents as a strategy to improve the effect of chemo-radiation in cervical cancer: in vitro and in vivo study. BMC Cancer. 2015;15:21. | ||
Jurado R, Lopez-Flores A, Alvarez A, Garcia-Lopez P. Cisplatin cytotoxicity is increased by mifepristone in cervical carcinoma: an in vitro and in vivo study. Oncol Rep. 2009;22(5):1237–1245. | ||
Li C, Ye H. Mifepristone sensitizing cisplatin for cervical adenocarcinoma HeLa cell sensitivity to chemotherapy and its mechanism. Eur J Gynaecol Oncol. 2013;34(2):142–147. | ||
Yokoyama M, Noguchi M, Nakao Y, Pater A, Iwasaka T. The tea polyphenol, (-)-epigallocatechin gallate effects on growth, apoptosis, and telomerase activity in cervical cell lines. Gynecol Oncol. 2004;92(1):197–204. | ||
Kilic U, Sahin K, Tuzcu M, et al. Enhancement of cisplatin sensitivity in human cervical cancer: epigallocatechin-3-gallate. Front Nutr. 2015;1:28. | ||
Li Y, Xing D, Chen Q, Chen WR. Enhancement of chemotherapeutic agent-induced apoptosis by inhibition of NF-kappaB using ursolic acid. Int J Cancer. 2010;127(2):462–473. | ||
Gupta RK, Singh N. Morinda citrifolia (Noni) alters oxidative stress marker and antioxidant activity in cervical cancer cell lines. Asian Pac J Cancer Prev. 2013;14(8):4603–4606. | ||
Gupta RK, Banerjee A, Pathak S, Sharma C, Singh N. Induction of mitochondrial-mediated apoptosis by Morinda citrifolia (Noni) in human cervical cancer cells. Asian Pac J Cancer Prev. 2013;14(1):237–242. | ||
He F, Wang Q, Zheng XL, et al. Wogonin potentiates cisplatin-induced cancer cell apoptosis through accumulation of intracellular reactive oxygen species. Oncol Rep. 2012;28(2):601–605. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.