")
Back to Journals » Clinical Ophthalmology » Volume 14
MOG-IgG- versus AQP4-IgG-Positive Optic Neuritis in Thailand: Clinical Characteristics and Long-Term Visual Outcomes Comparison
Authors Narongkhananukul C, Padungkiatsagul T , Jindahra P, Khongkhatithum C, Thampratankul L, Vanikieti K
Received 22 October 2020
Accepted for publication 12 November 2020
Published 26 November 2020 Volume 2020:14 Pages 4079—4088
DOI https://doi.org/10.2147/OPTH.S288224
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Chanomporn Narongkhananukul,1 Tanyatuth Padungkiatsagul,1 Panitha Jindahra,2 Chaiyos Khongkhatithum,3 Lunliya Thampratankul,3 Kavin Vanikieti1
1Department of Ophthalmology, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; 2Department of Medicine, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand; 3Department of Pediatrics, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Correspondence: Kavin Vanikieti Email [email protected]
Purpose: To compare demographic data, clinical and radiological characteristics, treatment, and long-term visual outcomes between myelin oligodendrocyte glycoprotein autoantibody-positive optic neuritis (MOG-IgG + ON) and aquaporin-4 autoantibody-positive optic neuritis (AQP4-IgG + ON) in Thailand.
Patients and Methods: We included individuals who were diagnosed with either MOG-IgG + ON or AQP4-IgG + ON over an 11-year period. Demographic data, clinical and radiological characteristics at ON presentation, treatment, and long-term visual outcomes were retrospectively collected.
Results: There were 16 patients (28 eyes) and 43 patients (59 eyes) in the MOG-IgG + ON and AQP4-IgG + ON groups, respectively. AQP4-IgG + ON occurred predominantly in female patients whereas MOG-IgG + ON-affected female patients and male patients equally (p < 0.001). Prior or concurrent non-ON demyelinating events were more often observed at AQP4-IgG + ON onset (p < 0.001). At ON presentation, bilaterality and the presence of optic disc edema were predominantly found in the MOG-IgG + ON group (bilaterality: 80% vs 8%, MOG-IgG + ON vs AQP4-IgG + ON patients, respectively (p < 0.001); presence of optic disc edema: 92.3% vs 36.6%, MOG-IgG + ON- vs AQP4-IgG + ON-affected eyes, respectively (p < 0.001)). There was no statistically significant difference in age at ON onset, nadir visual acuity (VA), presence of pain, segmental enhancement, and total enhanced segments of the anterior visual pathways. At the last follow-up, immunosuppressive drugs were used more often in the AQP4-IgG + ON group (43.7% vs 74.4%, MOG-IgG + ON vs AQP4-IgG + ON, respectively; p < 0.027). Remarkably better final VA was achieved in MOG-IgG + ON-affected eyes (median: 0.0 vs 0.4 logMAR, MOG-IgG + ON- vs AQP4-IgG + ON-affected eyes, respectively; p < 0.001).
Conclusion: Compared with AQP4-IgG + ON, MOG-IgG + ON tended to present with bilaterality and optic disc edema and demonstrated better visual outcomes.
Keywords: optic neuritis, myelin oligodendrocyte glycoprotein, aquaporin-4, neuromyelitis optica spectrum disorder, Thai
Introduction
Optic neuritis (ON) is an inflammation of the optic nerve, commonly associated with demyelinating diseases. In contrast with Caucasians, the prevalence of atypical ON in Asians is much higher than typical ON (multiple sclerosis-associated ON).1 A large number of patients with atypical ON were diagnosed with neuromyelitis optica (NMO). This was later redefined as neuromyelitis optica spectrum disorder (NMOSD), which is a severe inflammatory disease of the central nervous system (CNS). In the early 2000s, an autoantibody (immunoglobulin G (IgG)) against aquaporin-4 (AQP4), a water channel protein expressed in astrocytes predominantly in the CNS, especially in the optic nerve and spinal cord, was proposed as a pathogenic cause for NMOSD.2 However, a proportion of NMOSD-associated ON patients who were seronegative for AQP4-IgG showed remarkable distinction in terms of clinical characteristics and final visual outcomes. Such variations led to the search for other biomarkers to differentiate the new clinical entities among seronegative AQP4-IgG NMOSD patients. An IgG against myelin oligodendrocyte glycoprotein (MOG), which is the protein presenting at the outer surface of oligodendrocytic myelin sheaths, has been discovered and is proposed to be the pathogenic cause in seronegative AQP4-IgG NMOSD patients.3 Only recently, after the availability of a cell-based assay (CBA), which more reliably detects MOG-IgG, was this differentiation clarified.4 It is believed that AQP4-IgG primarily attacks the water channels of the astrocytes, causing astrocytopathy; however, MOG-IgG directly attacks the myelin and oligodendrocytes. Regarding ON, several studies have investigated the similarities and differences between these two IgGs and their related clinical and radiological characteristics, treatment, and long-term visual outcomes, but none directly compared these two entities in the Thai population. Therefore, our objective was to comprehensively compare demographic data, detailed clinical and radiological characteristics, treatment, and long-term visual outcomes between myelin oligodendrocyte glycoprotein autoantibody-positive (MOG-IgG +) and aquaporin-4 autoantibody-positive (AQP4-IgG +) ON in Thai patients.
Patients and Methods
This study was approved by the institutional review board of the Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand and followed the tenets of the Declaration of Helsinki. The patients were informed about the purpose of the study. Consent was obtained from the patients prior to study commencement. We retrospectively reviewed the electronic medical records of 16 patients who were diagnosed with MOG-IgG + ON and 43 patients who were diagnosed with AQP4-IgG + ON in the Neuro-Ophthalmology Clinic, Faculty of Medicine Ramathibodi Hospital between January 2010 and September 2020. MOG-IgG and AQP4-IgG serostatus were assessed by CBA. All patients showed seropositivity to either MOG-IgG or AQP4-IgG. Affected eyes with visually significant cataract and/or other ocular diseases affecting visual acuity (VA) were excluded from nadir VA at ON onset and final VA analysis.
The following data were collected from the patients’ electronic medical records: sex, age at ON onset, ON follow-up time, non-ON demyelinating events (prior to or simultaneous with ON onset), presence of antinuclear antibody (ANA), concurrent autoimmune diseases, and patients’ clinical characteristics at ON presentation, namely, laterality, presence of pain, nadir VA, and presence of optic disc edema. Magnetic resonance imaging (MRI) of the anterior visual pathway (AVP) performed at ON presentation was also evaluated. Each AVP scan was composed of five segments: orbital optic nerve, intracanalicular optic nerve, intracranial optic nerve, optic chiasm, and optic tract, and we assessed the presence of enhancement in each segment (Figure 1). In the acute phase, all patients in our cohort received 3–5 consecutive days of intravenous methylprednisolone (IVMP) 1 g/day (for adults) or 30 mg/kg/day (maximum 1 g/day) (for pediatrics) followed by slowly tapered prednisolone for 2–3 months as the first-line acute-phase treatment. Data for acute-phase treatment of each ON episode and maintenance phase treatment (immunosuppressive drugs used at the last follow-up) were also collected. Finally, we analyzed long-term visual outcomes, namely, final VA, presence of ON relapse, ON annualized relapse rate (ARR) (calculated as the ratio of the number of ON episodes and years since ON onset, excluding patients with ON follow-up time <1 year), and time from ON onset to the first ON relapse.
 |
Figure 1 (A) Each AVP scan was composed of five segments: orbital optic nerve, intracanalicular optic nerve, intracranial optic nerve, optic chiasm and optic tract. (B) Fat-suppressed T1-weighted with gadolinium contrast axial MRI showing enhancement of optic chiasm (arrow) and optic tract (arrowhead). |
Statistical Analysis
To compare differences between the two groups, for continuous data, we used the t-test for normally distributed data, and Wilcoxon’s rank-sum test or the Mann–Whitney U-test for non-normal distributions. Snellen VA was converted into a logarithm of the minimum angle of resolution (logMAR) equivalents for statistical analysis. The VA of counting fingers, hand motion, light perception, and no light perception were converted to 2.6, 2.7, 2.8, and 2.9 logMAR, respectively. For categorical data, Pearson’s chi-squared test and Fisher’s exact test were used as appropriate. P-values <0.05 were considered statistically significant, and all statistical analyses were performed using STATA software, version 16.0 (StataCorp LLC, College Station, TX, USA).
Results
Demographic Data
A total of 59 patients (87 eyes) were included in this study; 16 patients (28 eyes) and 43 patients (59 eyes) were MOG-IgG + and AQP4-IgG +, respectively, and all patients were Asian. The majority of our cohort was female (49 patients, 83%). In contrast to the AQP4-IgG + ON group in which female patients were highly predominant at 95.3%, the female: male ratio in the MOG-IgG + ON group was 1:1 (p < 0.001). Four patients in each group developed ON at ≤18 years of age, and these pediatric patients accounted for 25% and 9.3% of the MOG-IgG + ON and AQP4 IgG+ ON patients, respectively (p = 0.19). In the MOG-IgG + ON group, only one patient experienced non-ON demyelinating events prior to ON onset. In contrast, in the AQP4-IgG + ON group, 19 of 25 patients with available data (76%) had experienced non-ON demyelinating events prior to or simultaneous with ON onset; myelitis was the most common event (89.4%). This difference between the two groups was statistically significant (p < 0.001). There was no statistically significant difference in age at ON onset and ON follow-up time between the two groups. The presence of ANA was found in more than half of the patients in both groups (58.3% vs 63.8%, MOG-IgG + ON vs AQP4-IgG + ON patients, respectively; p = 0.74). In the AQP4-IgG + ON group, concurrent autoimmune diseases were identified in 11 patients (25.5%). These patients constituted those with Sjogren syndrome (four patients), systemic lupus erythematosus (three patients), Graves’ disease (three patients), and myasthenia gravis (one patient). However, none of the patients in the MOG-IgG + ON group had concurrent autoimmune disease (p = 0.05). A detailed summary of the patients’ demographic data is provided in Table 1.
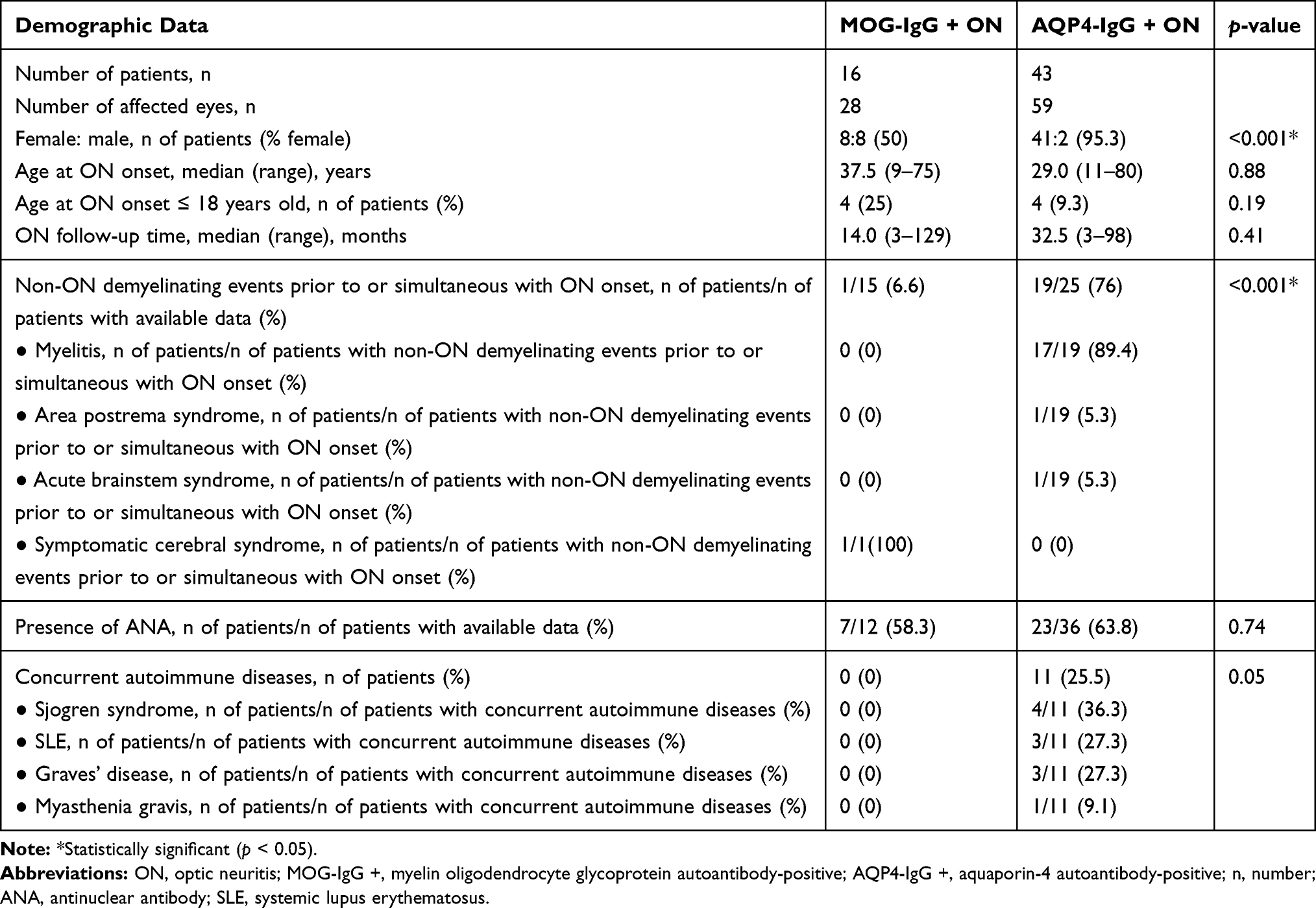 |
Table 1 Patients’ Demographic Data |
Clinical Characteristics at ON Presentation
Data regarding clinical characteristics at ON presentation were available for 15 patients (26 eyes) and 25 patients (30 eyes) in the MOG-IgG + ON and AQP4-IgG + ON group, respectively.
In contrast to the AQP4-IgG + ON group, bilaterality of ON at presentation was more common in patients in the MOG-IgG + ON group (80% vs 8%, MOG-IgG + ON vs AQP4-IgG + ON patients, respectively; p < 0.001).
Presence of pain was more frequently observed in MOG-IgG + ON-affected eyes at ON onset (92.3%) compared with AQP4-IgG + ON-affected eyes at ON onset (73.0%). However, this trend was not statistically significant (p = 0.08).
Both groups demonstrated poor nadir VA at ON onset without a statistically significant difference between the two groups (mean ± standard deviation (SD) 1.80 ± 0.91 vs 2.12 ± 0.87 logMAR, MOG-IgG + ON- vs AQP4-IgG + ON-affected eyes, respectively; p = 0.22). Likewise, more than three quarters of affected eyes in both groups demonstrated a nadir VA at ON onset of worse than or equal to 1.0 logMAR (worse than or equal to 20/200 measured by the Snellen chart) (76.9% vs 86.6%, MOG-IgG + ON- vs AQP4-IgG + ON-affected eyes, respectively; p = 0.48).
Interestingly, the presence of optic disc edema was found predominantly in MOG-IgG + ON-affected eyes at ON onset, and much less commonly in AQP4-IgG + ON-affected eyes (92.3% vs 36.6%, MOG-IgG + ON- vs AQP4-IgG + ON-affected eyes at ON onset, respectively; p < 0.001).
A detailed summary of patients’ clinical characteristics at ON presentation is provided in Table 2.
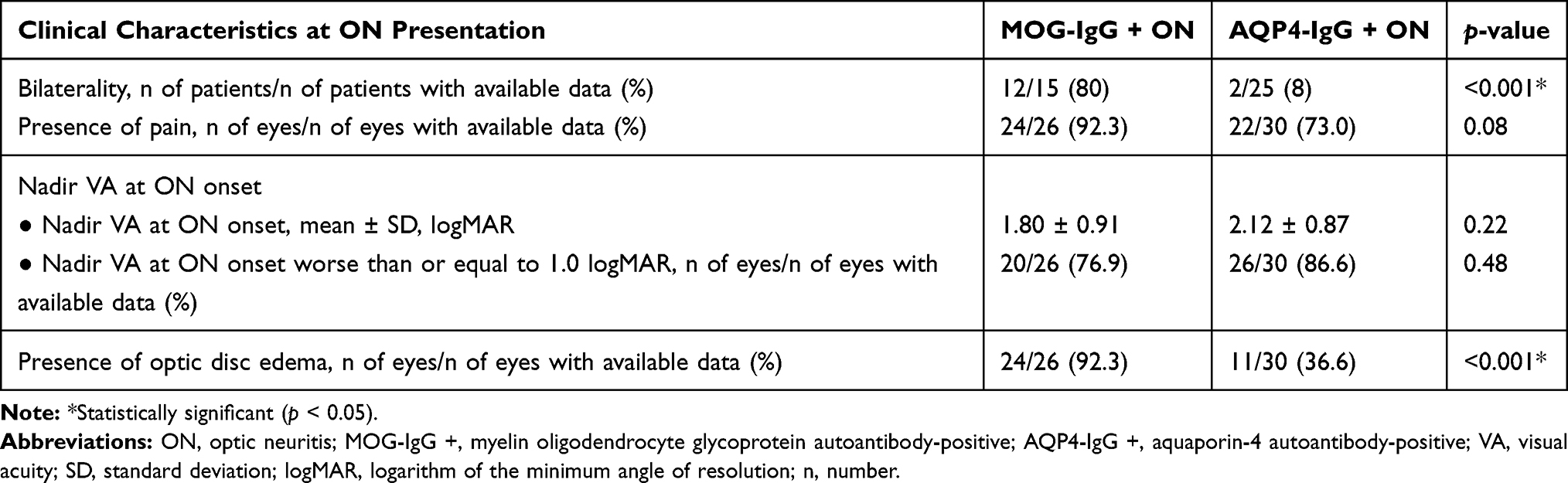 |
Table 2 Clinical Characteristics at ON Presentation |
MRI Characteristics at ON Presentation
At ON presentation, imaging data for 26 AVPs in MOG-IgG + ON-affected eyes (26 affected eyes) and 30 AVPs in AQP4-IgG + ON-affected eyes (30 affected eyes) were available for analysis. There was no statistically significant difference regarding segmental enhancement and total enhanced segments of AVPs between the two groups at ON presentation. In the MOG-IgG + ON group, there was a slightly higher proportion of AVPs demonstrating at least two or more consecutive enhanced segments compared with the AQP4-IgG + ON group; however, this trend was not statistically significant (92.3% vs 73.3%, MOG-IgG + ON- vs AQP4-IgG + ON-affected eyes, respectively, p = 0.06). A detailed summary of patients’ MRI characteristics at ON presentation is provided in Table 3.
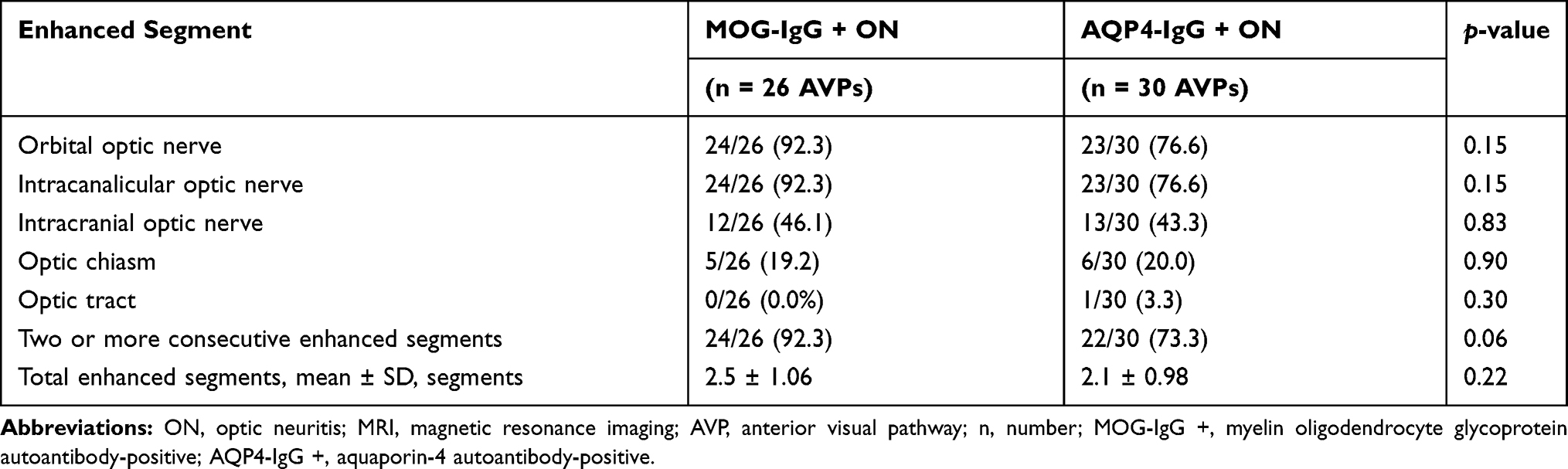 |
Table 3 MRI Characteristics at ON Presentation: Segmental Enhancement of AVPs During ON Presentation, n of AVPs with Each Enhanced Segment/n of Available AVP Images (%) |
Treatment
Acute Phase
There were 25 MOG-IgG + ON episodes and 80 AQP4-IgG + ON episodes treated acutely in our institution. In the acute phase, all patients in our cohort received 3–5 consecutive days of IVMP 1 g/day (for adults) or 30 mg/kg/day (maximum 1 g/day) (for pediatrics) followed by slowly tapered prednisolone for 2–3 months as the first-line acute-phase treatment, without a statistically significant difference in the time from ON symptom onset to IVMP treatment between the two groups (median 14 vs 7 days, MOG-IgG + ON vs AQP4-IgG + ON groups, respectively; p = 0.66).
As the second-line therapy, adjunctive plasma exchange (PLEX) was performed in 8.75% of AQP4-IgG + ON episodes, but in none of the MOG-IgG + ON episodes. However, this difference was not statistically significant (p = 0.19).
A detailed summary of the acute-phase treatment is provided in Table 4.
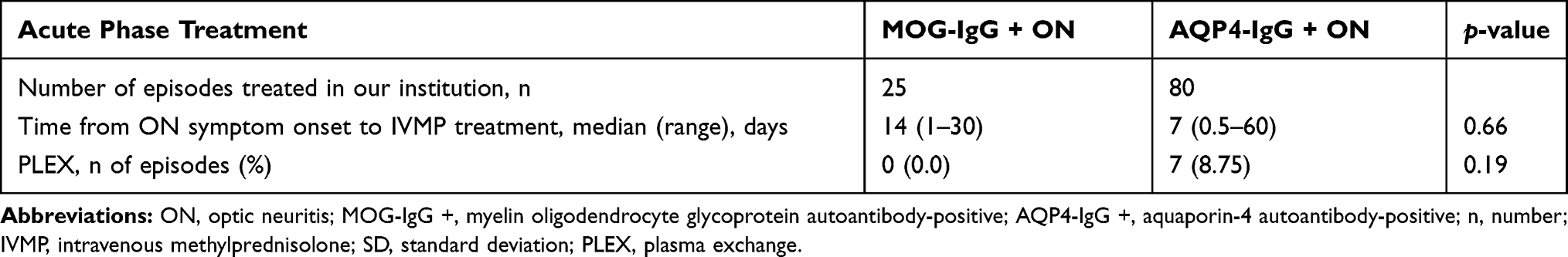 |
Table 4 Acute Phase Treatment |
Maintenance Phase
Notably, a much smaller percentage of patients receiving immunosuppressive drugs at the last follow-up was observed in 43.7% of the patients in the MOG-IgG + ON group compared with 74.4% of the patients in the AQP4-IgG + ON group (p = 0.027). Azathioprine and mycophenolate mofetil were the most common immunosuppressive drugs used in MOG-IgG + ON (57.1%) and AQP4-IgG + ON (43.8%) patients, respectively. A detailed summary of the maintenance phase treatment is provided in Table 5.
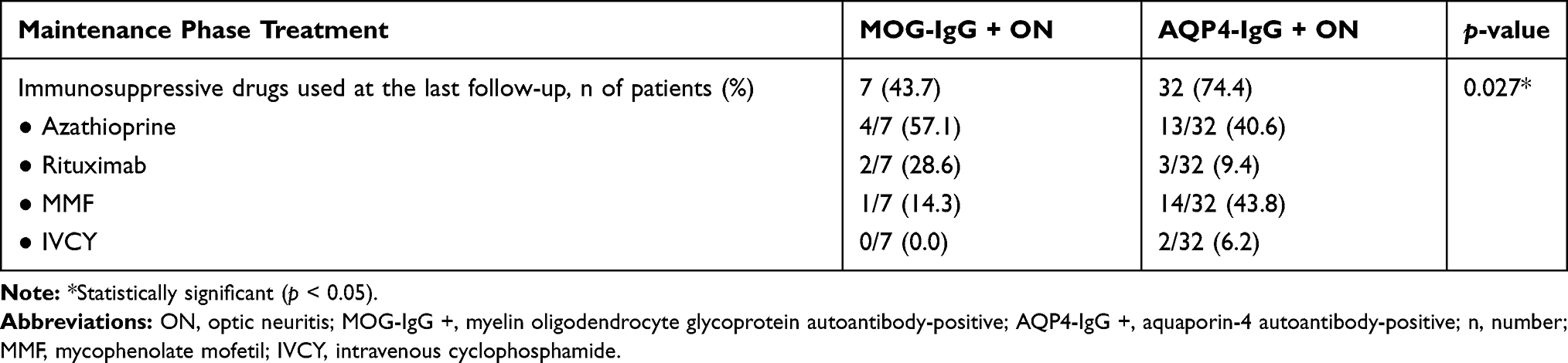 |
Table 5 Maintenance Phase Treatment: Immunosuppressive Drugs Used at the Last Follow-Up; n of Patients Receiving Each Immunosuppressive Drug/Total n of Patients Receiving Immunosuppressive Drugs (%) |
Long-Term Visual Outcomes
We excluded four affected eyes in the AQP4-IgG + ON group owing to the referral of the patients to their primary care hospitals. Therefore, 55 affected eyes remained in the AQP4-IgG + ON group to evaluate the final VA. To evaluate the final VA, which was one of the long-term visual outcomes, the last of the follow-up data was collected at least 3 months following the most recent ON episode. There was a remarkable difference in final VA between the two groups. In MOG-IgG + ON-affected eyes, the median final VA was 0.0 logMAR, compared with 0.4 logMAR observed in AQP4-IgG + ON-affected eyes (p < 0.001). Moreover, none of the MOG-IgG + ON-affected eyes achieved a final VA worse than or equal to 1.0 logMAR (worse than or equal to 20/200 measured by the Snellen chart), in contrast to 34.6% of AQP4-IgG + ON-affected eyes (p < 0.001). There were no statistically significant differences between MOG-IgG + ON and AQP4-IgG + ON patients in terms of the number of patients with ON relapse, ON ARR, and time from ON onset to first ON relapse. A detailed summary of patients’ long-term visual outcomes is provided in Table 6.
 |
Table 6 Long-Term Visual Outcomes |
Discussion
Records from 59 patients (87 eyes) constituting 16 patients (28 eyes) and 43 patients (59 eyes) who demonstrated seropositivity for MOG-IgG and AQP4-IgG, respectively, were enrolled in this study. To our knowledge, this is the first study to comprehensively compare demographic data, detailed clinical and radiological characteristics, treatment, and long-term visual outcomes between MOG-IgG + ON and AQP4-IgG + ON in the Thai population. Moreover, this study included the largest series of MOG-IgG + ON in Thai patients.
Demographic Data
In our study, we found an equal (1:1) distribution in the sex ratio of female: male in the MOG-IgG + ON patients. This finding was comparable to two previous large retrospective multicenter series of seropositive MOG-IgG patients.5,6 In contrast, a high predominance of females (95.3%) was observed in our AQP4-IgG + ON patients, which is consistent with findings in a previous AQP4-IgG + ON study.7 Moreover, we demonstrated a statistically significant difference between the two groups for this issue, which is consistent with previous reports conducted in both Asian and Caucasian populations.8–10
The median age at ON onset in MOG-IgG + ON patients was the mid-thirties, which was not statistically different from the age at ON onset in AQP4-IgG + ON patients. This result was comparable to previous studies.10,11
More than half of AQP4-IgG + ON patients experienced non-ON demyelinating events prior to or simultaneous with ON onset, with myelitis as the leading event. In contrast, we found only one MOG-IgG + ON patient who presented with concurrent symptomatic cerebral syndrome (seizure) at ON onset. This might imply that isolated ON is more likely the sole, or the first presentation, of MOG-antibody diseases (MOGAD). This finding was supported by several multicenter studies showing that isolated ON is often the first presentation of MOGAD.5,12 Moreover, in a large multicenter retrospective study comparing the clinical features and visual outcomes between Caucasian and Asian MOG-IgG + ON patients, the authors found that Asian patients were less likely to have non-ON demyelinating manifestations.6 This finding was comparable to findings in our study, in which all patients in the MOG-IgG + ON group were Asian. Therefore, we emphasized that ophthalmologists should be familiar with MOG-IgG + ON because patients usually presented with isolated ON, without other non-ON demyelinating disabilities, unlike AQP4-IgG + ON.
Although there was no statistically significant difference in the presence of ANA between the two groups (p = 0.74), one-quarter of AQP4-IgG + ON patients had concurrent autoimmune diseases compared with none of the MOG-IgG + ON patients. This difference was nearly statistically significant (p = 0.05), and this result was comparable to those in a previous study that revealed a lower rate of concurrent autoimmune diseases in MOGAD patients compared with seropositive AQP4-IgG patients.13
Clinical Characteristics at ON Presentation
In our study, the incidence of bilateral simultaneous ON was observed strikingly more frequently in the MOG-IgG + ON group compared with the AQP4-IgG + ON group (80% vs 8%, MOG-IgG + ON vs AQP4-IgG + ON patients, respectively; p < 0.001). This result is inconsistent with previous studies that showed no statistically significant difference in bilaterality at ON onset between the two groups.9,10,14
At ON presentation, severe visual loss was evident in both groups based on the average nadir VA of 1.80 logMAR and 2.12 logMAR in MOG-IgG + ON- and AQP4-IgG + ON-affected eyes, respectively. Moreover, more than three quarters of affected eyes in both groups exhibited a nadir VA at ON onset of worse than or equal to 1.0 logMAR (worse than or equal to 20/200 measured by the Snellen chart). However, without a statistically significant difference between the two groups for nadir VA at ON onset, this clinical characteristic could not help differentiate between the two etiologies. This finding was consistent with previous studies.9,14
Similarly, the presence of pain, one of the most frequent symptoms at ON presentation, was not statistically significantly different in our study and was a less useful tool in clinical differentiation between the two groups.
Interestingly, the number of eyes with optic disc edema was remarkably higher in MOG-IgG + ON-affected eyes compared with AQP4-IgG + ON-affected eyes (92.3% vs 36.6%, MOG-IgG + ON- vs AQP4-IgG + ON-affected eyes at ON onset, respectively; p < 0.001). This difference was fairly similar to previous studies that also found frequent optic disc edema among MOG-IgG + ON patients.9,14–16
MRI Characteristics at ON Presentation
In our study, orbital and intracanalicular optic nerves were the most commonly affected AVP segments in both groups. Moreover, there was no statistically significant difference in segmental enhancement of AVP between the two groups. These results were inconsistent with a previous study that demonstrated a higher tendency toward orbital optic nerve involvement in MOG-IgG + ON and intracranial optic nerve, optic chiasm, and optic tract involvement in AQP4-IgG + ON.11 However, the different methodology between our study and the previous study could have affected the results. In our study, we assessed only the presence of enhancement in each AVP segment, whereas the previous study evaluated the presence of T2 hyperintensity and/or contrast enhancement. Another possible explanation was that most of our patients underwent MRI examination 2–4 days following the initiation of IVMP, which could have interfered with the sensitivity of enhancement detection.
Two or more consecutive enhanced AVP segments were frequently observed in both groups. This was fairly comparable to a previous study that showed longitudinally extensive involvement of AVP in both MOG-IgG + ON and AQP4-IgG + ON.11 However, we did not assess the actual lesion length in millimeters, as in the previous study.
Treatment
Acute Phase
There was no statistically significant difference in time from ON symptom onset to IVMP treatment between the two groups. After excluding infection and other contraindications for corticosteroid treatment, high-dose IVMP was our first-line treatment in all patients with immune-mediated ON, either with MOG-IgG + ON or AQP4-IgG + ON. Our standard protocol of IVMP at 1 g/day (for adults) or 30 mg/kg/day (maximum 1 g/day) (for pediatrics) for 3 consecutive days was used in all patients, with an extension to 5 days if there was no clinical response within 3 days. Following IVMP treatment, slowly tapered prednisolone for 2–3 months was then initiated.
PLEX was our second-line adjunctive treatment, reserved only for patients who did not respond to IVMP. With a higher number of PLEX procedures performed in the AQP4-IgG + ON group compared with none in the MOG-IgG + ON group, despite the lack of statistical significance, this suggests that MOG-IgG + ON patients were more likely to achieve a satisfactory response to IVMP treatment compared with AQP4-IgG + ON patients.
Maintenance Phase
At the last follow-up, immunosuppressive drugs were used significantly more often in AQP4-IgG + ON patients compared with MOG-IgG + ON patients. This was because of the lack of standard treatment for MOG-IgG + ON, seroconversion of MOG-IgG at the 6-month follow-up in five MOG-IgG + ON patients, and the excellent visual prognosis of MOG-IgG + ON. These factors made the treating physicians feel less obliged to continue immunosuppressive drugs, especially in vulnerable and pediatric patients. Therefore, in our institution, immunosuppressive drugs were used only in patients with multiple ON relapses and/or persistent MOG-IgG during follow-up. In contrast, immunosuppressive drugs were compulsory for at least 5 years following ON onset in nearly all of the AQP4-IgG + ON patients.
Owing to the lack of prospective randomized controlled trials, there is no consensus regarding standard immunosuppressive drugs for MOG-IgG + ON and AQP4-IgG + ON. In our institution, overall, azathioprine was used as the primary treatment. Mycophenolate mofetil was the next immunosuppressive drug used in patients who did not respond well to azathioprine, or who had serious side effects; for example, drug-induced hepatitis. Medical expenses coverage was another factor in selecting appropriate immunosuppressive drugs for individual patients.
Long-Term Visual Outcomes
Despite the non-significant differences in nadir VA at ON onset, time from ON symptom onset to IVMP treatment, and ON ARR between the two groups, a remarkable distinction in final VA was demonstrated in our study, as the median final VA in MOG-IgG +ON-affected eyes was 0.0 logMAR in contrast to 0.4 logMAR in AQP4-IgG + ON-affected eyes. Moreover, none of the MOG-IgG + ON-affected eyes achieved a final VA of worse than or equal to 1.0 logMAR (worse than or equal to 20/200 measured by the Snellen chart) in contrast to 34.6% of AQP4-IgG + ON-affected eyes. This finding was consistent with several previous Asian and Caucasian studies that revealed better visual outcomes in MOG-IgG + ON compared with AQP4-IgG + ON.9,14,17 Several proposed theories tried to explain the mechanisms causing such different visual outcomes. Differences in specific proteins played an important role in the different mechanisms and outcomes. Kaneko et al reported the presence of high levels of myelin basic protein (MBP) in cerebrospinal fluid in both the MOG-IgG + and AQP4-IgG + groups, while high levels of glial fibrillary acidic protein (GFAP) were found only in the AQP4-IgG + group.18 This discovery indicated that there was myelin injury only in the MOG-IgG + group. In contrast, in the AQP4-IgG + group, the injury was not limited to myelin, but also affected the astrocytes, leading to greater damage at the cellular level in the optic nerve. Meanwhile, other studies in mice showed that MOG-IgG induced transient changes in myelin, with complete recovery.19,20
In our study, there was no statistically significant difference in ON ARR between the two groups. This was inconsistent with a previous exclusively Caucasian study that reported a higher ON ARR in 16 MOG-IgG + ON patients, compared with 16 AQP4-IgG + ON patients.21 Differences in ethnicity, number of AQP4-IgG + ON patients, and ON follow-up time between the two studies could have interfered with the results.
There were several strengths in our study. First, despite the relatively recent availability of MOG-IgG testing in Thailand, we were able to have the test performed in patients who were previously diagnosed with seronegative AQP4-IgG NMOSD ON or chronic relapsing inflammatory optic neuropathy (CRION), leading to a better understanding of long-term visual outcomes of MOG-IgG + ON. Second, the number of patients and affected eyes in our AQP4-IgG + ON group were quite large. Third, pediatric patients were also included in our study; therefore, our results are applicable to the pediatric population. Finally, to our knowledge, this is the first study to comprehensively compare various ophthalmic aspects between MOG-IgG + ON and AQP4-IgG + ON in the Thai population, and our study included the largest series of MOG-IgG + ON in Thai patients.
There were limitations in our study, and the first was the retrospective design, which resulted in some incomplete data. Second, we did not assess other aspects of visual function, such as color vision, contrast sensitivity, and visual field, or structural characteristics, such as retinal nerve fiber layer thickness and macular ganglion cell analysis.
Conclusions
We demonstrated an equal distribution in the female: male sex ratio in MOG-IgG + ON compared with a high predominance of females in the AQP4-IgG + ON group. Prior or concurrent non-ON demyelinating events were observed more frequently at AQP4-IgG + ON onset. At ON presentation, both groups shared the same clinical and MRI characteristics except for a higher proportion of bilaterality and the presence of optic disc edema observed in MOG-IgG + ON. Compared with AQP4-IgG + ON, better final visual outcomes were remarkably achieved in MOG-IgG + ON. These findings elucidated our hypothesis that MOG-IgG + ON constitutes a distinct and unique clinical entity compared with AQP4-IgG + ON. However, future prospective studies with larger numbers of patients are required to validate our results.
Consent for Publication
Not applicable.
Acknowledgments
The authors would thank Mr Nattawut Unwanatham, MSc (Statistics), and Miss Sasiporn Sitthisorn, MSc (Statistics), Department of Clinical Epidemiology and Biostatistics, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand for their advice on the statistical analyses. We thank Jane Charbonneau, DVM, from Edanz Group (https://en-author-services.edanzgroup.com/ac) for editing a draft of this manuscript.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Woung L, Chung H, Jou J, Wang K, Peng P. A comparison of optic neuritis in Asian and in Western countries. Neuroophthalmology. 2011;35(2):65–72.
2. Fujihara K. Neuromyelitis optica spectrum disorders. Curr Opin Neurol. 2019;32(3):385–394.
3. Peschl P, Bradl M, Höftberger R, Berger T, Reindl M. Myelin oligodendrocyte glycoprotein: deciphering a target in inflammatory demyelinating diseases. Front Immunol. 2017;8:529.
4. Waters P, Komorowski L, Woodhall M, et al. A multicenter comparison of MOG-IgG cell-based assays. Neurology. 2019;92(11):e1250–1255.
5. Cobo-Calvo A, Ruiz A, Maillart E, et al. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults. Neurology. 2018;90(21):e1858–1869.
6. Padungkiatsagul T, Chen J, Jindahra P, et al. Differences in clinical features of myelin oligodendrocyte glycoprotein antibody-associated optic neuritis in White and Asian race. Am J Ophthalmol. 2020;219:332–340.
7. Vanikieti K, Poonyathalang A, Jindahra P, Bouzika P, Rizzo J, Cestari D. Clinical characteristics and long-term visual outcome of optic neuritis in neuromyelitis optica spectrum disorder: a comparison between Thai and American-Caucasian cohorts. Mult Scler Relat Disord. 2017;17:87–91.
8. Zhao G, Chen Q, Huang Y, et al. Clinical characteristics of myelin oligodendrocyte glycoprotein seropositive optic neuritis: a cohort study in Shanghai, China. J Neurol. 2017;265(1):33–40.
9. Zhao Y, Tan S, Chan T, et al. Clinical features of demyelinating optic neuritis with seropositive myelin oligodendrocyte glycoprotein antibody in Chinese patients. Br J Ophthalmol. 2018;102(10):1372–1377.
10. Kitley J, Waters P, Woodhall M, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol. 2014;71(3):276–283.
11. Ramanathan S, Prelog K, Barnes E, et al. Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. Mult Scler. 2016;22(4):470–482.
12. Cobo‐Calvo A, Ruiz A, Rollot F, et al. Clinical features and risk of relapses in children and adults with MOGAD. Ann Neurol. 2020.
13. Ciotti J, Eby N, Wu G, Naismith R, Chahin S, Cross A. Clinical and laboratory features distinguishing MOG antibody disease from multiple sclerosis and AQP4 antibody-positive neuromyelitis optica. Mult Scler Relat Disord. 2020;45:102399.
14. Song H, Zhou H, Yang M, et al. Different characteristics of aquaporin-4 and myelin oligodendrocyte glycoprotein antibody-seropositive male optic neuritis in China. J Ophthalmol. 2019.
15. Akaishi T, Sato D, Nakashima I, et al. MRI and retinal abnormalities in isolated optic neuritis with myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies: a comparative study. J Neurol Neurosurg Psychiatry. 2015;87(4):446–448.
16. Chen JJ, Flanagan EP, Jitprapaikulsan J, et al. Myelin oligodendrocyte glycoprotein antibody-positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. 2018;195:8–15.
17. Sotirchos E, Filippatou A, Salama S, et al. MOG-IgG is associated with better visual outcomes after optic neuritis than AQP4-IgG seropositivity, despite similar severity of inner retinal layer thinning. Neurology. 2019;92(15 Supplement):
18. Kaneko K, Sato D, Nakashima I, et al. Myelin injury without astrocytopathy in neuroinflammatory disorders with MOG antibodies. J Neurol Neurosurg Psychiatry. 2016;87(11):1257–1259.
19. Dale RC, Tantsis EM, Merheb V, et al. Antibodies to MOG have a demyelination phenotype and affect oligodendrocyte cytoskeleton. Neurol Neuroimmunol Neuroinflamm. 2014;1(1):1–9.
20. Saadoun S, Waters P, Owens GP, Bennett JL, Vincent A, Papadopoulos MC. Neuromyelitis optica MOG-IgG causes reversible lesions in mouse brain. Acta Neuropathol Commun. 2014;2(1):35.
21. Pache F, Zimmermann H, Mikolajczak J, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 4: afferent visual system damage after optic neuritis in MOG-IgG-seropositive versus AQP4-IgG-seropositive patients. J Neuroinflammation. 2016;13(1):282.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.