")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
Modulation of the GABAergic pathway for the treatment of fragile X syndrome
Authors Lozano R, Hare E, Hagerman R
Received 3 January 2014
Accepted for publication 3 March 2014
Published 16 September 2014 Volume 2014:10 Pages 1769—1779
DOI https://doi.org/10.2147/NDT.S42919
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Reymundo Lozano,1,2 Emma B Hare,1,2 Randi J Hagerman1,2
1MIND Institute, 2Department of Pediatrics, UC Davis Medical Center, Sacramento, CA, USA
Abstract: Fragile X syndrome (FXS) is the most common genetic cause of intellectual disability and the most common single-gene cause of autism. It is caused by mutations on the fragile X mental retardation gene (FMR1) and lack of fragile X mental retardation protein, which in turn, leads to decreased inhibition of translation of many synaptic proteins. The metabotropic glutamate receptor (mGluR) hypothesis states that the neurological deficits in individuals with FXS are due mainly to downstream consequences of overstimulation of the mGluR pathway. The main efforts have focused on mGluR5 targeted treatments; however, investigation on the gamma-aminobutyric acid (GABA) system and its potential as a targeted treatment is less emphasized. The fragile X mouse models (Fmr1-knock out) show decreased GABA subunit receptors, decreased synthesis of GABA, increased catabolism of GABA, and overall decreased GABAergic input in many regions of the brain. Consequences of the reduced GABAergic input in FXS include oversensitivity to sensory stimuli, seizures, and anxiety. Deficits in the GABA receptors in different regions of the brain are associated with behavioral and attentional processing deficits linked to anxiety and autistic behaviors. The understanding of the neurobiology of FXS has led to the development of targeted treatments for the core behavioral features of FXS, which include social deficits, inattention, and anxiety. These symptoms are also observed in individuals with autism and other neurodevelopmental disorders, therefore the targeted treatments for FXS are leading the way in the treatment of other neurodevelopmental syndromes and autism. The GABAergic system in FXS represents a target for new treatments. Herein, we discuss the animal and human trials of GABAergic treatment in FXS. Arbaclofen and ganaxolone have been used in individuals with FXS. Other potential GABAergic treatments, such as riluzole, gaboxadol, tiagabine, and vigabatrin, will be also discussed. Further studies are needed to determine the safety and efficacy of GABAergic treatments for FXS.
Keywords: gamma-aminobutyric acid (GABA) system, targeted treatments, autism, ganaxolone, arbaclofen
Introduction
Fragile X syndrome (FXS) is the most common cause of inherited intellectual disability and largest single-gene cause of autism.1 The fragile X mental retardation gene (FMR1) located on the X chromosome at band q27.3 typically has 5–44 CGG repeats; however, this trinucleotide repeat length can expand to an unstable repeat length. 45-54 CGG repeats is considered a gray zone and may have some clinical involvement, including a higher rate of primary ovarian insufficiency compared to controls.2 Premutation carriers have trinucleotide repeats ranging from 55–200 CGGs in length. These are usually healthy individuals who are at risk of developing fragile X-associated tremor/ataxia syndrome, a neurodegenerative disorder seen in aging carriers.3 They are also at an increased risk for anxiety and mood disorders,4 immune-mediated disorders,5 migraine headaches,6 hypertension,7 and premature ovarian insufficiency,8 all of which range in severity and prevalence. Adult female carriers have evidence of gamma-aminobutyric acid (GABA) dysfunction.9
There are some carriers who may show an attention-deficit hyperactivity disorder (ADHD) and autism spectrum disorders (ASDs);10 however, the prevalence of those traits is far more common in individuals with the full mutation whose trinucleotide expansion is greater than 200 CGG repeats. The full mutation presents with a range of clinical features, including ADHD, ASDs, anxiety, intellectual disability, social avoidance, aggression, stereotyped behaviors, disrupted sleep patterns, epilepsy, macroorchidism, prominent ears, long faces, soft skin, and hyperextensible joints.11 Males with the full mutation are typically more severely affected than females due to the compensatory nature of the second X chromosome, which still translates the FMR1 gene leading to more normal fragile X mental retardation protein (FMRP) production. The prevalence of the full mutation is approximately one in 5,000 males12 and one in 8,000 females,13 whereas the premutation is far more common, at one in 130–250 females and one in 250–810 males.14
The phenotypic features found in both the premutation and full mutation are believed to be caused by the expanded trinucleotide sequences, and each child has a 50% risk of inheriting the defective X chromosome. The expansion of the trinucleotide sequence results in lowered FMRP levels. The premutation expansion results in a two- to eightfold increase in FMR1 mRNA levels, which lead to RNA toxicity accounting for phenotypic features; however, the FMRP levels often stay normal or become somewhat decreased, particularly after 120 CGG repeats.15,16 The full mutation, however, results in reduced or no FMRP production, which is found primarily in the brain and testes. In the brain, FMRP is crucial to synaptic development and refinement thus accounting for the psychiatric features of FXS, while its presence in the testes is needed for normal testicular size. The primary function of FMRP is to negatively regulate protein translation at the synapse,17 and it is estimated that FMRP binds up to 4%–8% of mRNA within the brain.18 This regulation is crucial in metabotropic glutamate receptors (mGluRs) 1 and 5, which become upregulated, leading to exaggerated long-term depression (LTD). This concept is the basis of the mGluR theory of FXS19 and the focus of many targeted treatments for FXS, such as the mGluR5 antagonists AFQ056 (mavoglurant; Novartis Pharmaceuticals, Basel, Switzerland) and RO4917523 (F Hoffmann-La Roche AG, Basel, Switzerland).
Additionally implicated in the dysregulation at the synapse is the GABA system, which is important in synaptic inhibition. An imbalance between the excitatory glutamatergic and the inhibitory GABAergic neurotransmission is proposed to cause the cognitive impartments, anxiety, and autism of FXS and several other neuropsychiatric and neurodevelopmental disorders.1,20,21 While two medications have been developed specifically for GABAergic modulation – arbaclofen (Seaside Therapeutics, Cambridge, MA, USA) and ganaxolone (Marinus Pharmaceuticals, Inc., New Haven, CT, USA) – this paper will also address GABAergic medications such as riluzole, gaboxadol, tiagabine, and vigabatrin. Before discussing the medications, a more in-depth focus on the GABA system and its dysregulation in FXS will follow.
Neurobiology
More than 3 decades of molecular research have led to a better understanding of the neurobiology of FXS and related disorders. FXS is caused by a dynamic mutation of more than 200 CGG trinucleotide repeats in the 5’ untranslated region on the FMR1 gene, which results in an absence of expression of FMRP. FMRP is a selective RNA-binding protein, found most abundantly in the CNS and testes, which regulates the expression of many mRNAs through inhibitory control at the synapse.22 The crucial role of FMRP in regulating the synthesis of synaptic proteins extends beyond the phenotype of FXS to other neurodevelopmental and neuropsychiatric disorders, in which FMRP may also be deficient.23
FMRP contains three main RNA-binding domains: two hnRNP K-homology domains and one arginine- and glycine-rich region of FMRP. In vitro FMRP is part of messenger ribonucleoparticles (structures that are involved in protein synthesis) and regulates dendritic transport of associated mRNAs.22 FMRP interacts with several cytoplasmic and nuclear proteins and has been found in granules containing translationally silent preinitiation complexes. In summary, FMRP regulates RNA transportation, stabilization, and translation, mainly at the synapse in neurons.
The activation of mGluR5 induces protein synthesis in the soma, axons, dendrites, and postsynaptic sites, as well as degradation and recycling of somatic and axonic proteins through the MAPK/ERK and mTOR pathways, which is required for LTD, a form of hippocampal synaptic plasticity that develops and consolidates long-term memories. In the Fmr1-knock out [KO] mice, LTD is significantly increased, and this leads to deficits in synaptic plasticity and weakening of synaptic connections.22 The mGluR theory, which suggests that psychiatric and neurological aspects of the syndrome are due to exaggerated downstream consequences of mGluR5 upregulation, was validated by genetic mouse studies in which rescue of several symptoms occurred when the Fmr1-KO mouse was crossed with the mGluR heterozygous mouse.24 The Fmr1-KO also shows hypothalamic excess of many synaptic proteins from increased protein translation rate and protein synthesis, whereas the GABA system is downregulated in the absence of FMRP, as described in the GABA neurobiology section.25
GABA neurobiology
The GABA system is the main inhibitory system in the brain. It works through two classes of GABA receptors: GABAA and GABAB. GABAA receptors are ligand-gated ion channels, whereas GABAB receptors are G protein-coupled receptors. GABAA receptors allow the flow of chloride ions across the membrane, which hyperpolarizes the neuron’s postsynaptic membrane and minimizes the effect of any coincident synaptic input. GABAB receptors hyperpolarize the neuron’s membrane by activating G-protein-coupled inwardly rectifying potassium channels.26 The GABA receptors are very diverse in their subunit composition and localization at the synapse and in regions of the brain; their effects are fast or slow persistent-tonic inhibition, depending on their localization and grade of stimulation.27
The GABA system is required for a balanced neuronal activation and network oscillations, direct flow of information, neural synchrony, and facilitating the movement of information in and between multiple brain areas involved in cognition.28–30 Since FXS is characterized by anxiety, hyperarousal, and epilepsy,31,32 recent studies33–36 have aimed to identify the defects in the inhibitory GABA system in the Fmr1-KO mouse.
GABAA
FMRP targets the mRNAs encoding eight different GABAA receptor subunits (α1, α3, α4, β1, β2, γ1, γ2, and δ), which were significantly reduced in the neocortex and cerebellum of Fmr1-KO mice, particularly the γ subunit, which represents extrasynaptic (perisynaptic) GABAA receptors.37,38 The Fmr1-KO mouse confirms a deficit in the production of GABAA receptors. Proteins required for GABA transport (eg, GABA transporter) and catabolism (eg, GABA transaminase [GABA-T], succinic semialdehyde [SSADH]) are also reduced.33,34 In addition, FMRP in the presynaptic side also regulates the expression of glutamic acid decarboxylase, which is the rate-limiting GABA-synthesis enzyme.33,35,37 Therefore, FMRP regulates the expression, metabolism, and catabolism of the GABAA receptors, and, in its absence, there is a decreased GABAergic input. The Fmr1-KO mouse exhibits reduced inhibitory postsynaptic currents in the amygdala35 and subicular neurons.36 Although FMRP inhibits the translation of many messages, it can also stimulate the translation of mRNAs, and the GABA system is downregulated in the absence of FMRP. Consequences of the reduced GABAA receptor expression in FXS likely include oversensitivity to sensory stimuli, seizures, and anxiety.
GABAB
GABAB is a metabotropic G protein-coupled receptor that regulates voltage-gated Ca2+ channels, G protein inwardly rectifying K+ channels, and adenylyl cyclase.26 Activation of the receptor triggers slow inhibitory postsynaptic currents, which reduce the neuronal excitability.26 Presynaptic GABAB receptors inhibit glutamate release, which further exacerbates the upregulation of the mGluR5 system; therefore, treatment with GABAB modulators has the potential to correct phenotypic deficits in FXS. When treated with a double-knockout of FMR1 and RGS4, male Fmr1-KO mice showed reduced susceptibility to audiogenic seizures.39 Since RGS4 is a regulator of G protein signaling and associated with GABAB receptors and inward-rectifying K+ channels, it has a therapeutic potential to regulate GABAB subunits in the treatment of FXS.39
Impacted regions of the brain
The amygdala, a component of the limbic system involved in emotional recognition and reaction, as well as fear processing, is one part of the brain considerably impacted by GABAergic dysregulation. Socioemotional impairments are prevalent in both premutation and full mutation patients, which warrants study of the amygdala as a basis for this impairment. Numerous studies have been performed assessing amygdala volume, activation in relation to gaze processing, and facial-emotional processing, which yield contradictory and inconsistent results due to varied protocols and populations (as summarized in Kim et al).40 Fear-specific activation of the amygdala was significantly reduced in patients with FXS compared to neurotypical controls, despite no differences in amygdala volume,40 which suggests biological impairment possibly at the synaptic level instead of altered brain size leading to emotional dysregulation. The amygdala’s phasic inhibitory postsynaptic currents, tonic inhibitory currents, reduced GABA release, and inhibitory synapses are considerably reduced in frequency and amplitude in the amygdala of Fmr1-KO mice.35 Due to the phenotypic socioemotional impairments seen in both the premutation and full mutation individuals alongside the biological impairments seen in the Fmr1-KO mouse, the amygdala is a point of study for gauging improvement with new treatments.
Further modifications in GABAergic activity are found in the cortex of the brain in FXS and, while monosynaptic GABAergic transmission is unaffected, there is a substantial deficit in local excitatory drive targeting fast-spiking inhibitory neurons in layer 4 of the somatosensory (barrel) cortex, which partially accounts for seizures, cognitive dysfunction, and sensory hypersensitivity of the FXS phenotype.41
A study assessing GABA-mediated cerebellar inhibition comparing healthy, asymptomatic women with and without the premutation found that women with the premutation show an absence of cerebellar inhibition over primary motor cortex as well as reduced GABAA-mediated intracortical and afferent inhibition.9 Even in asymptomatic carriers, there is still GABA dysregulation, which warrants the need for compounds with GABAergic modulation properties in premutation carriers. The available compounds are limited and mostly focus on antiepileptic properties; however, other mechanisms of action modified by such treatments may provide additional outlets for improving the phenotype in both carriers and individuals affected by the full mutation.
GABAergic treatments
After a better understanding of the neurobiology and neuropathogenesis of FXS, many compounds have been used as targeted treatments for FXS. Research has focused mainly on the development of mGluR5 antagonists, but better understanding of the GABAergic system in FXS has led to a new GABAergic approach and relevant targeted treatment. The GABA receptors in different regions of the brain are associated with some behavioral phenotypes in individuals with FXS. Particular attention has been devoted to correcting the amygdala-based symptoms. GABAergic agonists can be used for specific phenotypes including anxiety, autistic behavior, epilepsy, and cognitive impartment. GABA agonists have shown very limited efficacy in preliminary studies for these symptoms, but they have been well tolerated.42,43 The GABAergic treatments are a relatively new area and basic and translational research is limited in spite of their potential to treat FXS based on animal and small clinical studies.
Acamprosate
Acamprosate is a GABAA agonist approved for treatment of alcohol withdrawal. Excessive alcohol consumption over a long period of time changes the balance between the excitatory and inhibitory systems. Acamprosate helps people who have consumed large amounts of alcohol by stabilizing the excitatory/inhibitory balance in the brain, mainly by enhancing the function of GABAA receptors and, possibly, by its inhibitory effects on the mGluRs.44 Acamprosate does not prevent the withdrawal symptoms that people may experience when they stop drinking alcohol.
Clinically, a short report of three patients with FXS treated with acamprosate showed improvements in language and behavior,44 which led to other GABAergic targeted treatments and a second open-label 10-week trial of acamprosate (mean dose 1,054±422 mg/day) in 12 children and adolescents ages 6 to 17 years with FXS. The study showed improvements in social behavior and inattention/hyperactivity using multiple standard behavioral outcome measures. A Clinical Global Impressions-Improvement (CGI-I) scale score of “very much improved” or “much improved” was rated in nine of 12 (75%) subjects. No significant adverse effects were reported. Additionally, pre- and posttreatment blood biomarker analyses were performed by measuring brain-derived neurotrophic factor (BDNF) levels. A significant increase in the BDNF levels with treatment was described. However, treatment response did not correlate with change in the BDNF level.44
Ganaxolone
Ganaxolone (3a-hydroxy-3B-methyl analog of allopregnanolone) is a GABAA receptor agonist through allosteric modulation that has anticonvulsant, anxiolytic, and sedative effects.45 It is orally active and does not have hormonal effects. Neuroactive steroids like ganaxolone act most potently and effectively on GABAA receptors containing δ subunits. Ganaxolone is under development for treatment of seizure disorders and posttraumatic stress disorder. This pharmaceutical has been well tolerated, is safe in adults, children, and infants,46 and has been found to improve symptoms in humans and mouse models.46,47
Although many GABAA receptor subtypes are diminished in the Fmr1-KO mouse, there is evidence that extrasynaptic δ subunit containing GABAA receptors are especially affected.48 In the Fmr1-KO, ganaxolone has been shown to decrease audiogenic seizures.47 Similarly, studies in the dfmr mutant fly showed that GABAA agonists ameliorate the lethality phenotype of glutamate-containing food, neuropathology, excessive protein translation, and abnormal courtship behavior.49 The most frequently reported adverse events with ganaxolone in seizure studies are somnolence, convulsion, agitation, pharyngitis, otitis media, diarrhea, vomiting, cough, and pyrexia.50 A randomized, Phase II, double-blind, placebo-controlled crossover trial to investigate the efficacy of ganaxolone for the treatment of anxiety and attention deficits in children with FXS aged 6 to 17 years (http://www.ClinicalTrials.gov; NCT01725152)51 is currently under way. Ganaxolone should increase and normalize GABAA-mediated signaling – by boosting the signaling capacity of existing receptors – and improve behavior, particularly anxiety and attention.
Gaboxadol (THIP)
Tonic GABAA inhibition is associated with specific receptor subunits, particularly the relatively rare α4, α6, and δ subunits. The δ subunit-containing receptors are insensitive to benzodiazepine agonists,52,53 but highly sensitive to gaboxadol.54,55 Gaboxadol, the selective δ-GABAA super-agonist receptor, 4,5,6,7-tetrahydroisoxazolo(5,4-c)pyridin-3-ol (also known as THIP), has properties to generate tonic inhibition, namely activation by low concentrations of GABA through the activation of the extrasynaptic δ subunit-containing receptors.54,56
As previously mentioned, Fmr1-KO mouse studies have showed deficits in inhibitory transmission in the amygdala of the Fmr1-KO mouse, including reduction in the frequency and amplitude of phasic inhibitory postsynaptic currents and of tonic inhibitory currents, as well as a reduction in the number of inhibitory synapses and in neuronal hyperexcitability in principal neurons.35
A neuronal study has also shown significant increases in the action potential threshold in both wild-type and Fmr1-KO mice.57 Strikingly, the action potential threshold in Fmr1-KO mice in amygdala slices was restored to wild-type levels by THIP application. Thus, the electrophysiological abnormalities of neuronal hyperexcitability in the Fmr1-KO amygdala can be dramatically rescued by augmenting tonic inhibitory tone.35 A behavioral study in Fmr1-KO mice have shown that THIP significantly attenuated hyperactivity and reduced prepulse inhibition in a volume-dependent manner. However, THIP did not reverse the deficits in cued fear or startle response.58 Current studies show that enhancing GABAergic transmission can correct specific behavioral phenotypes of the Fmr1-KO mouse, further supporting focus on the GABAergic system and, specifically, tonic inhibition, which might be important for correcting or ameliorating specific behaviors in FXS.47,49,58
Gaboxadol reached a Phase III trial before its cessation because of side effects, such as hallucination and disorientation. Due to these safety concerns and lack of efficacy, work on the drug was discontinued in 2007; however, recent studies in animal models of ASD have shown that gaboxadol is effective in rescuing neurophysiological and behavioral deficits.58,59 Further studies in fragile X animal models are necessary to provide cumulative evidence in the efficacy and safety of gaboxadol. Currently, there are no studies in individuals with FXS.
Vigabatrin
Vigabatrin is an antiepileptic and analog of GABA (although not an agonist) that inhibits the catabolism of GABA by irreversibly inhibiting GABA transaminase. It has been found that the half-life of biologic activity is far longer than the elimination half-life,60,61 and there is no range of target concentrations because there was no difference between the serum concentration levels of responders and those of nonresponders.61 In addition, the duration of action is more a function of the GABA transaminase resynthesis rate.62 Vigabatrin has been approved by the US Food and Drug Administration (FDA) for use in patients with refractory complex partial seizures, but the rentinal toxicity of the medication limits it’s use to those who have not responded to other treatments.63 However, it is recommended when the benefits outweigh the side effects in individuals with intractable seizures. There are no studies in FXS animal models or clinical trials in individuals with FXS.
Arbaclofen
One compound shown to influence GABA regulation in the Fmr1-KO mouse is arbaclofen, a GABAB agonist developed by Seaside Therapeutics. Arbaclofen is a receptor agonist and active enantiomer of racemic baclofen, which presynaptically blocks glutamate release, thereby decreasing the overactivation of the glutamatergic pathways. In Fmr1-KO mice, arbaclofen corrected elevated protein synthesis in the hippocampus; reduced elevated AMPA (α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor internalization to wild-type values; decreased mRNA translation in the cortex; and corrected the increased spine density prevalent in the mouse phenotype.64
In pediatric, adolescent, and adult patients with FXS, arbaclofen was administered in a randomized, double-blind, placebo-controlled crossover study. The primary outcome measure, the Aberrant Behavior Checklist-Community Edition, a 58-item behavioral questionnaire, did not show reduction in any of the subscales compared to placebo42 and, due to resource limitations, Seaside Therapeutics had to terminate its arbaclofen program.65 However, a post hoc analysis saw a full study improvement on the Aberrant Behavior Checklist Social Avoidance subscale, which was an algorithm developed specifically to assess behavioral function in patients with FXS.66 In addition, a subgroup of 27 patients with more severe social impairment treated with arbaclofen showed improvement on all global measures, as well as on the Vineland Socialization subscale, which is a semi-structured parent interview that assesses adaptive behavior. There were also trends of improvement seen on parent-nominated problem behaviors in the visual analog scale as well as on multiple global measures.42 This demonstrates the need to use biological subtyping, particularly in a heterogeneous group of patients, such as those with ASD. For the subgroup of patients with FXS who did very well with arbaclofen, it became a problem to obtain the drug once the company folded, and the withdrawal from arbaclofen was very difficult for many families. Overall, arbaclofen was well tolerated, with sedation and headache being the most commonly reported side effects.42
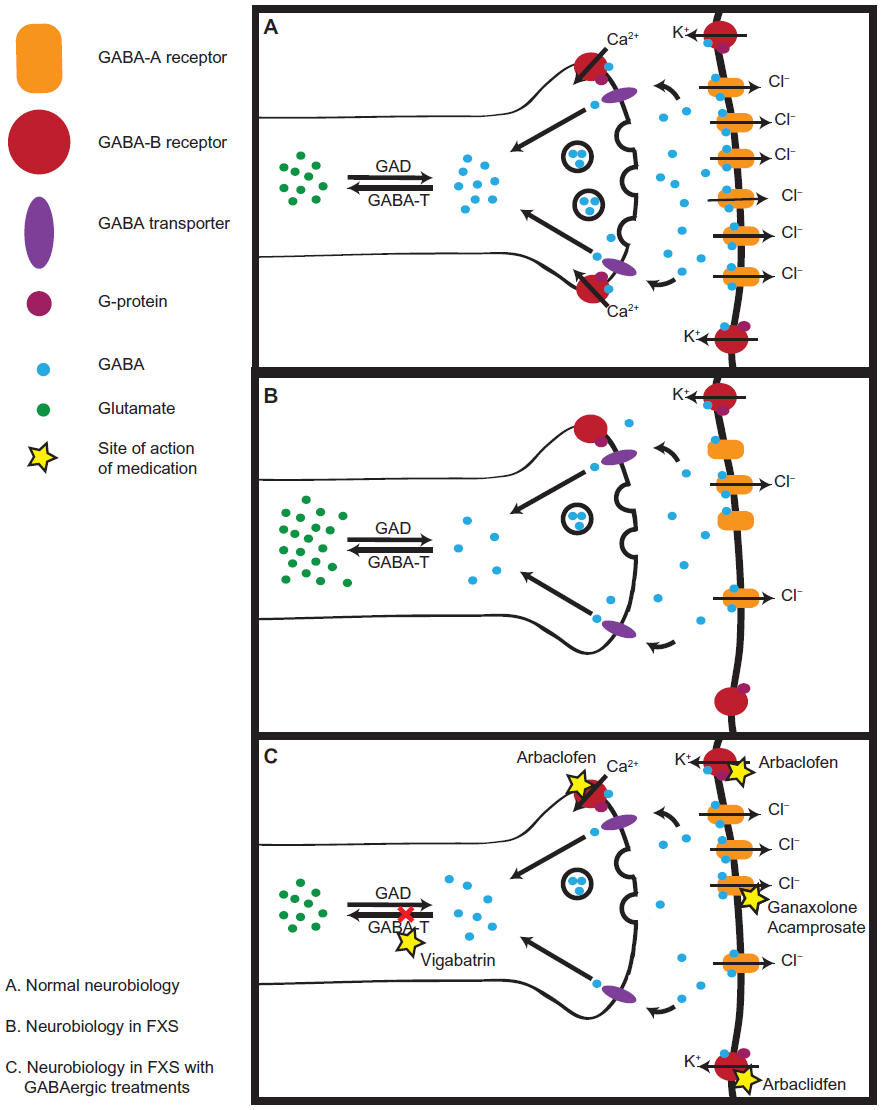 | Figure 1 Neurobiology in FXS. |
Riluzole
Riluzole is a medication approved for the treatment of amyotrophic lateral sclerosis, which blocks voltage-gated Na+ channels and selectively depresses glutamate over GABA release and which, at higher concentrations, can potently increase postsynaptic GABAA response in hippocampal neurons.67 Due to the GABAergic dysregulation and epilepsy shown in patients with FXS, it was proposed that riluzole could act as a beneficial treatment for FXS. An open-label study of riluzole in patients with FXS showed a clinical response in only one of six patients, with significant improvement as measured on the ADHD Rating Scale-IV.43 Riluzole administration was also associated with significant correction of ERK activation time in all subjects. Overall, the medication was well tolerated and showed non-clinically significant increases in liver function tests,43 a notable side effect of the medication.
Tiagabine
Tiagabine, a nipecotic acid derivative, is another antiepileptic that reduces neuronal and astrocytic uptake of GABA.68 Sound-induced convulsions in DBA/2 mice – which, notably, had no sedation or motor debilitation70 – were corrected when tiagabine was administered;69 however, there have not been any studies assessing the efficacy of tiagabine in treating FXS. While it may be used to control epilepsy in patients with FXS, other antiepileptic medications seem to have more potent treatment responses.71
Conclusion
Numerous GABAergic compounds are potential treatments for FXS; however, considerably more basic and translational research is needed. The current studies are mostly open-label treatment options with small sample sizes of patients in a wide age range. Larger, double-blind, placebo-controlled trials are needed to assess the efficacy of these treatments against the placebo in order to better establish the treatment profile of each medication. The largest double-blind, placebo-controlled study using a GABAergic compound in FXS used arbaclofen. Despite trends in a positive treatment response on secondary outcome measures, Seaside Therapeutics could not continue with additional studies focusing on the secondary outcome measures due to limited resources. This outcome brings to light the difficulty of establishing efficacy of targeted treatments in this population.42,72
A number of new treatments are also being studied in clinical trials after promising efficacy studies in the animal models of FXS.73 These include lovastatin, which lowers ERK phosphorylation in the mTOR pathway, and an insulin-like growth factor 1 analog made by Neuren (Neuren Pharmaceuticals Limited, Camberwell, VIC, Australia). Other treatment trials are in the pipeline but have yet to come to clinical trials.74
There are a number of barriers to new treatments, including the time and expense of carrying out the toxicity studies in animals and humans and subsequent multicenter human trials to demonstrate efficacy. Although FXS is a single-gene disorder, there is significant heterogeneity in clinical involvement and response to treatment. For many trials, approximately 30% respond well, but this may not be adequate to demonstrate overall efficacy, which prohibits FDA approval for marketing. The lack of biomarkers that would predict efficacy is greatly needed so that a “likely to respond subgroup” could be identified.
For example, the AFQ056 compound by Novartis appears to be most effective for those who are fully methylated, but additional biomarkers are needed.75 In addition, outcome measures that are quantitative and relate to central nervous system function or molecular changes and do not depend on questionnaires from the family would be useful to decreasing the placebo effect. Event-related potential paradigms of cognitive processing would also be useful, and we have seen a positive effect using the oddball paradigm and habituation task in children treated with minocycline in a controlled trial.76
The placebo effect is high, owing to the overwhelming need for effective interventions, which is further impacted by the difficulty of obtaining objective outcome measures that accurately assess a behavioral treatment response. A majority of the improvement measures were based on parent or caregiver response,42,66 which is subjective. More objective outcome measures are needed, not only for GABAergic medications, but for all clinical trials that focus on neurodevelopmental indications. Biomarkers relating specifically to GABA upregulation in patients who did improve on the arbaclofen medication would be a vital tool for establishing efficacy against placebo.
Combined clinical trials using two drugs or a drug with an intervention program may show positive outcomes for FDA approval. A combined clinical trial of lovastatin with language intervention for individuals with FXS has been supported by the National Institute of Child Health and Human Development. GABA downregulation and mGluR5 upregulation are thought to be the major problems in FXS, so interventions in both of these pathways will be critical for an overall treatment program. Ganaxolone is currently undergoing a clinical trial in FXS and is considered to have the best potential as a GABA agent because it specifically targets the GABAA pathway, which is most dysregulated in FXS. If efficacy in FXS is demonstrated and there is FDA approval for an FXS indication, then it will be studied in ASD and related disorders. Research needs to start focusing on multifaceted treatment options that combine multiple pharmacological agents and/or behavioral interventions due to the complex nature of the disease, but this may be difficult prior to FDA approval for each compound. Studies looking at behavioral and pharmacological interventions are planned for other targeted treatment options in FXS, including the mGluR5 antagonists and lovastatin. No studies are currently planned for multiple GABAergic medications or GABAergic medications with behavioral interventions, which would be highly useful and may provide more outcome measures with which to gauge treatment response.
In addition to difficulties in outcome measures, it should also be noted that reversing the behavioral and intellectual abilities in FXS is more difficult in adults because they are not typically in a learning program and because the neurobiological abnormalities may be less reversible over time. More intensive learning programs are required, although the improvements seen in the adult mouse with FXS after treatment with a long-acting mGluR5 antagonist were remarkable77 and gave hope to many of the families that have adult offspring with FXS. Owing to the process involved in obtaining FDA approval for medications, companies must establish safety and efficacy in the adult population prior to moving into younger age groups; as such, the low efficacy of adult studies prevents the medication from moving to the younger age groups, who are more likely to see more robust treatment responses.
It is important to remember that synaptic connections that are strengthened by either a GABA agonist or an mGluR5 antagonist also require an intensive learning environment to strengthen these connections. Although most school-aged children with FXS are receiving special education support in addition to speech and language therapy and occupational therapy, their programs can usually be enhanced by the use of digital learning programs that can be accessed on a tablet device. There are a variety of software applications that have been developed for individuals with ASD, and these programs are likely to also be helpful for those with FXS.78
There are many currently used medications that are helpful in FXS that will likely continue to be helpful even after GABA agonists and mGluR5 antagonists are more widely used clinically. These medications include sertraline, a selective serotonin reuptake inhibitor, that can be started at a young age and has been helpful in improving the language trajectory of toddlers with FXS.79 Another targeted treatment that can be effective at an early age is minocycline, which is an antibiotic that lowers the level of matrix metalloproteinase 9 and which is elevated in FXS.80,81 High levels of matrix metalloproteinase 9 interfere with the development of synaptic connections, and a controlled trial of minocycline in children between the ages of 3 and 17 years (doses ranging from 25 to 100 mg per day) recently demonstrated efficacy on the CGI-I in improving overall behavior and on the visual analog scale for improving mood and anxiety.80 Side effects of minocycline include graying of the permanent teeth if given before 8 years of age, and graying or darkening of tissue such as in the gums and nail beds at any age. On rare occasions, minocycline can cause a lupus-like syndrome with a rash or swollen joints or pseudotumor cerebri leading to a severe headache; if these problems occur, minocycline should be discontinued immediately.80 Once children with FXS reach 5 years of age, they usually have a positive response to a stimulant for their ADHD symptoms; for those who do not respond to a stimulant, then an alpha agonist such as guanfacine can be very helpful.32 Lastly, melatonin has also been helpful for sleep disturbances in FXS, particularly in young children.82 Overall, a GABA agonist will add significantly to the treatment regimen in children and adults with FXS, although we do not yet know if improvements will be seen only in behavior or if cognitive deficits will also improve with long-term use.
Acknowledgments
Funding for this chapter includes NICHD grant HD 036071, the Autism Research Training Program (MH073124), Department of Defense grant PR101054, support from the Health and Human Services Administration on Developmental Disabilities (grant 90DD05969), and the National Center for Advancing Translational Research (UL1 TR000002).
Disclosure
Dr Hagerman has received funding from Novartis Pharmaceuticals, F Hoffmann-La Roche, Seaside Therapeutics and Neuren Pharmaceuticals Limited to carry out treatment trials in fragile X syndrome and autism. She has also consulted with F Hoffmann-La Roche/Genentech, and Novartis Pharmaceuticals regarding treatment studies in fragile X syndrome. All authors have participated in clinical trials of ganaxolone and arbaclofen for fragile X syndrome.
References
Belmonte MK, Bourgeron T. Fragile X syndrome and autism at the intersection of genetic and neural networks. Nat Neurosci. 2006;9(10):1221–1225. | |
Bretherick KL, Fluker MR, Robinson WP. FMR1 repeat sizes in the gray zone and high end of the normal range are associated with premature ovarian failure. Hum Genet. 2005;117(4):376–382. | |
Hagerman R, Hagerman P. Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol. 2013;12(8):786–798. | |
Bourgeois JA, Seritan AL, Casillas EM, et al. Lifetime prevalence of mood and anxiety disorders in fragile X premutation carriers. J Clin Psychiatry. 2011;72(2):175–182. | |
Winarni TI, Chonchaiya W, Sumekar TA, et al. Immune-mediated disorders among women carriers of fragile X premutation alleles. Am J Med Genet A. 2012;158a(10):2473–2481. | |
Au J, Akins RS, Berkowitz-Sutherland L, et al. Prevalence and risk of migraine headaches in adult fragile X premutation carriers. Clin Genet. 2013;84(6):546–551. | |
Hamlin AA, Sukharev D, Campos L, et al. Hypertension in FMR1 premutation males with and without fragile X-associated tremor/ataxia syndrome (FXTAS). Am J Med Genet A. 2012;158A(6):1304–1309. | |
Sullivan SD, Welt C, Sherman S. FMR1 and the continuum of primary ovarian insufficiency. Semin Reprod Med. 2011;29(04):299–307. | |
Conde V, Palomar FJ, Lama MJ, et al. Abnormal GABA-mediated and cerebellar inhibition in women with the fragile X premutation. J Neurophysiol. 2013;109(5):1315–1322. | |
Farzin F, Perry H, Hessl D, et al. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006;27(Suppl 2):S137–S144. | |
Leigh MJS, Hagerman RJ, Hessl D. Fragile X Syndrome. In: Hansen RL, Rogers SJ, editors. Autism and Other Neurodevelopmental Disorders. Arlington: American Psychiatric Publishing; 2013:57–76. | |
Coffee B, Keith K, Albizua I, et al. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet. 2009;85(4):503–514. | |
Crawford DC, Acuña JM, Sherman SL. FMR1 and the fragile X syndrome: human genome epidemiology review. Genet Med. 2001;3(5):359–371. | |
Sorensen PL, Gane LW, Yarborough M, Hagerman RJ, Tassone F. Newborn screening and cascade testing for FMR1 mutations. Am J Med Genet A. 2013;161(1):59–69. | |
Allen EG, He W, Yadav-Shah M, Sherman SL. A study of the distributional characteristics of FMR1 transcript levels in 238 individuals. Hum Genet. 2004;114(5):439–447. | |
Ludwig AL, Espinal GM, Pretto DI. CNS expression of murine fragile X protein (FMRP) as a function of CGG-repeat size. Hum Mol Genet. Epub February 11, 2014. | |
Bhakar AL, Dölen G, Bear MF. The pathophysiology of fragile X (and what it teaches us about synapses). Annu Rev Neurosci. 2012;35:417–443. | |
Brown V, Jin P, Ceman S, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107(4):477–487. | |
Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27(7):370–377. | |
Moy SS, Nadler JJ. Advances in behavioral genetics: mouse models of autism. Mol Psychiatry. 2008;13(1):4–26. | |
Rubenstein J, Merzenich M. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003; 2(5):255–267. | |
Bagni C, Tassone F, Neri G, Hagerman R. Fragile X syndrome: causes, diagnosis, mechanisms, and therapeutics. J Clin Invest. 2012;122(12):4314–4322. | |
Fatemi S, Folsom T, Rooney R, Thuras P. mRNA and protein expression for novel GABAA receptors θ and ρ2 are altered in schizophrenia and mood disorders; relevance to FMRP-mGluR5 signaling pathway. Transl Psychiatry. 2013;3(6):e271. | |
Dölen G, Carpenter RL, Ocain TD, Bear MF. Mechanism-based approaches to treating fragile X. Pharmacol Ther. 2010;127(1):78–93. | |
Berry-Kravis E, Knox A, Hervey C. Targeted treatments for fragile X syndrome. J Neurodev Disord. 2011;3(3):193–210. | |
Padgett CL, Slesinger PA. GABAB receptor coupling to G-proteins and ion channels. Adv Pharmacol. 2010;58:123–147. | |
Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci. 2005;6(3):215–229. | |
Cardin JA, Carlén M, Meletis K, et al. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009; 459(7247):663–667. | |
Wang LW, Berry-Kravis E, Hagerman RJ. Fragile X: leading the way for targeted treatments in autism. Neurotherapeutics. 2010;7(3):264–274. | |
Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459(7247):698–702. | |
Berry-Kravis E, Grossman AW, Crnic LS, Greenough WT. Understanding fragile X syndrome. Current Paediatrics. 2002;12(4):316–324. | |
Hagerman RJ, Berry-Kravis E, Kaufmann WE, et al. Advances in the treatment of fragile X syndrome. Pediatrics. 2009;123(1):378–390. | |
Adusei DC, Pacey LK, Chen D, Hampson DR. Early developmental alterations in GABAergic protein expression in fragile X knockout mice. Neuropharmacology. 2010;59(3):167–171. | |
Liao CW, Lien C. Estimating intracellular Ca2+ concentrations and buffering in a dendritic inhibitory hippocampal interneuron. Neuroscience. 2009;164(4):1701–1711. | |
Olmos-Serrano JL, Paluszkiewicz SM, Martin BS, Kaufmann WE, Corbin JG, Huntsman MM. Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J Neurosci. 2010;30(29):9929–9938. | |
Curia G, Papouin T, Séguéla P, Avoli M. Downregulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome. Cereb Cortex. 2009;19(7):1515–1520. | |
D’Hulst C, Heulens I, Brouwer JR, et al. Expression of the GABAergic system in animal models for fragile X syndrome and fragile X associated tremor/ataxia syndrome (FXTAS). Brain Res. 2009;1253:176–183. | |
Gantois I, Vandesompele J, Speleman F, et al. Expression profiling suggests underexpression of the GABAA receptor subunit delta in the fragile X knockout mouse model. Neurobiol Dis. 2006;21(2):346–357. | |
Pacey LKK, Heximer SP, Hampson DR. Increased GABA(B) receptor-mediated signaling reduces the susceptibility of fragile x knockout mice to audiogenic seizures. Mol Pharmacol. 2009;76(1):18–24. | |
Kim SY, Burris J, Bassal F, et al. Fear-specific amygdala function in children and adolescents on the fragile x spectrum: a dosage response of the FMR1 gene. Cereb.Cortex. 2014;24(3):600–613. | |
Gibson JR, Bartley AF, Hays SA, Huber KM. Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J Neurophysiol. 2008;100(5):2615–2626. | |
Berry-Kravis EM, Hessl D, Rathmell B, et al. Effects of STX209 (arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Sci Transl Med. 2012;4(152):152ra127. | |
Erickson CA, Weng N, Weiler IJ, et al. Open-label riluzole in fragile X syndrome. Brain Res. 2011;1380:264–270. | |
Erickson CA, Wink LA, Ray B, et al. Impact of acamprosate on behavior and brain-derived neurotrophic factor: an open-label study in youth with fragile X syndrome. Psychopharmacology (Berl). 2013; 228(1):75–84. | |
Carter RB, Wood PL, Wieland S, et al. Characterization of the anticonvulsant properties of ganaxolone (CCD 1042; 3β-hydroxy-3β-methyl-5α-pregnan-20-one), a selective, high-affinity, steroid modulator of the γ-aminobutyric acidA receptor. J Pharmacol Exp Ther. 1997;280(3):1284–1295. | |
Kerrigan JF, Shields WD, Nelson TY, et al. Ganaxolone for treating intractable infantile spasms: a multicenter, open-label, add-on trial. Epilepsy Res. 2000;42(2):133–139. | |
Heulens I, D’Hulst C, Van Dam D, De Deyn PP, Kooy RF. Pharmacological treatment of fragile X syndrome with GABAergic drugs in a knockout mouse model. Behav Brain Res. 2012;229(1):244–249. | |
D’Hulst C, De Geest N, Reeve SP, et al. Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 2006;1121(1):238–245. | |
Chang S, Bray SM, Li Z, et al. Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat Chem Biol. 2008;4(4):256–263. | |
Pieribone VA, Tsai J, Soufflet C, et al. Clinical evaluation of ganaxolone in pediatric and adolescent patients with refractory epilepsy. Epilepsia. 2007;48(10):1870–1874. | |
Marinus Pharmaceuticals. Ganaxolone Treatment in Children With Fragile X Syndrome. Available from: http://clinicaltrials.gov/ct2/show/NCT01725152. NLM identifier: NCT01725152. Accessed March 24, 2014. | |
Nusser Z, Mody I. Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J Neurophysiol. 2002;87(5):2624–2628. | |
Cope DW, Hughes SW, Crunelli V. GABAA receptor-mediated tonic inhibition in thalamic neurons. J Neurosci. 2005;25(50):11553–11563. | |
Brown N, Kerby J, Bonnert T, Whiting P, Wafford K. Pharmacological characterization of a novel cell line expressing human alpha(4)beta(3)delta GABA(A) receptors. Br J Pharmacol. 2002;136(7):965–974. | |
Wohlfarth KM, Bianchi MT, Macdonald RL. Enhanced neurosteroid potentiation of ternary GABA(A) receptors containing the delta subunit. J Neurosci. 2002;22(5):1541–1549. | |
Haas KF, Macdonald RL. GABAA receptor subunit gamma2 and delta subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J Physiol. 1999;514(Pt 1):27–45. | |
Deng PY, Rotman Z, Blundon JA, et al. FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron. 2013;77(4):696–711. | |
Olmos-Serrano JL, Corbin JG, Burns MP. The GABA(A) receptor agonist THIP ameliorates specific behavioral deficits in the mouse model of fragile X syndrome. Dev Neurosci. 2011;33(5):395–403. | |
Egawa K, Kitagawa K, Inoue K, et al. Decreased tonic inhibition in cerebellar granule cells causes motor dysfunction in a mouse model of angelman syndrome. Sci Transl Med. 2012;4(163):163ra157. | |
Browne TR. Pharmacokinetics of antiepileptic drugs. Neurology. 1998;51(5 Suppl 4):S2–S7. | |
Browne T, Szabo G. New pharmacokinetic methods for the study of antiepileptic medications of the 1990s. Epilepsia. 1990;32:S66–S73. | |
Lindberger M, Luhr O, Johannessen SI, Larsson S, Tomson T. Serum concentrations and effects of gabapentin and vigabatrin: observations from a dose titration study. Ther Drug Monit. 2003;25(4):457–462. | |
Podboraczynska-Jodkoi K, Lubinski W, Hampel-Osipowicz E. [Retinal dysfunction in patients treated with vigabatrin]. Klin. Oczna. 2007;109(1–3):85–88. Polish. | |
Henderson C, Wijetunge L, Kinoshita MN, et al. Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci Transl Med. 2012; 4(152):152ra128. | |
Thomson Reuters. Key trial of Seaside autism drug fails to show benefit [press release]. New York, NY: Thomson Reuters; 2013 [May 1]. Available from: http://www.reuters.com/article/2013/05/01/us-autism-drug-idUSBRE9400NT20130501. Accessed May 05, 2014 | |
Sansone SM, Widaman KF, Hall SS, et al. Psychometric study of the Aberrant Behavior Checklist in Fragile X Syndrome and implications for targeted treatment. J Autism Dev Disord. 2012;42(7):1377–1392. | |
He Y, Benz A, Fu T, et al. Neuroprotective agent riluzole potentiates postsynaptic GABA(A) receptor function. Neuropharmacology. 2002;42(2):199–209. | |
Leach JP, Brodie MJ. New antiepileptic drugs – an explosion of activity. Seizure. 1995;4(1):5–17. | |
Suzdak PD, Jansen JA. A review of the preclinical pharmacology of tiagabine: a potent and selective anticonvulsant GABA uptake inhibitor. Epilepsia. 1995;36(6):612–626. | |
Nielsen EB, Suzdak PD, Andersen KE, Knutsen LJ, Sonnewald U, Braestrup C. Characterization of tiagabine (NO-328), a new potent and selective GABA uptake inhibitor. Eur J Pharmacol. 1991; 196(3):257–266. | |
Berry–Kravis E. Epilepsy in fragile X syndrome. Dev Med Child Neurol. 2002;44(11):724–728. | |
National Fragile X Foundation. Seaside Therapeutics Announces End of Arbaclofen (STX209) Extension Study [press release]. Walnut Creek, CA: National Fragile X Foundation; 2013 [May 16]. Available from: http://www.fragilex.org/2013/research/news-reports-and-commentaries/seaside-therapeutics-announces-end-of-arbaclofen-stx209-extension-study/. Accessed May 06, 2014. | |
Berry-Kravis E. Mechanism-based treatments in neurodevelopmental disorders: fragile X syndrome. Pediatr Neurol. 2014;50(4):297–302. | |
Braat S, Kooy RF. Fragile X syndrome neurobiology translates into rational therapy. Drug Discovery Today. 2014;19(4):510–519. | |
Jacquemont S, Curie A, des Portes V, et al. Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci Transl Med. 2011;3(64):64ra1. | |
Schneider A, Leigh MJ, Adams P, et al. Electrocortical changes associated with minocycline treatment in fragile X syndrome. J Psychopharmacol. 2013;27(10):956–963. | |
Michalon A, Sidorov M, Ballard TM, et al. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron. 2012;74(1):49–56. | |
Chen W. Multitouch tabletop technology for people with autism spectrum disorder: a review of the literature. Procedia Comput Sci. 2012;14:198–207. | |
Winarni TI, Schneider A, Borodyanskara M, Hagerman RJ. Early intervention combined with targeted treatment promotes cognitive and behavioral improvements in young children with fragile x syndrome. Case Rep Genet. 2012;2012:280813. | |
Leigh MJ, Nguyen DV, Mu Y, et al. A randomized double-blind, placebo-controlled trial of minocycline in children and adolescents with fragile x syndrome. J Dev Behav Pediatr. 2013;34(3):147–155. | |
Dziembowska M, Pretto DI, Janusz A, et al. High MMP-9 activity levels in fragile X syndrome are lowered by minocycline. Am J Med Genet A. 2013;161A(8):1897–1903. | |
Wirojanan J, Jacquemont S, Diaz R, et al. The efficacy of melatonin for sleep problems in children with autism, fragile X syndrome, or autism and fragile X syndrome. J Clin Sleep Med. 2009;5(2):145–150. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.