")
Back to Journals » Journal of Inflammation Research » Volume 14
Modulation of Sirt1-mTORC1 Pathway in Microglia Attenuates Retinal Ganglion Cell Loss After Optic Nerve Injury
Authors Mou Q, Yao K , Ye M, Zhao B, Hu Y, Lou X, Li H, Zhang H, Zhao Y
Received 16 September 2021
Accepted for publication 1 December 2021
Published 14 December 2021 Volume 2021:14 Pages 6857—6869
DOI https://doi.org/10.2147/JIR.S338815
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Qianxue Mou,1 Ke Yao,1 Meng Ye,1 Bowen Zhao,1 Yuanyuan Hu,1 Xiaotong Lou,1 Huixia Li,2 Hong Zhang,1 Yin Zhao1
1Department of Ophthalmology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430030, People’s Republic of China; 2Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Xi’an Jiaotong University Health Science Center, Xi’an, Shaanxi, People’s Republic of China
Correspondence: Hong Zhang
Department of Ophthalmology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430030, People’s Republic of China
Email [email protected]
Correspondence: Yin Zhao
Department of Ophthalmology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430030, People’s Republic of China
Email [email protected]
Purpose: Optic nerve injury (ONI) causes neuroinflammation and neurodegeneration leading to visual deficits. The response of microglia has emerged as an impactful component of etiology in neurodegeneration. This study aimed to investigate the effect of SIRT1-mTORC1 signaling pathway in microglia regulation after ONI.
Methods: Cx3Cr1-CreERT2/RaptorF/F and Cx3Cr1-CreERT2/Sirt1F/F mice were used to delete Raptor and Sirt1 in microglia, respectively. Optic nerve crush (ONC) model was established to mimic ONI. PLX5622, a highly specific inhibitor of the colony-stimulating factor 1 receptor (CSF1R), is used to eliminate microglia in optic nerve. Ionized calcium binding adaptor molecule 1 (Iba1) immunostaining was used to detect microglial activation. Retinal ganglion cells (RGCs) were quantified by Nissl staining and retinal whole-mount immunostaining with RNA-binding protein with multiple splicing (RBPMS). Axonal damage was valued by transmission electron microscopy (TEM).
Results: Microglial activation emerged on day 3 post ONC and was earlier than RGCs loss which occurred at day 5 after injury. Depleting microglia with PLX5622 could attenuate the loss of RGCs and axon damage after ONC. Gain- and loss-of-function studies revealed that SIRT1 determined the activation of microglia in optic nerve. In addition, microglia-specific deletion of Raptor resulted in decreased microglial activation. Interestingly, activating mTORC1 with CCT007093 could reverse the function of SIRT1 in regulating the process of microglial activation mediated RGCs loss.
Conclusion: Our study reveals a potential novel mechanism of SIRT1-mTORC1 pathway in microglia regulation, and indicates a therapeutic potential for the protection of RGCs in ONI.
Keywords: microglia, retinal ganglion cells, optic nerve injury, SIRT1, mTORC1
Introduction
Diverse pathological states cause degeneration of axons leading to irreversible vision loss, including ocular hypertension, trauma and optic neuritis.1,2 In these relative diseases, focal axon injury triggers a propagating axon loss and eventually neuronal death.3 Therefore, axonal injury is thought to be the critical early event for RGCs loss. Multiple glial cell types participate in the process of axonal degeneration after optic nerve injury (ONI), including oligodendroglia, astroglia and microglia.4–6 The microglia form a population of resident macrophages within the retina and optic nerve.6,7 In healthy retina, microglia are of vital importance in maintain the normal structure and functioning of the retina.8 Once activated by noxious insults, microglia can release inflammatory cytokines, such as TNF-α, IL-1β and reactive oxygen species, which could damage RGCs and promote neurodegeneration, and inappropriate phagocytosis by microglia can further accelerate RGCs loss.9,10 A previous study has shown that microglial activation can aggravate trauma-induced RGCs degeneration, which leads to RGCs loss.11 Thus, regulation of microglial activation enables microglia to function better as guardians during pathological processes.12 Besides immune activity, the microglia also participate in the development and maintenance of neural networks, as well as in neurovascular unit homeostasis.7,13,14 Noteworthy, genetic ablation and/or pharmacological blockade of microglia shows neuroprotective effect in neurodegenerative diseases.15,16
Sirtuin 1 (SIRT1), a nicotinamide adenine dinucleotide (NAD)-dependent deacetylase, deacetylates transcription factors, signaling molecules, and histones.17,18 Resveratrol enhances SIRT1 activity via promoting NADH oxidation to NAD+ and, possibly, by boosting SIRT1 affinity for NAD+ as well as acetylated substrate.18 Overexpression of SIRT1 in microglia protects against Aβ toxicity through the NF-κB pathway.19 Further study has confirmed that microglial Sirt1 deficiency induces aging- or tau-mediated memory deficits in mice.20 Mammalian target of rapamycin complex 1 (mTORC1) consists of mTOR, mammalian lethal with SEC13 protein 8 (mLST8), regulatory-associated protein of mTOR (raptor) and DEP domain-containing protein 6 (DEPTOR).21 Raptor is a specific member of mTORC1 and implicates in the recruitment process of mTORC1 to lysosome, where it encounters Rheb, a potent mTORC1 activator.22 mTORC1 was previously identified as a target of SIRT1. Previous study reported that SIRT1 inhibits S6K1 acetylation, and therefore activates mTORC1 signaling.23 Moreover, hepatocyte-specific deficiency of Sirt1 enhances mTORC1 signaling and exacerbates alcoholic fatty liver, inflammation, and liver damage in mice.24 Recent study reported that SIRT1 triggers mTORC1 signaling in bone development.25 However, the potential role of SIRT1-mTORC1 pathway in microglia and neurodegeneration disease remains unclear.
In this study, we aim to elucidate the role of SIRT1-mTORC1 pathway in microglia, especially in ONI induced neurodegeneration by using tamoxifen-inducible, microglia-specific Sirt1/Raptor knockout mice. Our observations suggest that SIRT1 determines the activation of microglia by SIRT1-mTORC1 signaling pathway.
Materials and Methods
Reagents and Antibodies
Nissl staining solution (G1036) was purchased from Servicebio (Wuhan, China). Corn oil was purchased from Maclin Biochemical (Shanghai, China). CCT007093 was purchased from MedChemExpress (New Jersey, USA). Resveratrol was purchased from Aladdin (Shanghai, China). Optimal cutting temperature compound (OCT) was purchased from Sakura Finetek (Torrance, USA). Paraformaldehyde (PFA) was purchased from Biosharp (Hefei, China). Antibodies: Rabbit anti-Iba1 (ab178846) and Rabbit anti RBPMS (ab152101) were purchased from Abcam (Cambridge, UK). Mouse anti IL-1β (#12242) were purchased from Cell Signaling Technology (Boston, USA). Alexa Fluor 594–conjugated goat anti-rabbit IgG (H+L) and Alexa Fluor 488–conjugated goat anti-mouse IgG (H+L) were purchased from Thermo Fisher Scientific (Waltham, USA).
Animals
All animal feeding and experiments were authorized by the Institutional Animal Research Committee of Tongji Medical Center and in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD, USA). The number of animal ethical approval was 300. Cx3cr1-CreERT2, RaptorF/F and Sirt1F/F mice were obtained from Jackson Labs. To specifically delete Raptor in microglia, Raptor F/F mice were crossed with Cx3cr1-CreERT2 mice to obtain Cx3cr1-CreERT2/ Raptor F/F mice. Similarly, Sirt1 F/F were crossed with Cx3cr1-CreERT2 mice to obtain Cx3cr1-CreERT2/ Sirt1 F/F mice whose floxed exon 4 of Sirt1 was deleted in microglia. Cx3cr1-CreERT2/ Raptor F/F mice and Cx3cr1-CreERT2/ Sirt1 F/F mice were under 28 days of tamoxifen pretreatment before experiments.
Optic Nerve Crush (ONC) Mice Model
Mice were anesthetized with sodium pentobarbital (20 mg/kg) and tissues surrounding the optic nerve were dissected carefully in order to expose the optic nerve. The crush site is around the optic nerve 5 mm behind the globe and crush time is 3s. N5 self-closing forceps were used to perform ONC. Then, the mice recovered at 37°C on a warming pad before returning to cages and time points of sacrificing were indicated in the text.
Grouping
The sample size was 8 per group. Animals were divided into the following groups:
Part 1: Group 0 d (post ONC 0 day); Group 3 d (post ONC 3 day); Group 5 d (post ONC 5 day); Group 7 d (post ONC 7 day); Group 14 d (post ONC 14 day).
Part 2: Group Vehicle+ONC (treated with standard chow and ONC); Group PLX+ONC (treated with standard chow containing PLX5622 and ONC).
Part 3: Group Vehicle (treated with corn oil); Group Res (treated with resveratrol); Group Vehicle+ONC (treated with corn oil and ONC); Group Res+ONC (treated with resveratrol and ONC).
Part 4: Group Sirt1(+/+) [WT mice without treatment]; Group Sirt1(±) [Sirt1(f/wt)Cx3Cr1cre(+) mice without treatment]; Group Sirt1(-/-) [Sirt1(f/f)Cx3Cr1cre(+) mice without treatment]; Group Sirt1(+/+)+ONC [WT mice treated with ONC]; Group Sirt1(±)+ONC [Sirt1(f/wt)Cx3Cr1cre(+) mice treated with ONC]; Group Sirt1(-/-) [Sirt1(f/f)Cx3Cr1cre(+) mice treated with ONC].
Part 5: Group Raptor(+/+) [WT mice without treatment]; Group Raptor(±) [Raptor(f/wt)Cx3Cr1cre(+) mice without treatment]; Group Raptor(-/-) [Raptor(f/f)Cx3Cr1cre(+) mice without treatment]; Group Raptor(+/+)+ONC [WT mice treated with ONC]; Group Raptor(±)+ONC [Raptor(f/wt)Cx3Cr1cre(+) mice treated with ONC]; Group Raptor(-/-) [Raptor(f/f)Cx3Cr1cre(+) mice treated with ONC].
Reagents Administration
PLX5622 (PLX) was formulated in standard chow at 1200 mg/kg. Mice were kept on PLX5622 diets for 3 weeks before experiment and maintained on their diets until sacrifice. Resveratrol (Res) powder was stored at −20°C and was dissolved in corn oil at 10 mg/mL before using. Mice were intragastrically administrated with resveratrol solution and the dosage was 100 mg/kg once a day. Treatment started at 3 days before ONC and the duration was 1 week. The concentration of CCT007093 (CCT) was 10 mg/mL and the storing temperature was −80°C. The dosage was 3.2 mg/kg and mice were intraperitoneally injected with CCT007093 twice a day. CCT007093 was used together with resveratrol.
Tissue Preparation
The eyes and optic nerve were harvested from animals after sacrifice and were soaked in 4% paraformaldehyde (PFA) at 4°C.
Paraffin Section
The eyes were postfixed for 24 h in 4% PFA, followed by paraffin embedding for obtaining 4-μm sections.
Frozen Section
After 24 h post-fixation in 4% PFA, optic nerve tissues were cryoprotected in 30% sucrose for 24 h, followed by being frozen in OCT on the freezing element of a Leica CM1950 cryostat microtome in order to acquire 6-μm sections.
Flattening of Mice Retina
After 24 h post-fixation in 4% PFA, the eyes were cut through the sclera just posterior to the ciliary body. The lens were removed and eyecups were fixed in 4% PFA for another 45 min in order to separate retina easily. To make the retina lay flat, 4 cuts were made toward the optic disc.
Immunofluorescence
Slices of optic nerve and whole retina tissues were permeabilized with 0.5% Triton X-100 for 15 min and then washed 3 times with PBS for 5 min. As for the eye tissue slices, they were dewaxed in environmentally safe clearing agent and gradient alcohol, washed with ddH2O and then soaked in trisodium citrate solution (pH = 6.0) for microwave antigen retrieval in 20 min. After above preparation, samples were blocked in donkey serum for 1 h at room temperature and incubated in diluted primary antibodies at 4°C overnight. Finally, these samples were incubated in diluted secondary antibodies at room temperature for 2 h. The images acquisition of optic nerve and retina slices were processed under a fluorescence microscope (OLYMPUS BX51) at 20× magnification. Iba1-positive area in optic nerve and RBPMS-positive cell number were quantified by ImageJ and Photoshop respectively.
Immunohistochemistry (IHC)
Immunohistochemistry was performed on optic nerve frozen sections. After blocking with 0.05% BSA, sections were incubated with primary antibodies overnight at 4°C. Subsequently, sections were washed and incubated with secondary antibodies for 1 h at room temperature (RT). Then, sections were washed and incubated with diluted streptavidin-biotin complex (SABC) at RT for 30 min. After being washed with PBS, sections were incubated with Horseradish Peroxidase (HRP) color developing agent. Finally, sections were washed with ddH2O and stained with hematoxylin followed by dehydration. IHC images were acquired by using light microscope (OLYMPUS BX53) at 20× magnification.
Nissl Staining
The eye tissue slices were dewaxed in the same way of immunofluorescence. After that, the slices were stained with Nissl staining solution for 5 min, washed by ddH2O until the water turned colorless and dried at 65°C. Retina Nissl staining images were acquired by using light microscope (OLYMPUS BX53) at 20× magnification. Retina RGCs number and length were obtained by ImageJ.
Transmission Electron Microscope (TEM) Imaging
Optic nerve tissue preparation for TEM and imaging procedure were done according to the previously reported method.26 Five fields of view in the central optic nerve and five fields of view in the ventral optic nerve at crush site were analyzed. Ten high-power images were generated. Total axons, axons with abnormal myelin, and areas of fields were calculated by ImageJ.
RGC Quantification
For Nissl staining, 5 retina sections were required per sample. The retina sections were parallel to axis oculi and contained optic nerve head. 6 images were generated per section (2 fields of view near optic nerve head, 2 fields of view in central retina and 2 fields of view in peripheral retina). Nissl cell in ganglion cell layer (GCL) and length of GCL were calculated by ImageJ and mean Nissl cell number per mm of 5 sections was one sample data. For retina flattening, 10 images were generated per sample (5 fields of view in central retina and 5 fields of view in peripheral retina). RBPMs positive cell and areas of fields were calculated by ImageJ.
Statistical Analysis
Data shown represents at least three independent experiments at least. Statistical software Prism9 (GraphPad) was used for data analysis. Two-tailed t tests or one-way ANOVA were used to assess differences between 2 or multiple groups, respectively. Error bars represent mean ± SEM. For all tests, P-values <0.05 were considered significant.
Results
Temporal Characteristics of RGCs Loss and Microglial Activation in ONC Model
We first assessed the cellular responses to optic nerve injury (ONI). To this end, we subjected the mice to crush injury which can mimic ONI-inducing neuroinflammation and neurodegeneration (Figure 1A). After the injury, mice were sacrificed at different time points (day 0, 3, 5, 7 after crush) (Figure 1A). Nissl staining was performed in the retinas of mice, which showed the number of RGCs decreased significantly on day 5 after the crush injury (Figure 1B and C). As shown in Figure 1C, RGCs number declined continuously at day 7 post injury but without statistical significance. But previous studies have reported that RGCs loss was progressive post ONC.27,28 Given that neurons, RGCs and displaced amacrine cells (dACs) comprise most of the ganglion cell layer (GCL) and microglia activate, migrate and proliferate towards the GCL post ONC, Nissl staining may not be specific enough for RGCs quantification.29,30 As RNA-binding protein with multiple splicing (RBPMS) reliably identifies RGCs, RGCs were also quantified by RBPMS.31 Although significant RGCs loss was observed on day 7 after crush injury via using RBPMS, the extent of RGCs loss on day 5 after injury was most significant, which was in line with Nissl staining (Figure 1D and E). To further evaluate RGCs injury directly, TEM of optic nerve was used to assess the damage of axons. Previous studies have reported that RGCs loss precedes the intraretinal axons clearance and traumatic injury could cause axon loss directly.32,33 The site selected for TEM analyzing was 0.5 mm post the crush site, and not only the number of total axons but also the proportion of axons with abnormal myelin were calculated. As shown in Figure 1F–H, total axons decreased and axons with abnormal myelin increased significantly at day 5 post ONC.
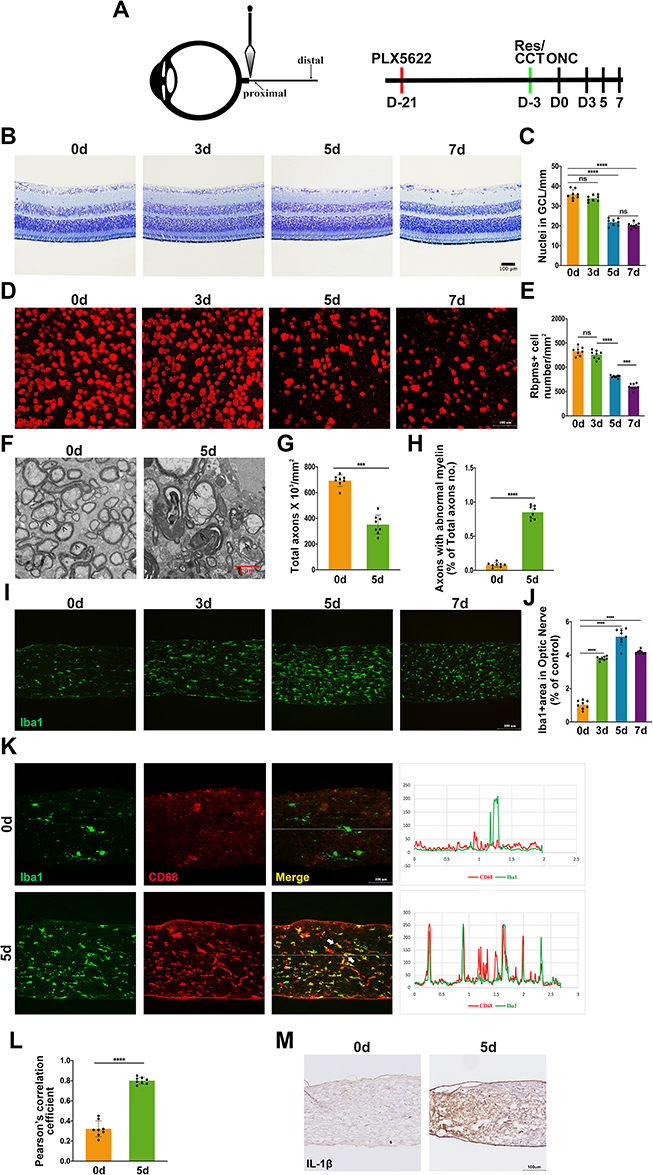 |
Figure 1 Optic nerve crush induces RGCs loss and microglia activation. (A) Optic nerve crush (ONC) paradigm and treatment scheme; (B–E) Retina Nissl staining and RBPMS immunofluorescence were conducted to assess RGCs loss. Representative images day 0, day 3, day 5, and day 7 after ONC are shown (B and D). Quantification of RGCs loss (C and E) by RGC-counting of time points in B and D after ONC (n = 8 mice per group); (F) TEM examination of axon damage at 0.5 mm behind crush injury site on day 0 and day 5 post ONC. Normal axon was indicated with arrow (>) and axons with abnormal myelin was indicated with (≫); (G–H) Quantification of axon damage was conducted by calculating total axons x 103/mm2 (G) and proportion of axons with abnormal myelin (H) under conditions of F (n = 8 mice per group); (I) Iba1+ area in proximal optic nerve was measured for microglial activation. Representative images of Iba1+ area in on day 0, day 3, day 5, and day 7 post injury; (J) Quantification of Iba1+ area in proximal optic nerve from the results of D (n = 8 mice per group); (K) Both Iba1 and CD68 were used to further evaluate microglial activation on day 5 post ONC. Co-localization between Iba1 and CD68 was indicated with white arrow; (L) Quantification of co-localization between Iba1 and CD68 under conditions of K (n = 8 mice per group). (M) Microglial activation on day 0 and 5 post ONC detected by IL-1β. Data are presented as the mean ± SEM. ***P < 0.001, ****P < 0.001. |
We then investigated whether microglia participated in ONC-induced ONI. Immunofluorescence showed that Iba1-positive area increased on day 3 after ONC, reached peak at day 5 post injury and started to decrease 2 days later (Figure 1I and J) at 0.5 mm post the crush site, which were not detected at the distal site (data not shown). When microglial cells activate, they change their ramified morphology to fusiform or ameboid and exaggerate amounts of inflammatory mediators.8 Thus, only detecting Iba1 is not sufficient to quantify microglial activation. Previous research reported that the reactivity of Iba1 correlated with the level of CD86 during microglial activation.34 Hence, CD68 and IL-1β were also used to quantify microglia activation. On day 5 post ONC, the extent of the co-localization between Iba1 and CD68 and the level of IL-1β increased significantly, which indicated microglial activation (Figure 1K–N). Taken together, our data suggested that microglial activation started earlier than RGCs loss after ONC.
Microglial Ablation Attenuated RGCs Loss After ONC
As the above results indicated that both significant RGCs loss and microglial activation peak occurred on day 5 after the crush injury, we therefore selected this time point in all further experiments of this study. To examine the role of microglia in RGCs loss post injury, we depleted microglia conditionally with PLX5622. After ONC, PLX-treated mice represented microglia absence (Figure 2A and B). And RGCs loss was attenuated when microglia were depleted (Figure 2C–D). TEM images also revealed that axon damage was less severe in PLX-treated group (Figure 2E–G). These findings collectively suggested that microglia as one component at least did take part in the process of RGCs loss.
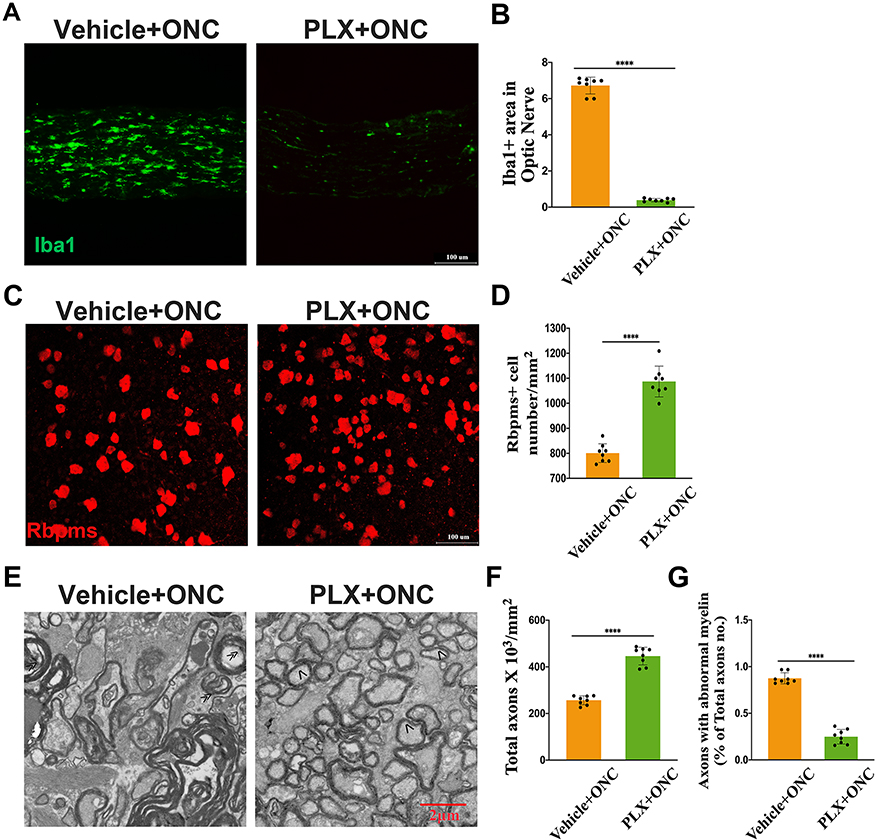 |
Figure 2 Microglial activation participates in RGCs loss after ONC. (A) Representative images of Iba1+ area in proximal optic nerve. Tissue samples were from Vehicle- and PLX- group after ONC; (B) Quantitative analysis of Iba1+ area under conditions represented in A (n = 8 mice per group); (C and D) Representative RBPMS+ immunofluorescence images (C) of PLX and Vehicle chow-fed mice post injury. Quantification of RGCs loss (D) from the results of C (n = 8 mice per group); (E) Representative images of axon damage measured by TEM at crush injury site of Vehicle- and PLX- group. Normal axon was indicated with arrow (>) and axons with abnormal myelin was indicated with (≫); (F and G) Quantification of axon damage at crush injury site of Vehicle- and PLX- group (n = 8 mice per group). Data are presented as the mean ± SEM. ****P < 0.001. |
SIRT1 Exhibited RGCs Protection via Down-Regulating Microglial Activation
As SIRT1 was implicated in Aβ induced microglial activation, we assumed that SIRT1 could regulate microglia activation in ONI. Firstly, we examined the cellular responses to resveratrol in adult wildtype C57BL/6J mice. There were no differences of microglial activation and RGCs number between Res and Vehicle group (Figure 3A–D). However, under ONC lesion, Res dampened microglial activation and RGCs loss (Figure 3E–J). In addition, less severe axon damage was observed in Res-treated mice (Figure 3K–M).
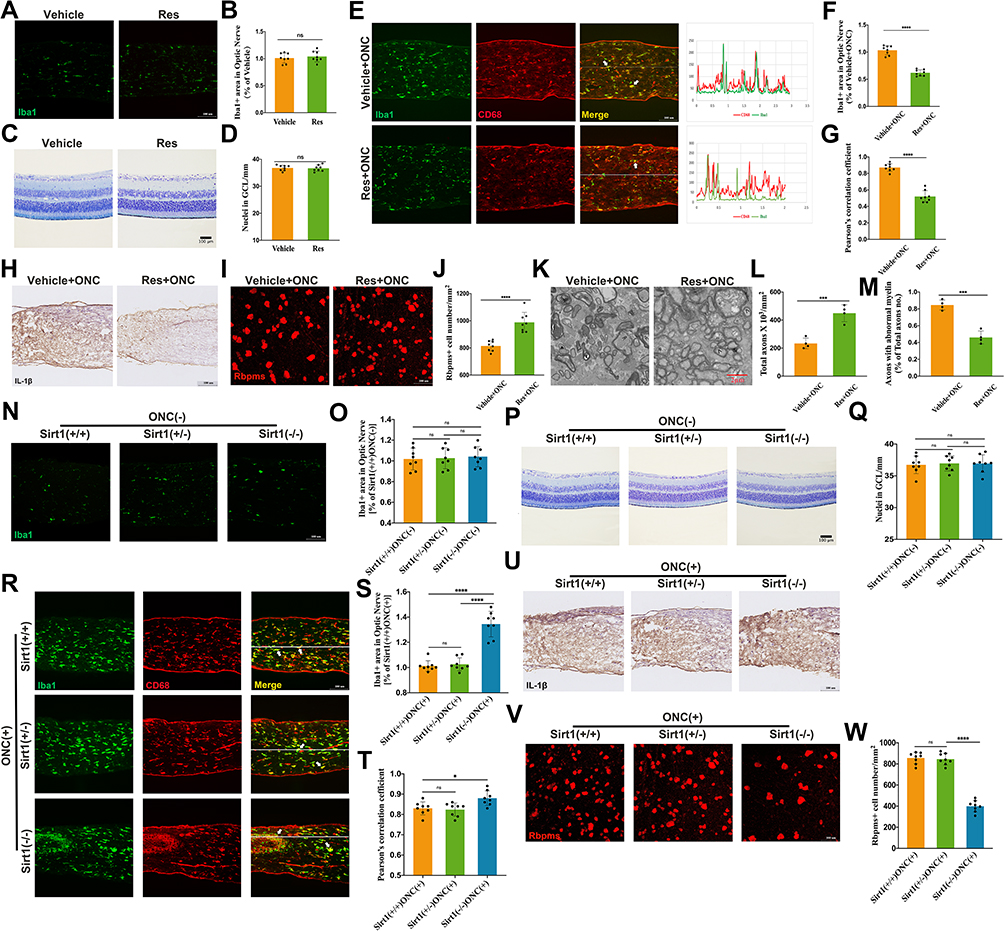 |
Figure 3 SIRT1 reduces RGCs loss by microglia inhibition. (A) Representative images of Iba1+ area in proximal optic nerve of Vehicle- and Res- group without ONC; (B) Quantitative analysis of Iba1+ area in proximal optic nerve under conditions represented in A (n = 8 mice per group); (C) Representative images of retina Nissl staining of Vehicle- and Res- group without crush injury; (D) Quantitative analysis of RGCs loss of the 2 groups in C (n = 8 mice per group); (E–H) Activation of Sirt1 inhibited Microglial activation (E and H). Co-localization between Iba1 and CD68 was indicated with white arrow. Quantitative analysis of Iba1+ area (F) and co-localization between Iba1 and CD68 (G) under conditions represented in E (n = 8 mice per group); (I)RBPMs+ cell in retina was counted for examining RGCs loss. Representative images of RBPMs+ cell in retina of Vehicle- and Res- group after crush injury; (J) Quantification of RGCs loss by counting RBPMs+ cell in retinas from Vehicle- and Res- group after ONC (n = 8 mice per group); (K) Representative TEM images of axon damage at 0.5 mm behind crush injury site of Vehicle- and Res- group after crush injury. Normal axon was indicated with arrow (>) and axons with abnormal myelin was indicated with (≫); (L and M) Quantification of axon damage of the two groups in K post crush (n = 8 mice per group); (N–Q) Microglia Sirt1 deficiency did not influence microglial activation and RGCs number without injury (N and P). Quantification of microglial activation (O) and RGC number (Q) under conditions in N and P (n = 8/genotype); (R–W) Microglial Sirt1 deficiency increased RGCs (V) loss via aggravating microglial activation (R and U). Co-localization between Iba1 and CD68 was indicated with white arrow. Quantification of microglial activation (S and T) and RGCs loss (W) from the results of R and V (n = 8/genotype). Data are presented as the mean ± SEM. *P < 0.05, ***P < 0.001, ****P < 0.0001. |
To confirm these results, Cx3cr1-CreERT2/ Sirt1F/F mice were used to genetically reduce levels of microglial Sirt1. Microglial Sirt1 deficiency did not influence microglial activation and RGCs number without injury (Figure 3N–Q). Interestingly, when the crush injury happened, more severe microglial activation and RGCs loss were observed in microglial Sirt1 deficiency mice (Figure 3R–W). However, TEM images revealed that more severe axon loss was not observed in microglial Sirt1 deficiency mice (data not shown). Given that ONC is severe trauma and Sirt1 is protective, the results of TEM were not opposite to previous results. Therefore, these findings revealed that SIRT1 could attenuate RGCs loss by inhibiting microglial activation post ONC.
mTORC1 Deficiency Reduced RGCs Loss Through Blocking Microglial Activation
mTORC1 is a key component in sensing nutrients, growth factors, and cellular energy, inhibiting autophagy and stimulating cell growth. Therefore, we needed to establish the effects of mTORC1 in microglial activation. We obtained Cx3cr1-CreERT2/RaptorF/F mice to genetically reduce mTORC1 in microglia without altering other mTOR complexes. Microglial mTORC1 deficiency had no effect on microglial activation and RGCs number without ONC (Figure 4A–D). However, microglial activation and RGCs loss decreased after ONC in Raptor knockout mice (Figure 4E–J). As shown in Figure 4K–M, TEM images revealed similar results. Taken together, these findings indicated that microglial mTORC1 deficiency reduced RGC loss by exerting an inhibitory effect on microglial activation.
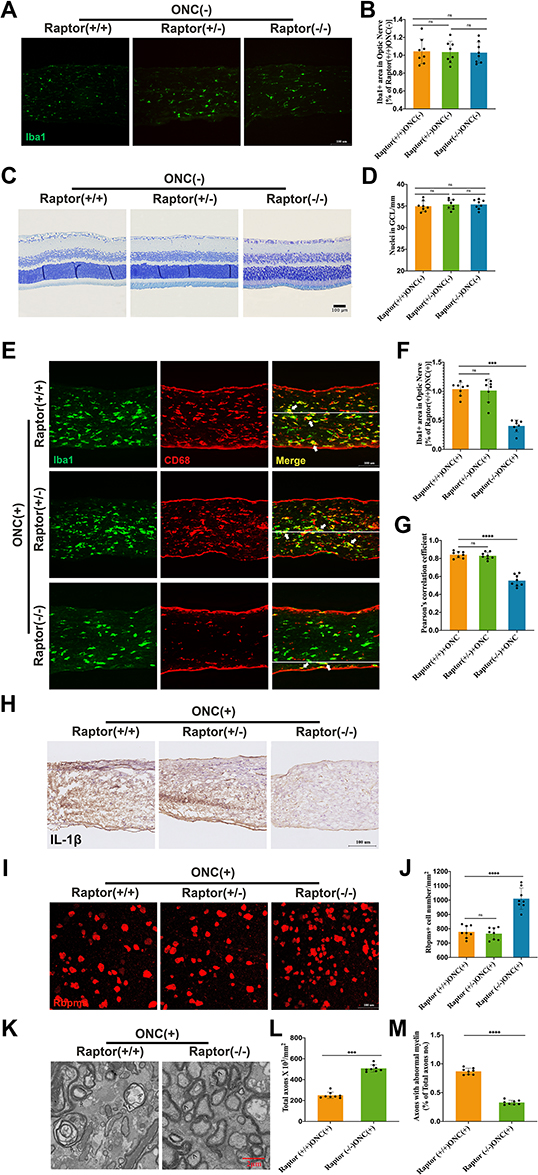 |
Figure 4 Microglial mTORC1 deficiency attenuates RGCs loss via microglial activation inhibition. (A) Representative images of Iba1+ area in proximal optic nerve of RaptorF/F, Cx3Cr1-CreERT2/ RaptorF/WT and Cx3Cr1-CreERT2/ RaptorF/F mice without injury; (B) Quantification of Iba1+ area under conditions represented in A (n = 8/genotype); (C) Representative images of retina Nissl staining of RaptorF/F, Cx3Cr1-CreERT2/ RaptorF/WT and Cx3Cr1-CreERT2/ RaptorF/F mice without injury; (D) Quantitative analysis of RGCs loss from the results of C (n = 8/genotype); (E–H) Genetically reducing mTORC1 in microglia reduced RGCs loss after ONC by inhibiting microglial activation (E and H). Co-localization between Iba1 and CD68 was indicated with white arrow. Quantification of Iba1+ area (F), and co-localization between Iba1 and CD68 (G) from the results of E (n = 8/genotype); (I) Representative images of RBPMs+ cell in retinas of RaptorF/F, Cx3Cr1-CreERT2/ RaptorF/WT and Cx3Cr1-CreERT2/ RaptorF/F mice; (J) Quantification of RGCs loss by counting RBPMs cell in retinas under conditions represented in I (n = 8/genotype); (K–M) Representative TEM images reveal that mTORC1 deficiency attenuated axons damage (K). Normal axon was indicated with arrow (>) and axons with abnormal myelin was indicated with (≫). Quantification of axon damage (L and M) under conditions represented in K (n = 8/genotype). Data are presented as the mean ± SEM. ***P < 0.001, ****P < 0.0001. |
SIRT1-mTORC1 Signaling Pathway Implicated in Microglia-Mediated RGCs Loss After ONC
Since previous studies reported that mTORC1 was one downstream substrate of SIRT1, whether SIRT1 regulates microglia activation via mTORC1 remained unclear. CCT007093 was used in Res-treated mice. As shown in Figure 5A–D, CCT had no effect on microglial activation and RGCs number without ONC. CCT-treatment reversed the effect of Res on microglial activation, and RGCs number is less in CCT-treated mice after ONC (Figure 5E–J). Consistently, axon damage became worse when Res-treated mice were subjected to CCT (Figure 5K–M). These results collectively indicated that mTORC1 served as a substrate of SIRT1 and its activation could reverse the benefits of SIRT1 after ONC.
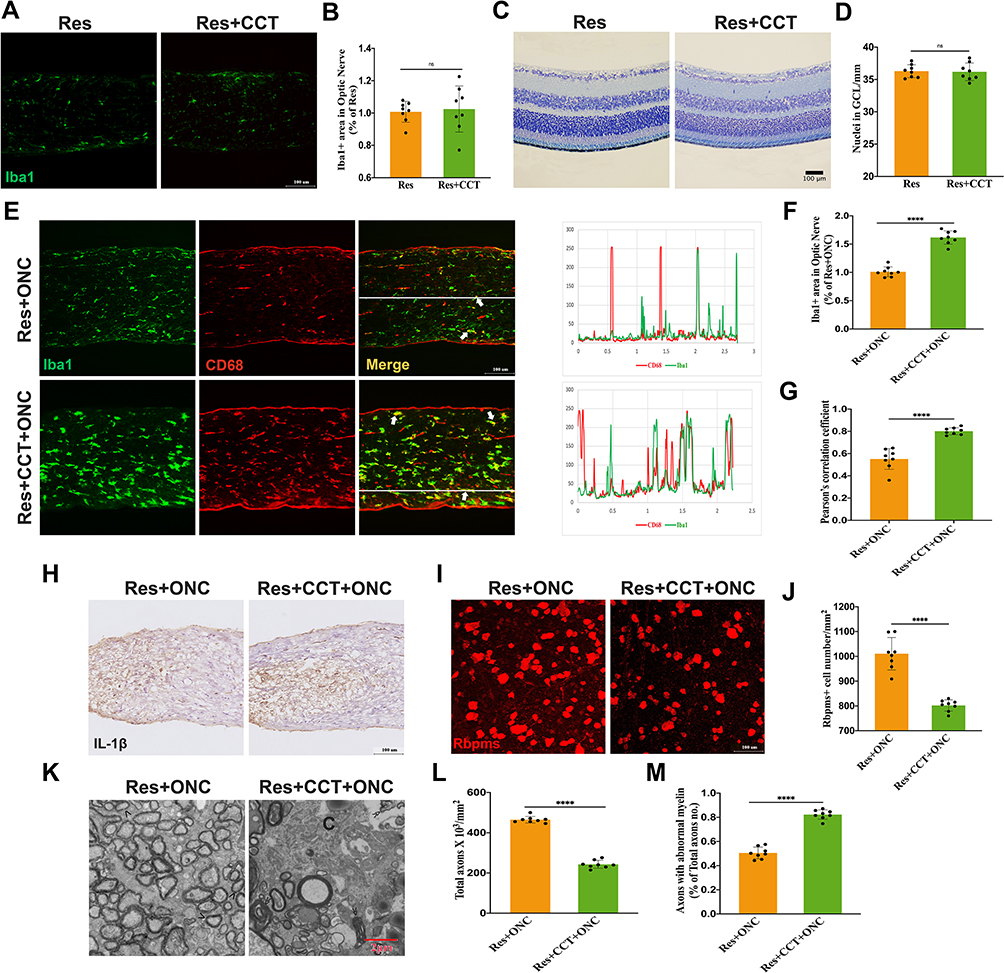 |
Figure 5 SIRT1-mTORC1 axis is involved in microglia-mediated RGCs loss. (A–D) Res (A) and CCT (C) administration had no effect on microglial activation and RGCs number without ONC. Quantification of Iba1+ area (B) and RGC number (D) under conditions represented in A and C (n = 8 mice per group); (E-J) mTORC1 activation induced by CCT could reverse the effect of sirt1 on inhibiting microglial activation (E and H) and reducing RGCs loss (I and J) post injury. Co-localization between Iba1 and CD68 was indicated with white arrow. Quantification of Iba1+ area (F), co-localization between Iba1 and CD68 (G), and RGCs number (J) (n = 8 mice per group). (K–M) Representative images of TEM (K) and quantification (L and M) of axon damage at 0.5 mm behind crush injury site. Normal axon was indicated with arrow (>) and axons with abnormal myelin was indicated with (≫) (n = 8 mice per group). Data are presented as the mean ± SEM.****P < 0.0001. |
Discussion
This study demonstrated that depletion of microglia could delay RGCs damage and loss at early ONI. The SIRT1-mTORC1 pathway determined microglia activation, therefore attenuated RGCs damage and promoted RGCs survival. These results suggest that SIRT1-mTORC1 pathway could be considered as a research target for microglia regulation and ONI relative diseases.
Microglia are principal innate immune cells of myeloid origin in the retina and optic nerve, and therefore monitor and regulate neuroinflammation in various ocular diseases.35 ONI triggers microglia to turn into surveillance mode, and migrate to injury sites.36 Although early microglial responses encompass reparative elements to remove pathogenic factors, this protective state cannot sustain over prolonged periods. Experimental evidence suggests that chronic release of proinflammatory cytokines and reactive oxygen species from microglia may cause progressive neurodegeneration. Recently, a colony stimulating factor 1 receptor (CSF1R) inhibitor, PLX5622, was developed to deplete >95% of all microglia in the central nervous system.37 In Alzheimer’s disease (AD), several groups have identified that PLX5622 could improve cognition in 3xTg-AD, APP-PS1 transgenic and 5xFAD mice model.38–41 In traumatic brain injury (TBI), recent studies also have reported that PLX5622 treatment limited TBI-associated neuropathology.42,43 All these data suggested that depletion of microglia with PLX5622 could be beneficial in neurodegeneration.
In order to investigate the role of microglia in ONI, we performed optic nerve crush lesions on adult C57BL/6J mice. Interestingly, we found that the microglial activation was earlier than RGCs soma loss after lesion. Depletion of microglia with PLX5622 could partially rescue RGCs loss. We believe that at early ONI, depletion of microglia is beneficial for RGCs loss delay. However, a previous study reported that eliminating microglia with PLX5622 could not rescue RGCs degeneration on 7 to 21 days after lesion.44 We speculated that the different outcome is due to several reasons. Firstly, our results suggested that at early stage of injury (5 days or less), depletion of microglia could protect axonal loss. Hilla and colleagues examined the function of microglia for a longer period. Secondly, microglia communicate with RGCs by secreting factors. A representative example of a microglia-RGCs interaction is the fractalkine and its G protein coupled receptor CX3CR1.45,46 We speculated that at different lesion stages, the microglia-RGCs interaction is dynamic. Finally, a recent study reported that genetic ablation of microglia protects RGCs against NMDA injury. They proved that TNFα (secreted by microglia) and its receptor on RGCs is the key cytotoxic factor for microglia-RGCs interaction.47 However, undergoing a longer injury period, for example 7 to 21 days after ONI, the secretion of TNFα might be different.
SIRT1 protects against neurodegeneration by suppression of microglia activation, and mainly through HMGB1/NF-κB/IL-1β inflammation signal pathway.20,48 The mammalian target of rapamycin (mTOR) signaling pathway serves as a central regulator of cell metabolism, and mTOR complex 1 (mTORC1) is composed of mTOR and Raptor. As a regulatory protein, Raptor determines the stability and function of mTORC1.49 Molecular mechanism studies have identified that SIRT1 negatively regulates mTORC1 signaling mainly through two pathways. Ghosh and colleagues have reported that SIRT1 negatively regulates mTORC1 through the TSC1/2 complex.50 Furthermore, Hong and members reported that inhibition of SIRT1 could induce p70 ribosomal S6 kinase (S6K1) acetylation and inhibit mTORC1-dependent S6K1 phosphorylation.23 In this study, we found that microglia specific Sirt1 knockout could induce more microglia activation and aggravate RGCs degeneration. Inhibition of mTORC1 with specific Raptor knockout could reduce microglia activation, in addition, activation of mTORC1 with CCT could partially rescue the outcome induced by SIRT1. These results are the first identified evidence of SIRT1-mTORC1 signaling in innate immune cells which was independent of the canonical HMGB1/NF-κB/IL-1β inflammation signal pathway. Further studies should identify the role of SIRT1-mTORC1 signaling in long-term effect of microglia regulation and neuroprotection. Besides, we should investigate the molecular pathway to define how SIRT1 regulates mTORC1 signaling in microglia.
Conclusion
Our findings identify a novel treatment strategy for RGCs loss induced by ONI mice model. The SIRT1-mTORC1 signaling serves as a potential target in microglia regulation and neuroprotection. These findings provide a novel target for ONI relative gene therapy.
Abbreviations
ONI, optic nerve injury; ONC, optic nerve crush; CSF1R, colony-stimulating factor 1 receptor; Iba1, ionized calcium binding adaptor molecule 1; RGCs, retinal ganglion cells; RBPMS, RNA-binding protein with multiple splicing; TEM, transmission electron microscopy; OCT, optimal cutting temperature compound; PLX, PLX5622; Res, resveratrol; CCT, CCT007093; PFA, paraformaldehyde; dACs, displaced amacrine cells; GCL, ganglion cell layer.
Acknowledgments
The current studies were supported by funding from the National Natural Science Foundation of China Grant Nos. 31800868, Tongji Hospital (HUST) Foundation for Excellent Young Scientist Grant No. 2020YQ18 (to Dr Yin Zhao) and National Natural Science Foundation of China Grant Nos. 82070965 (to Dr Hong Zhang).
Disclosure
The authors report no biomedical financial interests or potential conflicts of interest in this work.
References
1. Weinreb RN, Leung CK, Crowston JG, et al. Primary open-angle glaucoma. Nature Reviews. Disease Primers. 2016;2(1):16067. doi:10.1038/nrdp.2016.67
2. Shah SM, Khanna CL. Ophthalmic Emergencies for the Clinician. Mayo Clin proc. 2020;95(5):1050–1058. doi:10.1016/j.mayocp.2020.03.018
3. Williams PR, Benowitz LI, Goldberg JL, He Z. Axon Regeneration in the Mammalian Optic Nerve. Ann Rev Vision Sci. 2020;6:195–213. doi:10.1146/annurev-vision-022720-094953
4. Ransom BR, Orkand RK. Glial-neuronal interactions in non-synaptic areas of the brain: studies in the optic nerve. Trends Neurosci. 1996;19(8):352–358. doi:10.1016/0166-2236(96)10045-X
5. Dezawa M, Adachi-Usami E. Role of Schwann cells in retinal ganglion cell axon regeneration. Prog Retin Eye Res. 2000;19(2):171–204. doi:10.1016/S1350-9462(99)00010-5
6. Yazdankhah M, Shang P, Ghosh S, et al. Role of glia in optic nerve. Prog Retin Eye Res. 2021;81:100886. doi:10.1016/j.preteyeres.2020.100886
7. Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi:10.1016/j.neuron.2012.03.026
8. Rashid K, Akhtar-Schaefer I, Langmann T. Microglia in Retinal Degeneration. Front Immunol. 2019;10:1975. doi:10.3389/fimmu.2019.01975
9. Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–1394. doi:10.1038/nn1997
10. Huang Z, Zhou T, Sun X, et al. Necroptosis in microglia contributes to neuroinflammation and retinal degeneration through TLR4 activation. Cell Death Differ. 2018;25(1):180–189. doi:10.1038/cdd.2017.141
11. Cai XF, Lin S, Geng Z, et al. Integrin CD11b Deficiency Aggravates Retinal Microglial Activation and RGCs Degeneration After Acute Optic Nerve Injury. Neurochem Res. 2020;45(5):1072–1085. doi:10.1007/s11064-020-02984-6
12. Zhao X, Sun R, Luo X, Wang F, Sun X. The Interaction Between Microglia and Macroglia in Glaucoma. Front Neurosci. 2021;15:610788. doi:10.3389/fnins.2021.610788
13. Cheadle L, Rivera SA, Phelps JS, et al. Sensory Experience Engages Microglia to Shape Neural Connectivity through a Non-Phagocytic Mechanism. Neuron. 2020;108(3):451–468.e459. doi:10.1016/j.neuron.2020.08.002
14. Zarb Y, Sridhar S. Microglia control small vessel calcification via TREM2. Sci Adv. 2021;7(9):545.
15. Guerrero A, De Strooper B, Arancibia-Cárcamo IL. Cellular senescence at the crossroads of inflammation and Alzheimer’s disease. Trends Neurosci. 2021;44(9):714–727. doi:10.1016/j.tins.2021.06.007
16. Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nature Reviews. Neurology. 2021;17(3):157–172. doi:10.1038/s41582-020-00435-y
17. Wan X, Garg NJ. Sirtuin Control of Mitochondrial Dysfunction, Oxidative Stress, and Inflammation in Chagas Disease Models. Front Cell Infect Microbiol. 2021;11:693051. doi:10.3389/fcimb.2021.693051
18. Gomes BAQ, Silva JPB, Romeiro CFR. Neuroprotective Mechanisms of Resveratrol in Alzheimer’s Disease: role of SIRT1. Oxidative Medicine and Cellular Longevity. 2018;2018:8152373. doi:10.1155/2018/8152373
19. Chen J, Zhou Y, Mueller-Steiner S, et al. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J Biol Chem. 2005;280(48):40364–40374. doi:10.1074/jbc.M509329200
20. Cho SH, Chen JA, Sayed F. SIRT1 deficiency in microglia contributes to cognitive decline in aging and neurodegeneration via epigenetic regulation of IL-1β. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2015;35(2):807–818. doi:10.1523/JNEUROSCI.2939-14.2015
21. Murugan AK. mTOR: role in cancer, metastasis and drug resistance. Semin Cancer Biol. 2019;59:92–111. doi:10.1016/j.semcancer.2019.07.003
22. Dunlop EA, Tee AR. Mammalian target of rapamycin complex 1: signalling inputs, substrates and feedback mechanisms. Cell Signal. 2009;21(6):827–835. doi:10.1016/j.cellsig.2009.01.012
23. Hong S, Zhao B, Lombard DB, Fingar DC, Inoki K. Cross-talk between sirtuin and mammalian target of rapamycin complex 1 (mTORC1) signaling in the regulation of S6 kinase 1 (S6K1) phosphorylation. J Biol Chem. 2014;289(19):13132–13141. doi:10.1074/jbc.M113.520734
24. Yin H, Hu M, Liang X, et al. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology. 2014;146(3):801–811. doi:10.1053/j.gastro.2013.11.008
25. Jin X, Kang X, Zhao L, et al. Cartilage Ablation of Sirt1 Causes Inhibition of Growth Plate Chondrogenesis by Hyperactivation of mTORC1 Signaling. Endocrinology. 2019;160(12):3001–3017. doi:10.1210/en.2019-00427
26. Giacci MK, Bartlett CA, Huynh M, Kilburn MR, Dunlop SA, Fitzgerald M. Three dimensional electron microscopy reveals changing axonal and myelin morphology along normal and partially injured optic nerves. Scientific Reports. 2018;8(1):3979. doi:10.1038/s41598-018-22361-2
27. Galindo-Romero C, Avilés-Trigueros M, Jiménez-López M, et al. Axotomy-induced retinal ganglion cell death in adult mice: quantitative and topographic time course analyses. Exp Eye Res. 2011;92(5):377–387. doi:10.1016/j.exer.2011.02.008
28. Nadal-Nicolás FM, Jiménez-López M, Sobrado-Calvo P, et al. Brn3a as a marker of retinal ganglion cells: qualitative and quantitative time course studies in naive and optic nerve-injured retinas. Invest Ophthalmol Vis Sci. 2009;50(8):3860–3868. doi:10.1167/iovs.08-3267
29. Nadal-Nicolás FM, Sobrado-Calvo P, Jiménez-López M, Vidal-Sanz M, Agudo-Barriuso M. Long-Term Effect of Optic Nerve Axotomy on the Retinal Ganglion Cell Layer. Invest Ophthalmol Vis Sci. 2015;56(10):6095–6112. doi:10.1167/iovs.15-17195
30. Nadal-Nicolás FM, Jiménez-López M, Salinas-Navarro M, Sobrado-Calvo P, Vidal-Sanz M, Agudo-Barriuso M. Microglial dynamics after axotomy-induced retinal ganglion cell death. J Neuroinflammation. 2017;14(1):218. doi:10.1186/s12974-017-0982-7
31. Rodriguez AR, de Sevilla Müller LP, Brecha NC. The RNA binding protein RBPMS is a selective marker of ganglion cells in the mammalian retina. J Comp Neurol. 2014;522(6):1411–1443. doi:10.1002/cne.23521
32. Jecrois ES, Zheng W, Bornhorst M, et al. Treatment during a developmental window prevents NF1-associated optic pathway gliomas by targeting Erk-dependent migrating glial progenitors. Dev Cell. 2021;56(20):2871–2885.e2876. doi:10.1016/j.devcel.2021.08.004
33. Rovere G, Nadal-Nicolás FM, Agudo-Barriuso M, et al. Comparison of Retinal Nerve Fiber Layer Thinning and Retinal Ganglion Cell Loss After Optic Nerve Transection in Adult Albino Rats. Invest Ophthalmol Vis Sci. 2015;56(8):4487–4498. doi:10.1167/iovs.15-17145
34. Cao P, Chen C, Liu A, et al. Early-life inflammation promotes depressive symptoms in adolescence via microglial engulfment of dendritic spines. Neuron. 2021;109(16):2573–2589.e2579. doi:10.1016/j.neuron.2021.06.012
35. Yu C, Roubeix C, Sennlaub F, Saban DR. Microglia versus Monocytes: distinct Roles in Degenerative Diseases of the Retina. Trends Neurosci. 2020;43(6):433–449. doi:10.1016/j.tins.2020.03.012
36. Heuss ND, Pierson MJ, Roehrich H, et al. Optic nerve as a source of activated retinal microglia post-injury. Acta Neuropathologica Communications. 2018;6(1):66. doi:10.1186/s40478-018-0571-8
37. Jing L, Hou L, Zhang D, et al. Microglial Activation Mediates Noradrenergic Locus Coeruleus Neurodegeneration via Complement Receptor 3 in a Rotenone-Induced Parkinson’s Disease Mouse Model. J Inflamm Res. 2021;14:1341–1356. doi:10.2147/JIR.S299927
38. Dagher NN, Najafi AR, Kayala KM, et al. Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J Neuroinflammation. 2015;12:139. doi:10.1186/s12974-015-0366-9
39. Unger MS, Schernthaner P, Marschallinger J, Mrowetz H, Aigner L. Microglia prevent peripheral immune cell invasion and promote an anti-inflammatory environment in the brain of APP-PS1 transgenic mice. J Neuroinflammation. 2018;15(1):274. doi:10.1186/s12974-018-1304-4
40. Spangenberg E, Severson PL, Hohsfield LA, et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nature Communications. 2019;10(1):3758. doi:10.1038/s41467-019-11674-z
41. Casali BT, MacPherson KP, Reed-Geaghan EG, Landreth GE. Microglia depletion rapidly and reversibly alters amyloid pathology by modification of plaque compaction and morphologies. Neurobiol Dis. 2020;142:104956. doi:10.1016/j.nbd.2020.104956
42. Henry RJ, Ritzel RM, Barrett JP, et al. Microglial Depletion with CSF1R Inhibitor During Chronic Phase of Experimental Traumatic Brain Injury Reduces Neurodegeneration and Neurological Deficits. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2020;40(14):2960–2974. doi:10.1523/JNEUROSCI.2402-19.2020
43. Dziabis JE, Quan N, Eiferman DS, et al. Traumatic Brain Injury Causes Chronic Cortical Inflammation and Neuronal Dysfunction Mediated by Microglia. J Neurosci. 2021;41(7):1597–1616. doi:10.1523/JNEUROSCI.2469-20.2020
44. Hilla AM, Diekmann H, Fischer D. Microglia Are Irrelevant for Neuronal Degeneration and Axon Regeneration after Acute Injury. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2017;37(25):6113–6124. doi:10.1523/JNEUROSCI.0584-17.2017
45. Vessey KA, Waugh M, Jobling AI, et al. Assessment of Retinal Function and Morphology in Aging Ccl2 Knockout Mice. Invest Ophthalmol Vis Sci. 2015;56(2):1238–1252. doi:10.1167/iovs.14-15334
46. Zhang Y, Zhao L, Wang X. Repopulating retinal microglia restore endogenous organization and function under CX3CL1-CX3CR1 regulation. Science Advances. 2018;4(3):eaap8492. doi:10.1126/sciadv.aap8492
47. Takeda A, Shinozaki Y, Kashiwagi K, Ohno N, Eto K, Wake H. Microglia mediate non-cell-autonomous cell death of retinal ganglion cells. Glia. 2018;66(11):2366–2384. doi:10.1002/glia.23475
48. Chen X, Chen C, Fan S, et al. Omega-3 polyunsaturated fatty acid attenuates the inflammatory response by modulating microglia polarization through SIRT1-mediated deacetylation of the HMGB1/NF-κB pathway following experimental traumatic brain injury. J Neuroinflammation. 2018;15(1):116. doi:10.1186/s12974-018-1151-3
49. Li K, Wei X, Zhang L, Chi H, Yang J. Raptor/mTORC1 Acts as a Modulatory Center to Regulate Anti-bacterial Immune Response in Rockfish. Front Immunol. 2019;10:2953. doi:10.3389/fimmu.2019.02953
50. Ghosh HS, McBurney M, Robbins PD. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS One. 2010;5(2):e9199. doi:10.1371/journal.pone.0009199
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.