")
Back to Archived Journals » Gastrointestinal Cancer: Targets and Therapy » Volume 8
Models of intrahepatic cholangiocarcinoma: novel tools and therapeutic applications
Authors Hill MA, Alexander WB, Hezel AF
Received 18 April 2017
Accepted for publication 25 October 2017
Published 19 February 2018 Volume 2018:8 Pages 1—11
DOI https://doi.org/10.2147/GICTT.S115757
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Eileen O'Reilly
Margaret A Hill,1,2,* William B Alexander,1,2,* Aram F Hezel1,2
1Department of Biomedical Genetics, 2Department of Medicine, Hematology/Oncology, Wilmot Cancer Institute, University of Rochester Medical Center, Rochester, NY, USA
*These authors contributed equally to this work
Abstract: Intrahepatic cholangiocarcinoma (iCCA) is the second most common hepatic malignancy, and a number of recent studies have identified an increasing trend in incidence and mortality. As an aggressive disease characterized by early metastasis, surgical resection is not an option for most patients, and chemotherapy has limited benefit. Thus, the prognosis is extremely poor, warranting the development of novel models to improve detection and treatment strategies for this lethal cancer. In this regard, significant technological advancements have provided key tools to model and study iCCA. Furthermore, these technologies are addressing the need for models that can readily be adapted to address different genetic contexts, an important consideration for genetically diverse cancers such as iCCA. In this review, we outline these various available tools, discussing specifically how they have been employed to study iCCA while highlighting important therapeutic implications. Finally, we discuss novel strategies utilizing patient-derived tumor tissue which have promising translational applications.
Keywords: cholangiocarcinoma, biliary cancer, models, cell of origin, Cre-Lox, sleeping beauty
Introduction
Intrahepatic cholangiocarcinoma (iCCA) is an extremely lethal cancer with limited therapeutic options; therefore, novel approaches to early detection and treatment are desperately needed.1 Key to this is the development of models that accurately recapitulate critical aspects of human iCCA to both better understand the underlying biology of the disease and provide systems in which new therapeutic strategies can be explored. Furthermore, iCCA is a genetically diverse cancer, and designing flexible models that can readily be adapted to reflect this genetic complexity is of paramount importance.
In this review, we discuss the various strategies that have been employed to model iCCA. Throughout, we highlight important biological insights made possible by the specific models and also discuss related therapeutic implications. In deciding how to best utilize these models and how to approach the development of new models, there are a number of important considerations, which are outlined in Box 1. Balancing these often requires a combination of complementary approaches, which is evident in the crossover of technologies in some of the sections of this review. To date, Cre-Lox-based genetically engineered mouse models (GEMMs) have proven to be an invaluable resource for studying iCCA, and therefore, we devote significant space to describing their utility and contributions. We further discuss novel approaches that have been applied to modeling iCCA using non-germline-based technologies. Finally, we focus on recent developments that rely on patient-derived tumor tissue, patient-derived tumor xenografts (PDTXs), and organoids, which have significant potential for translational applications.
 | Box 1 Considerations for the development and utilization of iCCA models Abbreviation: iCCA, intrahepatic cholangiocarcinoma. |
Chemical and surgical models
Historically, rodent models of liver carcinogenesis have heavily relied upon administering exogenous toxic agents that induce injury and/or carcinogenesis, in part due to the liver conveniently being the central organ for toxin metabolism. In addition to being extremely practical immunocompetent in vivo models, these agents can also recapitulate features of premalignant and malignant disease, which may not accompany other models that depend strictly on genetic perturbations, such as ductular reactions or fibrosis. A wide array of liver injury agents exist.2,3 Those that have been utilized to study cholangiocarcinoma (CCA) in particular are listed in Table 1. Briefly, the nitrosamines, diethylnitrosamine and dimethylnitrosamine, and thioacetamide (TAA) have both been used as carcinogens, as they can reliably lead to cancer without additional agents or engineered mutations.13,16–18 It is important to note that hepatocellular carcinoma (HCC) may be the dominant tumor type arising in carcinogenic models. Most often, an additional oncogenic event, such as liver fluke infection or biliary-specific deletion of a tumor suppressor, is needed to promote iCCA development.17,18 Although such extreme carcinogenic injury is not frequently recognized epidemiologically among human iCCA, tumors arising in this context are likely more genetically diverse and, therefore, can be useful for study when the genetic context is of minimal importance. Carbon tetrachloride (CCl4), a less severe agent, induces liver fibrosis and cirrhosis, relevant features of human CCA, and, in a Tp53-null setting, promotes iCCA development.19 Finally, bile duct ligation (BDL) and 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) are used to induce cholestatic injury and ductular reactions, a common feature of liver fluke infections and early iCCA development.13,20–22 When deciding which chemical agents to use, one must consider both the context in which the toxin is to be used and which aspects of injury are most important for the study. For example, if a defined genetic context is desired, a carcinogenic injury model that has an unpredictable genetic progression should be discouraged. In other studies, groups may want to induce a specific feature of liver injury for which a chemical or surgical model is best suited. For example, in an extended experiment assessing the impact of ductular reactions on iCCA development, a flexible dietary model like DDC may be preferable to a surgical model BDL, which is more suitable for short-term experiments. These various models continue to play a key role in the study of liver-related malignancies, and in particular, integrating them with other advanced genetic tools, as addressed in the following sections, has shed much light on the development and biology of iCCA.
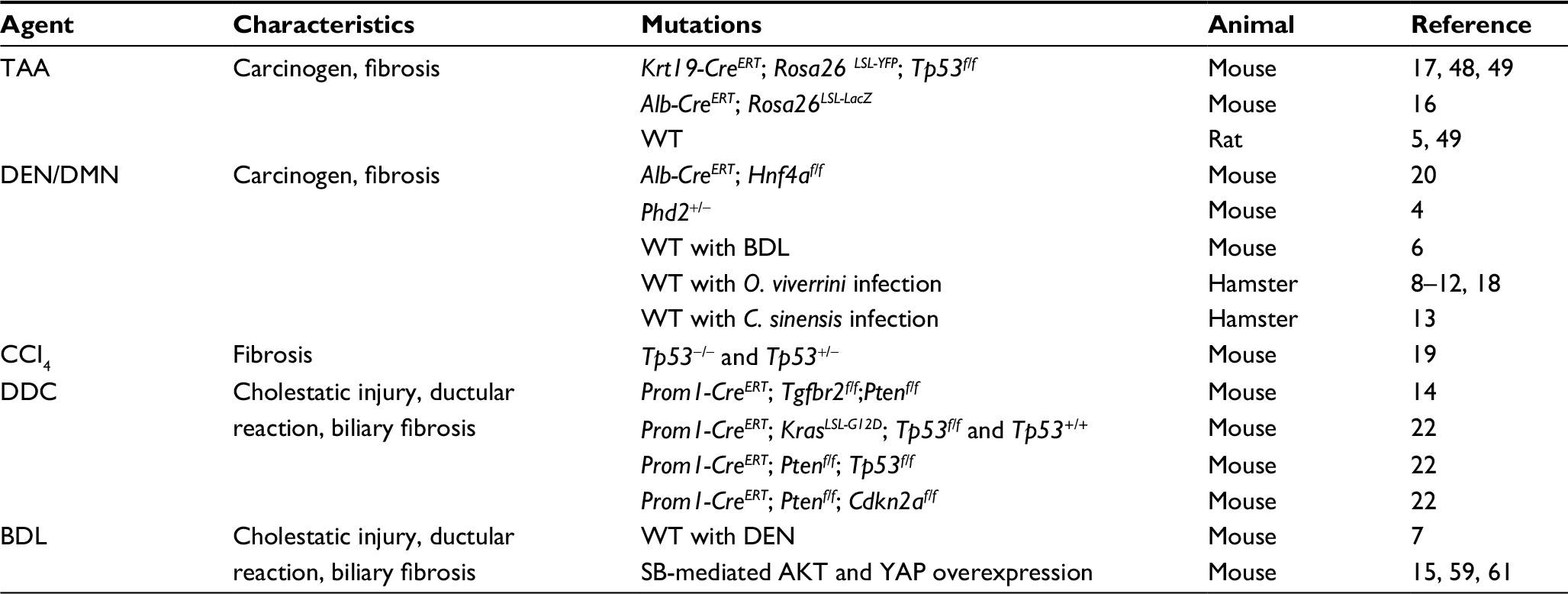 | Table 1 Summary of injury and carcinogen models Abbreviations: BDL, bile duct ligation; CCI4, carbon tetrachloride; C. sinensis, Clonorchis sinensis; DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; DEN, diethylnitrosamine; DMN, dimethylnitrosamine; O. viverrini, Opisthorchis viverrini; SB, sleeping beauty; TAA, thioacetamide; WT, wild type. |
Cre-Lox-based models
Conditional GEMMs that utilize Cre-Lox technology have been one of the most powerful tools available to understand the development and progression of cancer.23 In this review, we outline how this technology has been utilized to study the liver and liver-related malignancies and highlight a number of important considerations (Table 2). Cre-Lox technology is a conditional system that uses a Cre-recombinase allele to excise DNA between two LoxP sites oriented in the same direction. Depending on the location of LoxP sites, it can be used to both inactivate and activate genes. Engineered alleles for oncogenic Kras and Tp53 provide examples and have been used cooperatively in models of iCCA. The Trp53flox allele has LoxP sites engineered in introns 1 and 10, leading to deletion of exons 2–10 upon Cre-mediated recombination, thereby inactivating Tp53.36 Alternatively, an oncogenic allele can be rendered conditional by inserting a Stop codon flanked by a LoxP site upstream. The KrasLSL-G12D allele is composed of the constitutively active Kras mutant which in the absence of Cre-recombinase is not transcribed due to the upstream Lox-Stop-Lox (LSL) element; however, upon Cre-mediated recombination, the Stop codon is excised and the oncogenic allele is expressed.37
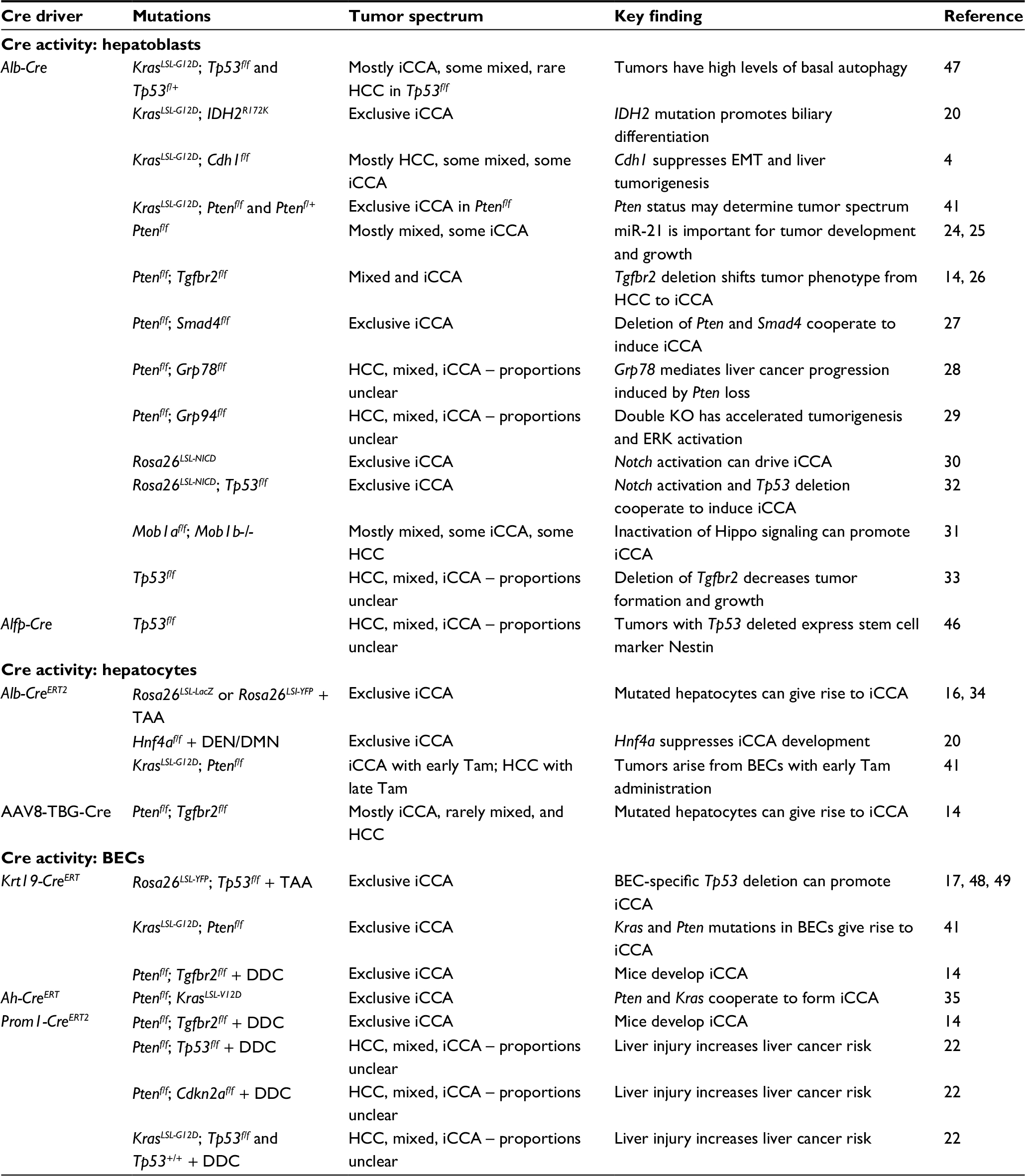 | Table 2 Summary of iCCA Cre-Lox GEMMs Abbreviations: AAV8, Adeno-associated virus 8; BECs, biliary epithelial cells; DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; EMT, epithelial-mesenchymal transition; GEMMs, genetically engineered mouse models; HCC, hepatocellular carcinoma; iCCA, intrahepatic cholangiocarcinoma; NICD; Notch1 intracellular domain; Tam, tamoxifen. |
Tissue specificity and temporal control can be achieved with this system by placing the Cre under the control of a cell- or tissue-specific promoter, thereby generating LoxP site recombination exclusively in that lineage (Figure 1). Alternatively, viruses that have tissue-specific tropisms can be used to deliver Cre-recombinase. Adeno-associated virus 8 (AAV8) is a notable example that has a hepatocyte-specific tropism and is therefore frequently used to target hepatocytes while excluding biliary epithelial cells (BECs).38 In addition, the CreERT system, a fusion of Cre and the tamoxifen-inducible domain of the estrogen receptor (ER), enables spatiotemporal control, which has been crucial for the generation of BEC-specific Cre alleles and manipulating genes in the adult mouse that would otherwise be necessary for normal development.21,39
 | Figure 1 Schematic of Cre driver activity within the liver. Note: *Specificity depends on when tamoxifen is administered. Abbreviation: BEC, biliary epithelial cell. |
Further specificity can be achieved with tetracycline (Tet) expression systems that come in two forms, Tet-on or Tet-off, depending on whether promoter function is enabled or disabled by exogenous Tet administration. Both systems require the addition of either a tTA or rtTA allele, respectively. A significant advantage to using a Tet expression system is that induction via Tet is reversible (unlike CreERT), and combining this system with Cre-Lox technology through addition of an LSL between the promoter and start codon of the Tet-controlled transactivator can enable tissue specificity as well. One such example is the Tet-IDH2LSL-R140Q allele that when combined with Rosa26LSL-rtTA and Alb-Cre restricts Tet-inducible expression of mutant IDH1 to the liver.20 Another useful tool that can be combined with Cre-Lox is the flippase (FLP)- recombinase target (FRT) system, which functions analogously: an FLP recombinase excises DNA between two FRT sequences. It has been employed in the liver in combination with Cre-Lox to enable highly specific labeling of SOX9-positive hepatocytes, a unique compartment with potential to regenerate hepatocytes and BECs following injury.40 Thus, these systems, which allow for broad or specific manipulation of organs and cell types, are essential tools for modeling the various aspects of the human disease in mice.
There are several important considerations that investigators should bear in mind in applying these mutagenesis strategies. When using a Cre that is active in the liver progenitor population, such as the Alb-Cre allele, both liver cell types will be affected, limiting conclusions that can be drawn about the cell of origin. In addition, when using CreERT systems, the timing of tamoxifen administration may be critical. Alb-CreERT, for example, can label a small proportion of BECs, potentially confounding lineage tracing and cell of origin experiments.40,41 Also, all of the listed BEC-specific Cre drivers label epithelial cells across multiple organs including the lung, pancreas, and colon; in the absence of additional liver-specific oncogenic challenges, these experimental animals often develop tumors outside of the liver.41 Nevertheless, significant advancements have been made in iCCA biology using the Cre-Lox system. In this review, we discuss more specifically how Cre-Lox-based GEMMs have advanced our understanding regarding three related topics: the cell of origin, the function of common mutations, and processes important for tumor development and maintenance.
Cell of origin
Although the BEC has historically been the assumed cell of origin for iCCA, several observations suggest a more complex origin, including the existence of mixed HCC–iCCA tumors, an association with chronic hepatocellular injury, and the recently recognized capacity for hepatocytes to transdifferentiate into BECs.1,42 To test the possibility that hepatocytes can be the cell of origin for iCCA, one experiment lineage traced the hepatocyte compartment with an AAV8-Ttr-Cre and a Rosa26LSL-YFP reporter.43 Using sleeping beauty (SB) transposase (to be discussed) to express AKT and a constitutively active Notch1 intracellular domain (NICD) in the liver, iCCAs developed that expressed the YFP marker, indicating a hepatocyte origin. In another experiment, hepatocytes and BECs were lineage traced using the Alb-CreERT and Krt19-CreERT, respectively, with the Rosa26LSL-lacZ reporter.16 Following iCCA induction via the carcinogen TAA, iCCAs with β-gal activity were present only in the Alb-CreERT cohort, similarly illustrating hepatocytes as a possible cell of origin. On the other hand, when Krt19-CreERT is used to delete Tp53 and label BECs, thereby predisposing BECs to oncogenesis, it was demonstrated that TAA administration induces iCCA that is of biliary origin.17 Finally, in another key experiment demonstrating BECs as a cell of origin, induction of Kras and Pten mutations in BECs via Krt19-CreERT promoted iCCA development.41 Notably, mice in the latter experiment also exhibited significant extrahepatic tumor burden, illustrating a critical limitation of alleles such as Krt19-CreERT and Hnf1b-CreERT that are active in extrahepatic epithelial compartments as well. Altogether, the current evidence supports the notion that both hepatocytes and BECs can be a cell of origin in iCCA depending on the context (Figure 2). Understanding distinguishing biological features of BEC-derived and hepatocyte-derived iCCAs may prove to be relevant in the development of future therapeutic strategies.
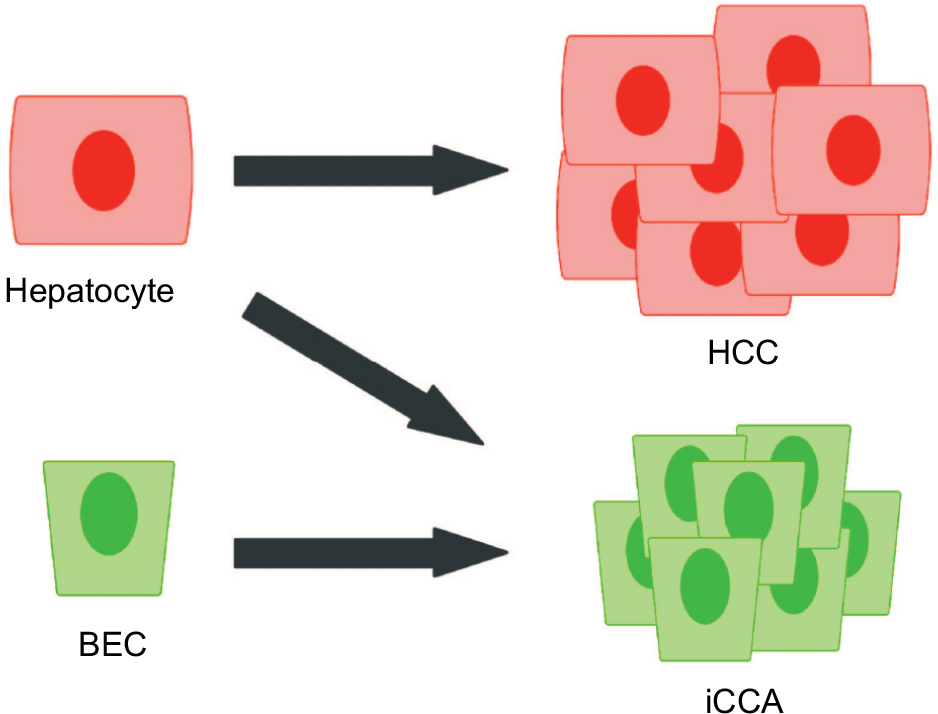 | Figure 2 Current model of the cells of origin in primary liver cancers. Abbreviations: BEC, biliary epithelial cell; HCC, hepatocellular carcinoma; iCCA, intrahepatic cholangiocarcinoma. |
Function of common mutations
In recent large-scale sequencing studies, the genetic landscape of iCCA has come into view. In non-liver fluke-related iCCA, the most common mutations are in IDH1/2 (22%), BAP1 (22%), TP53 (7%), KRAS (7%), and ARID1A (7%). Liver fluke-related iCCA, on the other hand, is distinguished by mutations in TP53 (45%), KRAS (19%), SMAD4 (16%), ARID1A (12%), and KMT2C (11%).44,45 Cre-Lox-based GEMMs have proven useful in discerning the functions of some of these mutations in iCCA. To explore the function of IDH1/2 mutations in iCCA, Alb-Cre was utilized to enable Tet-inducible expression of oncogenic IDH1/2 mutations in the liver, demonstrating that mutant IDH1/2 function to inhibit hepatocyte differentiation via epigenetic silencing of the major regulator of hepatocyte identity, Hnf4a, a key driver of hepatocyte differentiation.20 In a similar theme of mutations that impact cellular differentiation, Tp53 deletion in an Alfp-Cre-driven model leads to poorly differentiated liver tumors that have high expression of the stem cell marker Nestin and can be further directed toward a hepatocellular or biliary fate with additional lineage-specific mutations. Thus, Tp53 is an important suppressor of liver cell plasticity.46 Finally, Alb-Cre-driven KrasG12D expression and Tp53 deletion was shown to promote development of iCCAs that depend on autophagy, an important downstream process in many Kras-driven tumors.47 Despite significant progress in the past decade, much remains to be explored regarding the roles of these and many other oncogenic mutations found in iCCA.
Critical processes for tumor development and maintenance
Cre-Lox-based GEMMs are an essential tool to study and model the development of cancer in ways that accurately recapitulate the natural disease progression and tumor microenvironment. In addition to studying the importance of downstream pathways using genetic strategies, these models provide the opportunity to test potential therapeutic interventions in a relevant in vivo setting. In this regard, one group showed that iCCA in the Krt19-CreERT;Tp53f/f TAA model, a rat TAA model, and xenograft models are all sensitive to both macrophage-inhibiting therapies and Wnt-inhibiting therapies.48 The same research group again employed the Krt19-CreERT;Tp53f/f TAA and rat TAA models to demonstrate that the Notch pathway may be an additional therapeutic target.49 One strategy put the mouse model on a Notch3 null genetic background, finding that there is comparably minimal tumor development in this setting. Furthermore, pharmacologic inhibition of Notch signaling in the rat model similarly reduced tumor formation. These results suggest that further development of strategies to target these processes may be worthwhile. Finally, although not illustrated in the aforementioned examples, another clear advantage to GEMMs is the presence of an intact immune system. This important feature has been exploited in GEMMs of other malignancies such as pancreatic cancer to test T-cell-directed immunotherapies such as promising anti-PD-1/PD-L1 therapies.50
Novel Cre-Lox tools
Despite the many advantages offered by the Cre-Lox-based GEMMs described earlier, the newly discovered breadth of mutations in this disease and the need to rapidly evaluate different combinations of mutations warrant the development of more efficient modeling strategies. To address this need, there have been a number of recent efforts to generate non-germline Cre-Lox models. One approach involves the culture of embryonic hepatoblasts from Alb-Cre KrasLSL-G12D Tp53LSL-R172H mice, which can then be orthotopically transplanted into a syngeneic host to generate iCCA within several months.51 These mutant hepatoblasts can be further manipulated to validate other oncogenic drivers and evaluate their potential as a therapeutic target. For example, to determine the impact of a newly identified fusion gene, FIG-ROS1, on iCCA growth, a Tet-inducible FIG-ROS1 gene was introduced into the hepatoblast-allograft model, dramatically accelerating tumor development. Furthermore, turning off FIG-ROS1 expression via doxycycline withdrawal resulted in dramatically slower tumor progression. A similar strategy could express shRNAs to determine the impact of knocking down putative tumor suppressors or putative tumor-promoting genes. In addition to the improved flexibility and ease of generation in comparison to traditional GEMMs, this approach has the additional advantage of not generating mutations throughout the entire organ, thus maintaining an environment that consists of cells not genetically predisposed to cancer, more akin to human tumors.
Another promising strategy has been developed to study pancreatic cancer and represents a system that could be adapted to study liver malignancies as well.52 Embryonic stem cells (ESCs) were established that contain a pancreas-specific driver (Pdx-Cre or p48-Cre) and the KrasLSL-G12D allele. In addition to these features, the cells harbor a “recombinase-mediated cassette exchange” strategy for rapid exchange of Tet-inducible shRNA- or cDNA-expressing constructs accompanied by a fluorescent reporter and a Cre-dependent Tet-transactivator accompanied by a different fluorescent reporter. The ESCs can be manipulated in vitro to introduce the shRNA/cDNAs of interest and subsequently used to generate chimeric mice for study. This flexible technology enables a variety of questions to be asked, illustrated by the following examples. First, by introducing a doxycycline-inducible shRNA-targeting Pten, they demonstrated that knockdown of a tumor suppressor can accelerate Kras-induced tumorigenesis. Furthermore, doxycycline withdrawal, resulting in reexpression of Pten, causes a dramatic decrease in tumor burden and an increase in survival. To determine the role of Myc in pancreatic tumor development, they employed tandem shRNA technology to introduce an shRNA targeting Tp53, which similarly accelerates tumorigenesis, and an shRNA targeting either Myc or a control shRNA. Compared to the control shRNA, tumor development in the Myc knockdown cohort was significantly impaired, confirming a role for Myc in promoting tumor development.
Thus, these technologies maintain many of the advantages of traditional germline GEMMs: the ability to make temporal- and tissue-specific genetic perturbations in an immune-competent environment, thereby generating cancers that closely follow the histopathologic progression of human disease. However, they offer flexible strategies to bypass the extensive breeding required to address similar questions with germline GEMMs.
SB models
Given the limitations associated with Cre-Lox GEMMs, a number of alternative approaches have been developed. The mouse liver is readily accessible via hydrodynamic tail vein injection (HTVI) for delivery of plasmids, but due to the transient nature of traditional expression plasmids, sustained expression of transcripts for oncogenic studies is a challenge.53 Thus, the SB transposon system has found particular relevance in enabling stable integration of transgenes in the mouse liver (Table 3). The SB transposon is a cut and paste element, which was created based on the inactive Tc1/mariner superfamily of transposons in fish. The transposase recognizes inverted repeat (IR) sequences that flank a DNA sequence, excise that DNA, and insert it at thymine and adenine sites elsewhere in the genome. The endogenous SB transposon is a single element, where the SB transposase gene is flanked by IR/direct repeat (DR) sequences. In order to use this system experimentally, the SB transposase and IR/DR sequences have been separated where the IR/DR sequences flank the gene of interest and are often introduced as unique vectors.
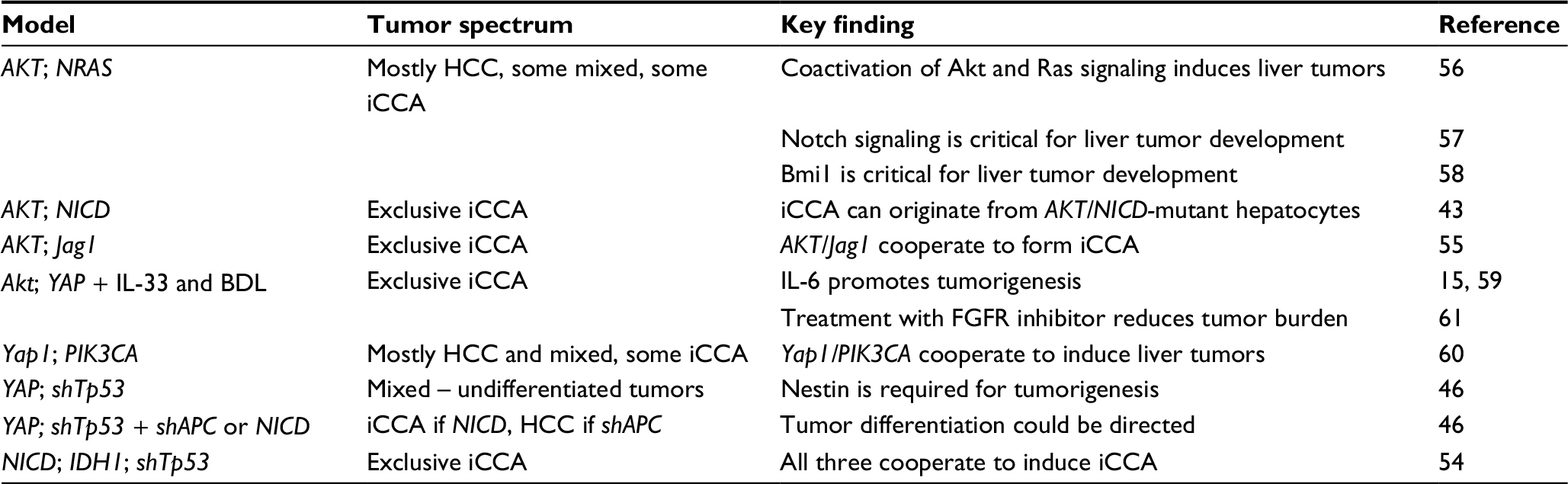 | Table 3 Summary of iCCA SB models Abbreviations: BDL, bile duct ligation; HCC, hepatocellular carcinoma; iCCA, intrahepatic cholangiocarcinoma; IDH1, IDH1R132C; NICD, Notchl intracellular domain; PIK3CA, PIK3CAH1047R; YAP, YAPS127A; SB, sleeping beauty. |
To drive iCCA development, a number of studies have employed an activated form of Notch signaling in combination with various other perturbations including activation of PI3K signaling, activation of Yap signaling, mutant IDH1 expression, and Tp53 knockdown.43,46,54,55 Notch-independent models have also been generated, including coactivation of Yap/PI3K signaling and mutant Kras/PI3K signaling.15,46,56–61 While tail vein injection is believed to largely target hepatocytes, targeting the biliary tract specifically was achieved by injecting plasmids directly into the gallbladder following bile-duct ligation, thereby preventing spillover into the gut. It was further demonstrated that Yap and Akt expression in this setting cooperate to induce iCCA in a small proportion of mice, a phenotype that significantly increased when IL-33 is systemically administered.
SB technology has also been adapted for forward genetic mutagenesis screens to identify genes that are critical in regulating liver cancer development.62,63 This is achieved using a Cre-Lox-regulated SB transposase and a T2/onc transposon which can lead to expression of an oncogene or inactivation of a tumor suppressor. In an SB mutagenesis screen using livers predisposed to tumor development via expression of dominant negative Tp53 (Tp53LSL-R270H), sequencing of the resulting tumors revealed genes that are known HCC drivers such as Egfr and Met, as well as a number of novel genes not previously implicated in cancer. Another experiment used Myc overexpression to predispose the liver to tumorigenesis and a similar SB mutagenesis strategy to identify Ncoa2 as a novel tumor suppressor. Currently, these screens in the liver have focused on HCC; thus, similar approaches remain to be employed in a setting that will lead to iCCA.
CRISPR/Cas9 models
Another especially promising gene-editing tool is the clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9) system.64 Like SB, CRISPR/Cas9 offers an efficient method to introduce oncogene expression, but it has additional flexibility to inactivate genes such as tumor suppressors and repair genes with point mutations. In brief, the Cas9 nuclease is directed to a target sequence via a single-guide RNA (sgRNA), where double-strand breaks are repaired by error-prone nonhomologous end joining (NHEJ), often resulting in insertion/deletions (indels) that can disrupt a locus through introduction of reading frame shifts. Alternatively, investigators can use homology-directed repair (HDR) to create specific mutations through use of a DNA template to guide homologous recombination (HR).
To date, there are three hallmark studies that illustrate various ways in which CRISPR/Cas9 technology can be applied to the murine liver. One group used HTVI to introduce a single vector containing both Cas9 and sgRNAs to target Pten and Tp53.65 Phenocopying the comparable Cre-Lox model, loss of Pten alone led to up-regulation of AKT, and loss of both Pten and Tp53 resulted in iCCA. Investigators also showed that gain-of-function mutations can be introduced through HDR, paving way for CRISPR/Cas9 modeling of oncogenic mutations commonly used in GEMMs, such as KrasG12D. Another group, which also sought to disrupt tumor suppressors, attempted to achieve long-term in vivo CRISPR/Cas9 activity by combining it with the SB transposon system to incorporate Cas9 and the sgRNAs into the genome.66 Although very few tumors had stable integration of the CRISPR/Cas9 vectors, this study established an effective strategy to model mutations in large gene sets in the mouse liver, which is particularly advantageous for verifying tumor-causing mutations (ie, driver versus passenger mutations). Finally, CRISPR/Cas9 was used to repair a disease-causing point mutation in the Fah gene, that causes hereditary tyrosinemia type I in mice.67 Investigators were able to successfully mend the mutation and also demonstrate that the mice regained weight, signifying alleviation of the disease burden. An interesting therapeutic application related to this is the possibility of repairing point mutations important for tumorigenesis. Currently, however, it should be noted that this strategy corrects mutations in a very small proportion of cells, which may be best suited for instances where the repaired cells have a selective growth advantage. Thus, although promising, much improvement is needed regarding the delivery and repair efficiency of this system for it to be applied therapeutically to reverse cancer causing mutations.
Patient-derived models
The utility of studying human cancer cell lines in the culture dish and as xenografts is well established, and they will continue to be critical in understanding the biology of the disease and in developing therapeutic strategies. In this review, we highlight novel approaches that can offer some advantages over these traditional models.
PDTXs
The applications of PDTXs have been rapidly expanding, from studying biological processes to predicting the therapeutic response of patient samples.68,69 Unlike cell line xenografts where cultured cell lines are injected into immune-deficient mice, PDTXs are implanted tumor fragments taken from biopsy, maintaining key features of the tumor lost in two-dimensional (2D) culturing such as stroma, architecture, and heterogeneity. Preserving these qualities has allowed for accurate prediction of therapeutic sensitivities in numerous other cancers.70–72 Recently, multiple groups demonstrated successful PDTX models of CCA. A KRAS-mutant iCCA model was generated and shown to retain many features of the original tumor, including immunoreactivity and miRNA expression.73 Another group showed an IDH1-mutant PDTX displayed sensitivity to dasatinib, a multi-tyrosine kinase inhibitor shown to have efficacy on IDH1-mutant iCCA cells in vitro.74 Similarly, two PDTX models of iCCA were treated with an FGFR inhibitor, and the YAP-driven model displayed significantly reduced growth with treatment, supporting the group’s in vitro finding of a feed-forward loop between YAP and FGFR signaling.61 Some key limitations nevertheless exist for PDTX models. First, there is substantial variability in the success rate for creating PDTXs both among different cancers as well as between laboratories. Relatedly, the tumors that do successfully take in mice likely represent only the highly aggressive tumors. Finally, extensive time is required for the development of PDTXs, up to several months for the establishment of the primary graft, which is an especially important factor if being used to predict therapeutic response of a patient’s tumor.
Organoids
A growing interest has developed regarding the utility of “organoid” models, a culture system in which cells are grown in a three-dimensional matrix and organize into epithelial-like structures that resemble the tissue of origin.75–77 Although liver cancer models remain to be adapted for this system, organoids have shown promise in a related gastrointestinal malignancy, pancreatic cancer. For example, one group fully characterized organoids derived from both normal, premalignant, and malignant pancreatic tissue from murine and human sources, demonstrating that organoids display morphological features and express markers that reflect the disease stage. Furthermore, orthotopic transplantation of these organoids fully recapitulates the disease spectrum. In addition to more closely resembling the native environment of the cells, a distinct advantage of organoids over 2D culture methods is the ability to retain more heterogeneity under these conditions due to the minimal selective pressure. It is also possible that this system may also more readily support growth of stroma to more closely recapitulate the native environment. Finally, a key advantage is that organoids can be prepared from tissue biopsies with relative ease and within a short time frame, such that therapeutic testing can be evaluated on organoids in parallel with the patient’s treatment. These properties suggest that there is significant application for personalized medicine. Thus, it remains to be determined how predictive organoids are for therapeutic responses.
Conclusion
Models of iCCA have progressed significantly over the past decade. Recent genomic advancements have enabled genetic characterization of hepatic malignancies and have fueled the rapid development of sophisticated animal models, from conditional GEMMs to refined somatic gene-editing tools. These advanced systems will enable a more thorough understanding of the development and progression of the disease and will provide flexible platforms for the evaluation of new treatments in a precise manner.
Disclosure
The authors report no conflicts of interest in this work.
References
Banales JM, Cardinale V, Carpino G, et al. Expert consensus document: cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol. 2016;13(5):261–280. | ||
Liu Y, Meyer C, Xu C, et al. Animal models of chronic liver diseases. Am J Physiol Gastrointest Liver Physiol. 2013;304(5):G449–G468. | ||
De Minicis S, Kisseleva T, Francis H, et al. Liver carcinogenesis: rodent models of hepatocarcinoma and cholangiocarcinoma. Dig Liver Dis. 2013;45(6):450–459. | ||
Nakagawa H, Hikiba Y, Hirata Y, et al. Loss of liver E-cadherin induces sclerosing cholangitis and promotes carcinogenesis. Proc Natl Acad Sci U S A. 2014;111(3):1090–1095. | ||
Moriconi F, Ramadori P, Schultze FC, et al. Characterization of the erythropoietin/erythropoietin receptor axis in a rat model of liver damage and cholangiocarcinoma development. Histochem Cell Biol. 2013;139(3):473–485. | ||
Heindryckx F, Bogaerts E, Coulon SH, et al. Inhibition of the placental growth factor decreases burden of cholangiocarcinoma and hepatocellular carcinoma in a transgenic mouse model. Eur J Gastroenterol Hepatol. 2012;24(9):1020–1032. | ||
Yang H, Li TW, Peng J, et al. A mouse model of cholestasis-associated cholangiocarcinoma and transcription factors involved in progression. Gastroenterology. 2011;141(1):378–88, 388.e1–4. | ||
Sudsarn P, Boonmars T, Ruangjirachuporn W, et al. Combination of praziquantel and aspirin minimizes liver pathology of hamster Opisthorchis viverrini infection associated cholangiocarcinoma. Pathol Oncol Res. 2016;22(1):57–65. | ||
Yothaisong S, Thanee M, Namwat N, et al. Opisthorchis viverrini infection activates the PI3K/AKT/PTEN and Wnt/beta-catenin signaling pathways in a Cholangiocarcinogenesis model. Asian Pac J Cancer Prev. 2014;15(23):10463–10468. | ||
Wonkchalee N, Boonmars T, Laummaunwai P, et al. A combination of praziquantel and the traditional medicinal plant Thunbergia laurifolia on Opisthorchis viverrini infection and cholangiocarcinoma in a hamster model. Parasitol Res. 2013;112(12):4211–4219. | ||
Laothong U, Pinlaor P, Boonsiri P, et al. Melatonin inhibits cholangiocarcinoma and reduces liver injury in Opisthorchis viverrini-infected and N-nitrosodimethylamine-treated hamsters. J Pineal Res. 2013;55(3):257–266. | ||
Chusorn P, Namwat N, Loilome W, et al. Overexpression of microRNA-21 regulating PDCD4 during tumorigenesis of liver fluke-associated cholangiocarcinoma contributes to tumor growth and metastasis. Tumour Biol. 2013;34(3):1579–1588. | ||
Lee J-H, Rim H-J, Sell S. Heterogeneity of the “oval-cell” response in the hamster liver during cholangiocarcinogenesis following Clonorchis sinensis infection and dimethylnitrosamine treatment. J Hepatol. 1997;26(6):1313–1323. | ||
Mu X, Pradere J-P, Affò S, et al. Epithelial transforming growth factor-β signaling does not contribute to liver fibrosis but protects mice from cholangiocarcinoma. Gastroenterology. 2016;150(3):720–733. | ||
Li J, Razumilava N, Gores GJ, et al. Biliary repair and carcinogenesis are mediated by IL-33-dependent cholangiocyte proliferation. J Clin Invest. 2014;124(7):3241–3251. | ||
Sekiya S, Suzuki A. Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. J Clin Invest. 2012;122(11):3914–3918. | ||
Guest RV, Boulter L, Kendall TJ, et al. Cell lineage tracing reveals a biliary origin of intrahepatic cholangiocarcinoma. Cancer Res. 2014;74(4):1005–1010. | ||
Thamavit W, Bhamarapravati N, Sahaphong S, Vajrasthira S, Angsubhakorn S. Effects of dimethylnitrosamine on induction of cholangiocarcinoma in Opisthorchis viverrini-infected Syrian golden hamsters. Cancer Res. 1978;38(12):4634–4639. | ||
Farazi PA, Zeisberg M, Glickman J, Zhang Y, Kalluri R, DePinho RA. Chronic bile duct injury associated with fibrotic matrix microenvironment provokes cholangiocarcinoma in p53-deficient mice. Cancer Res. 2006;66(13):6622–6627. | ||
Saha SK, Parachoniak CA, Ghanta KS, et al. Mutant IDH inhibits HNF-4a to block hepatocyte differentiation and promote biliary cancer. Nature. 2014:513(7516):110–114. | ||
Jörs S, Jeliazkova P, Ringelhan M, et al. Lineage fate of ductular reactions in liver injury and carcinogenesis. J Clin Invest. 2015;125(6):2445–2457. | ||
Zhu L, Finkelstein D, Gao C, et al. Multi-organ mapping of cancer risk. Cell. 2016;166(5):1132–1146.e7. | ||
Frese KK, Tuveson DA. Maximizing mouse cancer models. Nat Rev Cancer. 2007;7(9):645–658. | ||
Zhang J, Jiao J, Cermelli S, et al. miR-21 inhibition reduces liver fibrosis and prevents tumor development by inducing apoptosis of CD24+ progenitor cells. Cancer Res. 2015;75(9):1859–1867. | ||
Galicia VA, He L, Dang H, et al. Expansion of hepatic tumor progenitor cells in Pten-null mice requires liver injury and is reversed by loss of AKT2. Gastroenterology. 2010;139(6):2170–2182. | ||
Morris SM, Carter KT, Baek JY, et al. TGF-beta signaling alters the pattern of liver tumorigenesis induced by Pten inactivation. Oncogene. 2015;34(25):3273–3282. | ||
Xu X, Kobayashi S, Qiao W, et al. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption of Smad4 and Pten in mice. J Clin Invest. 2006;116(7):1843–1852. | ||
Chen WT, Zhu G, Pfaffenbach K, Kanel G, Stiles B, Lee AS. GRP78 as a regulator of liver steatosis and cancer progression mediated by loss of the tumor suppressor PTEN. Oncogene. 2014;33(42):4997–5005. | ||
Chen WT, Tseng CC, Pfaffenbach K, et al. Liver-specific knockout of GRP94 in mice disrupts cell adhesion, activates liver progenitor cells, and accelerates liver tumorigenesis. Hepatology. 2014;59(3):947–957. | ||
Zender S, Nickeleit I, Wuestefeld T, et al. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell. 2013;23(6):784–795. | ||
Nishio M, Sugimachi K, Goto H, et al. Dysregulated YAP1/TAZ and TGF-beta signaling mediate hepatocarcinogenesis in Mob1a/1b-deficient mice. Proc Natl Acad Sci U S A. 2016;113(1):E71–E80. | ||
El Khatib M, Bozko P, Palagani V, Malek NP, Wilkens L, Plentz RR. Activation of Notch signaling is required for cholangiocarcinoma progression and is enhanced by inactivation of p53 in vivo. PLoS One. 2013;8(10):e77433. | ||
Morris SM, Baek JY, Koszarek A, Kanngurn S, Knoblaugh SE, Grady WM. Transforming growth factor-beta signaling promotes hepatocarcinogenesis induced by p53 loss. Hepatology. 2012;55(1):121–131. | ||
Terada M, Horisawa K, Miura S, et al. Kupffer cells induce Notch-mediated hepatocyte conversion in a common mouse model of intrahepatic cholangiocarcinoma. Sci Rep. 2016;6:34691. | ||
Marsh V, Davies EJ, Williams GT, Clarke AR. PTEN loss and KRAS activation cooperate in murine biliary tract malignancies. J Pathol. 2013;230(2):165–173. | ||
Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29(4):418–425. | ||
Jackson EL, Willis N, Mercer K, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15(24):3243–3248. | ||
Yanger K, Zong Y, Maggs LR, et al. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev. 2013;27(7):719–724. | ||
Yanger K, Knigin D, Zong Y, et al. Adult hepatocytes are generated by self-duplication rather than stem cell differentiation. Cell Stem Cell. 2014;15(3):340–349. | ||
Font-Burgada J, Shalapour S, Ramaswamy S, et al. Hybrid periportal hepatocytes regenerate the injured liver without giving rise to cancer. Cell. 2015;162(4):766–779. | ||
Ikenoue T, Terakado Y, Nakagawa H, et al. A novel mouse model of intrahepatic cholangiocarcinoma induced by liver-specific Kras activation and Pten deletion. Sci Rep. 2016;6:23899. | ||
Marquardt JU, Andersen JB, Thorgeirsson SS. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat Rev Cancer. 2015;15(11):653–667. | ||
Fan B, Malato Y, Calvisi DF, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest. 2012;122(8):2911–2915. | ||
Chan-on W, Nairismägi M-L, Ong CK, et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat Genet. 2013;45(12):1474–1478. | ||
Nakamura H, Arai Y, Totoki Y, et al. Genomic spectra of biliary tract cancer. Nat Genet. 2015;47(9):1003–1010. | ||
Tschaharganeh DF, Xue W, Calvisi DF, et al. p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell. 2014;158(3):579–592. | ||
O’Dell MR, Huang JL, Whitney-Miller CL, et al. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 2012;72(6):1557–1567. | ||
Boulter L, Guest RV, Kendall TJ, et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J Clin Invest. 2015;125(3):1269–1285. | ||
Guest RV, Boulter L, Dwyer BJ, et al. Notch3 drives development and progression of cholangiocarcinoma. Proc Natl Acad Sci U S A. 2016; 113(43):12250–12255. | ||
Winograd R, Byrne KT, Evans RA, et al. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res. 2015;3:399–411. | ||
Saborowski A, Saborowski M, Davare MA, Druker BJ, Klimstra DS, Lowe SW. Mouse model of intrahepatic cholangiocarcinoma validates FIG–ROS as a potent fusion oncogene and therapeutic target. Proc Natl Acad Sci U S A. 2013;110(48):19513–19518. | ||
Saborowski M, Saborowski A, Morris JP, et al. A modular and flexible ESC-based mouse model of pancreatic cancer. Genes Dev. 2014;28(1):85–97. | ||
Ju HL, Han KH, Lee JD, Ro SW. Transgenic mouse models generated by hydrodynamic transfection for genetic studies of liver cancer and preclinical testing of anti-cancer therapy. Int J Cancer. 2016;138(7):1601–1608. | ||
Ding N, Che L, Li XL, et al. Oncogenic potential of IDH1R132C mutant in cholangiocarcinoma development in mice. World J Gastroenterol. 2016;22(6):2071–2080. | ||
Che L, Fan B, Pilo MG, et al. Jagged 1 is a major Notch ligand along cholangiocarcinoma development in mice and humans. Oncogenesis. 2016;5(12):e274. | ||
Ho C, Wang C, Mattu S, et al. AKT (v-akt murine thymoma viral oncogene homolog 1) and N-Ras (neuroblastoma ras viral oncogene homolog) coactivation in the mouse liver promotes rapid carcinogenesis by way of mTOR (mammalian target of rapamycin complex 1), FOXM1 (forkhead box M1)/SKP2, and c-Myc pathways. Hepatology. 2012;55(3):833–845. | ||
Huntzicker EG, Hotzel K, Choy L, et al. Differential effects of targeting Notch receptors in a mouse model of liver cancer. Hepatology. 2015;61(3):942–952. | ||
Fan L, Xu C, Wang C, et al. Bmi1 is required for hepatic progenitor cell expansion and liver tumor development. PLoS One. 2012;7(9):e46472. | ||
Yamada D, Rizvi S, Razumilava N, et al. IL-33 facilitates oncogene-induced cholangiocarcinoma in mice by an interleukin-6-sensitive mechanism. Hepatology. 2015;61(5):1627–1642. | ||
Li X, Tao J, Cigliano A, et al. Co-activation of PIK3CA and Yap promotes development of hepatocellular and cholangiocellular tumors in mouse and human liver. Oncotarget. 2015;6(12):10102–10115. | ||
Rizvi S, Yamada D, Hirsova P, et al. A hippo and fibroblast growth factor receptor autocrine pathway in cholangiocarcinoma. J Biol Chem. 2016;291(15):8031–8047. | ||
O’Donnell KA, Keng VW, York B, et al. A Sleeping Beauty mutagenesis screen reveals a tumor suppressor role for Ncoa2/Src-2 in liver cancer. Proc Natl Acad Sci U S A. 2012;109(21):E1377–E1386. | ||
Keng VW, Villanueva A, Chiang DY, et al. A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma. Nat Biotechnol. 2009;27(3):264–274. | ||
Sanchez-Rivera FJ, Jacks T. Applications of the CRISPR-Cas9 system in cancer biology. Nat Rev Cancer. 2015;15(7):387–395. | ||
Xue W, Chen S, Yin H, et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature. 2014;514(7522):380–384. | ||
Weber J, Ollinger R, Friedrich M, et al. CRISPR/Cas9 somatic multiplex-mutagenesis for high-throughput functional cancer genomics in mice. Proc Natl Acad Sci U S A. 2015;112(45):13982–13987. | ||
Yin H, Xue W, Chen S, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol. 2014;32(6):551–553. | ||
Aparicio S, Hidalgo M, Kung AL. Examining the utility of patient-derived xenograft mouse models. Nat Rev Cancer. 2015;15(5):311–316. | ||
Hidalgo M, Amant F, Biankin AV, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4(9):998–1013. | ||
Fichtner I, Slisow W, Gill J, et al. Anticancer drug response and expression of molecular markers in early-passage xenotransplanted colon carcinomas. Eur J Cancer. 2004;40(2):298–307. | ||
Fichtner I, Rolff J, Soong R, et al. Establishment of patient-derived non-small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clin Cancer Res. 2008;14(20):6456–6468. | ||
Nemati F, Bras-Goncalves R, Fontaine JJ, et al. Preclinical assessment of cisplatin-based therapy versus docetaxel-based therapy on a panel of human non-small-cell lung cancer xenografts. Anticancer Drugs. 2009;20(10):932–940. | ||
Cavalloni G, Peraldo-Neia C, Sassi F, et al. Establishment of a patient-derived intrahepatic cholangiocarcinoma xenograft model with KRAS mutation. BMC Cancer. 2016;16:90. | ||
Saha SK, Gordan JD, Kleinstiver BP, et al. Isocitrate dehydrogenase mutations confer dasatinib hypersensitivity and SRC dependence in intrahepatic cholangiocarcinoma. Cancer Discov. 2016;6(7):727–739. | ||
Baker LA, Tiriac H, Clevers H, Tuveson DA. Modeling pancreatic cancer with organoids. Trends Cancer. 2016;2(4):176–190. | ||
Boj SF, Hwang CI, Baker LA, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160(1–2):324–338. | ||
Huang L, Holtzinger A, Jagan I, et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat Med. 2015;21(11):1364–1371. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.