")
Back to Journals » Drug Design, Development and Therapy » Volume 12
MMI-0100 ameliorates lung inflammation in a mouse model of acute respiratory distress syndrome by reducing endothelial expression of ICAM-1
Authors He B, Geng S, Zhou W, Rui Y, Mu X, Zhang C, You Q , Su X
Received 19 September 2018
Accepted for publication 23 November 2018
Published 13 December 2018 Volume 2018:12 Pages 4253—4260
DOI https://doi.org/10.2147/DDDT.S188095
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Binchan He,1,* Shen Geng,2,* Wei Zhou,1 Yuwen Rui,1 Xianmin Mu,3 Chen Zhang,3 Qiang You,3 Xin Su1,2
1Department of Respiratory and Critical Care Medicine, Jinling Hospital, Nanjing University School of Medicine, Nanjing 210002, China; 2Department of Respiratory and Critical Care Medicine, Jinling Hospital, Southern Medical University, Guangdong 510000, China; 3Department of Biotherapy, Second Affiliated Hospital, Nanjing Medical University, Nanjing 210002, Jiangsu, China
*These authors contributed equally to this work
Purpose: ICAM-1 plays a critical role in the development of acute respiratory distress syndrome (ARDS). MK2 regulates the expression of ICAM-1 in human pulmonary microvascular endothelial cells. To explore whether the inhibition of MK2 activation has the same effect in experimental animals, MMI-0100, a peptide-mediated inhibitor of MK2, was used to verify whether MMI-0100 can ameliorate lung inflammation in a mouse model of ARDS by reducing endothelial expression of ICAM-1.
Methods: In this study, C57BL/6 mice were randomly divided into three groups: a control group, an lipopolysaccharides (LPS) group, and an LPS plus MMI-0100 group. Mice were killed 24 hours after the administration of LPS and MMI-0100. The mouse lung tissue histopathology, wet/dry weight ratio (W/D), and the neutrophil count were used to measure the severity of lung inflammation in mice. The pulmonary microvascular endothelial cells (PMVECs) of the mice were isolated. The mRNA expression of ICAM-1 in mouse PMVECs was determined using RT-PCR, and the protein expression of MK2 and ICAM-1 in mouse PMVECs was analyzed using Western blotting and immunohistochemistry.
Results: We found that the level of phosphorylated MK2 in the LPS plus MMI-0100 group was reduced. Compared with the LPS group, the LPS plus MMI-0100 group of mice showed less severe inflammation, including a lower W/D and neutrophil count. The mRNA and protein expression of ICAM-1 in the LPS group was significantly higher than in the control group in mouse PMVECs, and the ICAM-1 level was reduced after the administration of MMI-0100.
Conclusion: These data indicate that MMI-0100 ameliorates lung inflammation in a mouse model of ARDS by reducing endothelial expression of ICAM-1.
Keywords: pulmonary microvascular endothelial cell, inflammation, acute respiratory distress syndrome
Introduction
Acute respiratory distress syndrome (ARDS) is a clinical syndrome with high morbidity and mortality. ARDS can be induced by direct or indirect injury to the lung, including sepsis, pneumonia, aspiration of gastric contents, and major trauma.1,2 The lung injury is caused primarily by neutrophil-dependent damage to the endothelial and epithelial barriers of the lung.
ICAM-1 is a cell surface glycoprotein. Studies have shown that ICAM-1 plays an important role in the neutrophil influx into the lung.3 One study showed that following TNF-α stimulation, the vascular permeability and the neutrophil count in the lung are both reduced by more than 70% in mice upon knockout of ICAM-1.4,5 ICAM-1 is also a biomarker used to assess the severity and outcome in trauma-induced ARDS.6
MK2 is a downstream kinase of p38 MAPK.7,8 MK2 is phosphorylated by p38 MAPK and can regulate the expression of ICAM-1 and IL-8 post-transcriptionally without influencing NF-κB activation or ICAM-1 or IL-8 mRNA transcription in human pulmonary microvascular endothelial cells (HPMECs).9 MMI-0100, the cell-permeant peptide inhibitor of MK2, can reduce inflammatory cytokine expression and inhibit myofibroblast activation.10
Our previous study suggested that MK2 regulates TNF-α-induced ICAM-1 expression by stabilization of ICAM-1 mRNA in HPMECs.11,12 This phenomenon, observed in the cell model, must be confirmed in an animal model. Furthermore, we need to determine whether any of these pathways can affect lung inflammation in ARDS. The purpose of this work was to verify whether MMI-0100 can ameliorate lung inflammation in a mouse model of ARDS by reducing endothelial expression of ICAM-1.
Materials and methods
Animal model
Male wild-type C57BL/6 mice (6–8 weeks old, weighing 20–25 g) were obtained from the Nanjing Biomedical Research Institute of Nanjing University. All of the experimental procedures involving animals were approved by the Jinling Hospital Animal Care Committee and strictly followed the international recommendations for the use and care of animals (National Institutes of Health). All of the mice were housed in bright rooms with appropriate temperature (20°C–25°C), humidity (40%–70%), and 12 hours light and 12 hours dark cycle. The mice had ad libitum access to food and water. All of the mice were allowed to adapt to the experimental environment at least 1 week before experimentation, and were monitored once a day.
Establishment of the model of lipopolysaccharides (LPS)-induced ARDS
For each experiment, mice were randomly divided into three groups: a control group (n=5), an intraperitoneal LPS treatment group (LPS group) (n=5), and an LPS plus MMI-0100 group (n=5). The mouse experiments were repeated three times. To induce ARDS, mice in the LPS group received LPS (5 mg/kg; LPS from Escherichia coli O111:B4; Sigma-Aldrich Co., St Louis, MO, USA) diluted in PBS through intraperitoneal injection, whereas the LPS plus MMI-0100 group of mice received LPS injection and MMI-0100 (50 μg/kg) at 0 hour and then received MMI-0100 (50 μg/kg) after 6 hours, 12 hours, and 18 hours. Animals in the control group were given PBS intraperitoneally without LPS. After administration, all mice were monitored four times a day (at 6, 12, 18, and 24 hours) to observe their behavior. All of the mice were killed by sharp cervical dislocation after 24 hours.
Histopathologic study
At 24 hours after administration of LPS or PBS, the mice were killed, and then the upper lobe of the right lung of each mouse was harvested and fixed with 10% buffered formalin for 24 hours, embedded in paraffin, and then sectioned. Lung tissues were observed and recorded by pathologists in a blinded fashion under a light microscope after being stained with H&E.
Measurement of mouse lung wet/dry weight ratios (W/Ds)
After being killed, the middle lobe of the right lung of each mouse was retrieved, weighed quickly to record the wet weight, and then dried in an oven at 85°C for 24 hours. The dry weight was recorded. The W/D was determined to quantify lung tissue edema.
Neutrophil count
Lung tissues were perfused with PBS for 10 minutes through the right ventricle to wash out the blood cells in the pulmonary vessels. This was followed by instillation of collagenase (Sigma-Aldrich Co.) diluted in HBSS buffer. Then the tissue was cut into pieces, digested for 30 minutes at 37°C, pulverized, washed with RPMI 1640 medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) using a strainer, resuspended in 35% Percoll (Sigma-Aldrich Co.), centrifuged at 4°C (800 g) for 15 minutes, fixed in Red Blood Cell Lysis Buffer (Beyotime Institute of Biotechnology, Jiangsu, China) for 3 minutes, and centrifuged at 4°C (800 g) for 8 minutes to remove red blood cells. Then, the cells were resuspended again and stained with CD11b+-labeled and Ly6G+-labeled antibodies (eBioscience, Thermo Fisher Scientific) for 30 minutes at 25°C. The number of neutrophils was determined using a flow cytometer counting cells stained with the antibodies CD11b+ and Ly6G+.
Collection of murine PMVECs
After being killed, the lung tissues were incubated with collagenase (Sigma-Aldrich Co.) diluted in 1× HBSS buffer, cut into pieces, digested for 30 minutes at 37°C, and then they were pulverized and washed with RPMI 1640 through a strainer. Samples were resuspended in 35% Percoll (Sigma-Aldrich Co.), centrifuged at 4°C (800 g) for 15 minutes, fixed in Red Blood Cell Lysis Buffer (Beyotime Institute of Biotechnology) for 5 minutes, and centrifuged at 4°C (800 g) for 8 minutes to remove red blood cells. Then, the cells were resuspended again and stained with CD45-PE-labeled (BD Biosciences, San Jose, CA, USA) and CD31-APC-labeled antibodies (Miltenyi Biotec Technology, Bergisch Gladbach, Germany) for 30 minutes at 25°C. The mouse lung microvascular endothelial cells were collected with CD45− and CD31+ immunomagnetic beads for positive selection.
mRNAs of ICAM-1 by qRT-PCR
RNA was isolated from mouse PMVECs using TRIzol (Beyotime Institute of Biotechnology). RNA was eluted in RNase-free water and stored at −80°C. Total RNA was reverse transcribed to cDNA using a PrimeScript™ RT Master Mix (Perfect Real Time) cDNA synthesis kit (Takara, Dalian, China), according to the manufacturer’s protocol. The qPCR was performed on a StepOne Plus system (Life Technologies, Thermo Fisher Scientific) using SYBR Premix Ex Taq (Takara). Amplification conditions were: 95°C for 30 seconds, 40 cycles of 95°C for 5 seconds, and 60°C for 31 seconds. Melting curve analyses were used to confirm the specificity of the primers. Relative expression values were normalized using an internal GAPDH control. The primers used for the amplifications are shown in Table 1.
 | Table 1 Primer sequences for qRT-PCR |
Western blot analysis
The total proteins and cytoplasmic and nuclear proteins were extracted from mouse PMVECs using Total and Cytoplasmic Protein Extraction Kits (Beyotime Institute of Biotechnology) according to the manufacturer’s protocol. Then, equal amounts of protein were loaded in each well on SDS-polyacrylamide gels (12%) and transferred onto polyvinylidene fluoride membranes. The membranes were subjected to blocking with 5% skim milk (Sigma-Aldrich Co.) at room temperature for 2 hours and incubated overnight at 4°C with rabbit polyclonal antibodies specific for MK2 (Cell Signaling Technology, Danvers, MA, USA) (1:1,000 dilution), phosphorylated MK2 (p-MK2) (Cell Signaling Technology) (1:1,000 dilution), ICAM-1 (Santa Cruz Biotechnology Inc., Dallas, TX, USA) (1:400 dilution), and β-actin (1:3,000) (Santa Cruz Biotechnology Inc.). Then, the membranes were incubated with relevant peroxidase-conjugated secondary antibodies (Bioss, Beijing, China) (1:10,000) at 20°C for 2 hours and washed with TBS-T four times. Immunoreactive proteins were detected using an enhanced chemiluminescence Western blot kit (Beyotime Institute of Biotechnology) following the manufacturer’s instructions. Bands were imaged by exposing the film to light.
Immunohistochemical study
To evaluate the effect of MMI-0100 on the expression of p-MK2 and ICAM-1, the lung tissues were collected and fixed with 10% formalin. Immunohistochemistry was performed following the manufacturer’s protocol.
Statistical analysis
Values are denoted as mean ± SEM of at least three independent experiments. Statistical analysis was performed using SPSS 19.0 (IBM Corporation, Armonk, NY, USA). ANOVA and independent Student’s t-tests were used to analyze the differences between the different time-points within the same treatment group and among different groups. In this study, a probability value of <0.05 was considered to be statistically significant.
Results
Administration of MMI-0100 ameliorated LPS-induced ARDS
Six hours after the injection of LPS, mice started to exhibit crawling or prostration, they were inactive, and they often clustered together. These behaviors were most apparent at 24 hours. The LPS plus MMI-0100 group of mice showed less crawling or prostration than the LPS group of mice. The PBS mice did not show these behaviors. Photomicrographs revealed severe histopathological changes in the LPS group (Figure 1A, LPS group), such as interstitial hyperemia and edema, alveolar collapse, and infiltration of inflammatory cells. In the control mice, no abnormal histopathological lesions were observed (Figure 1A, control group). The LPS plus MMI-0100 group had less alveolar edema and alveolar wall thickness than the LPS group (Figure 1A, LPS plus MMI-0100 group). To explore whether MMI-0100 induces any histopathological changes in lung tissue, we administered mice with PBS at 0 hour and MMI-0100 (50 μg/kg) at 0 hour, 6 hours, 12 hours, and 18 hours. We found that 24 hours later, lung H&E staining showed that just like the PBS group of mice, the PBS plus MMI-0100 mice had no abnormal histopathological lesions (Figure 1A, PBS plus MMI-0100 group). The W/D of the LPS group was significantly higher than in the other groups (Figure 1B, P<0.05). To further compare the inflammatory condition among the three groups, we measured neutrophil counts in the lung homogenates. The flow cytometry analysis revealed that the number of neutrophils in lung homogenates was higher in the LPS group than the other groups (Figure 1C and D, P<0.05).
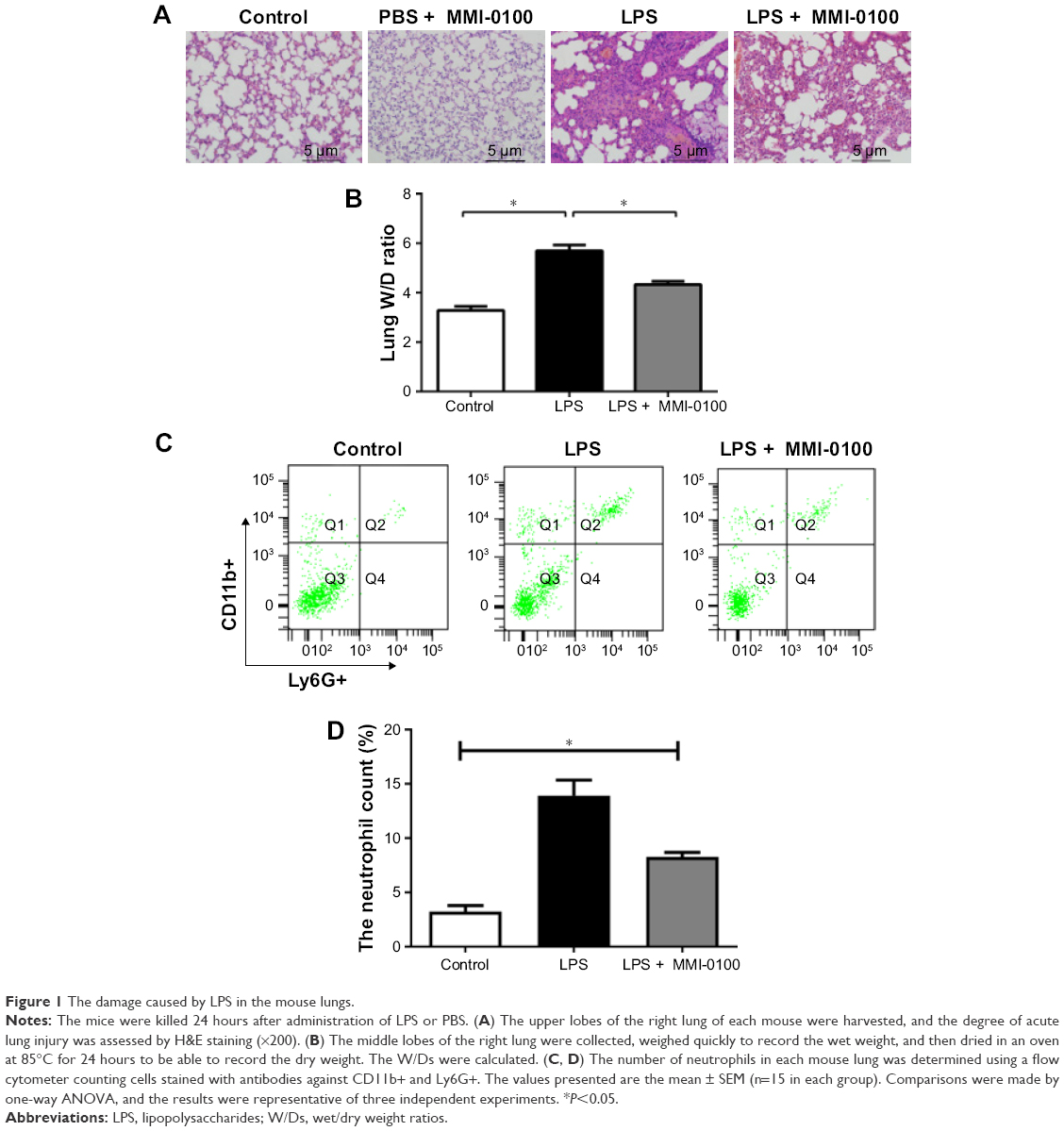 | Figure 1 The damage caused by LPS in the mouse lungs. |
Administration of MMI-0100 reduced the expression of ICAM-1 on the membranes of PMVECs in mice with LPS-induced ARDS
Previous research has shown that endothelial cells play a central role in inflammatory conditions. In addition, several studies have also shown that LPS mediates pulmonary inflammatory responses partly by increasing the expression of ICAM-1 on the surface of endothelial cells, promoting the migration and adhesion of leukocytes.5 To further investigate the importance of mouse PMVECs in ARDS, these cells were collected by immunomagnetic separation. Compared with the corresponding levels in the control group, the mRNA and protein expression levels of ICAM-1 in the LPS group increased significantly (Figure 2A–C, P<0.05) in mouse PMVECs. To investigate whether the reduction of lung neutrophil infiltration in mice with LPS-induced ARDS after administration of MMI-0100 was due to the reduction of ICAM-1 on the membrane of PMVECs in these mice, ICAM-1 mRNA and protein levels were measured. Compared with LPS group, the mRNA and protein expression of ICAM-1 in the LPS plus MMI-0100 group decreased significantly (Figure 2A–C, P<0.05) in mouse PMVECs. Similarly, the flow cytometry with the antibody CD54+ showed the same trend (Figure 3, P<0.05).
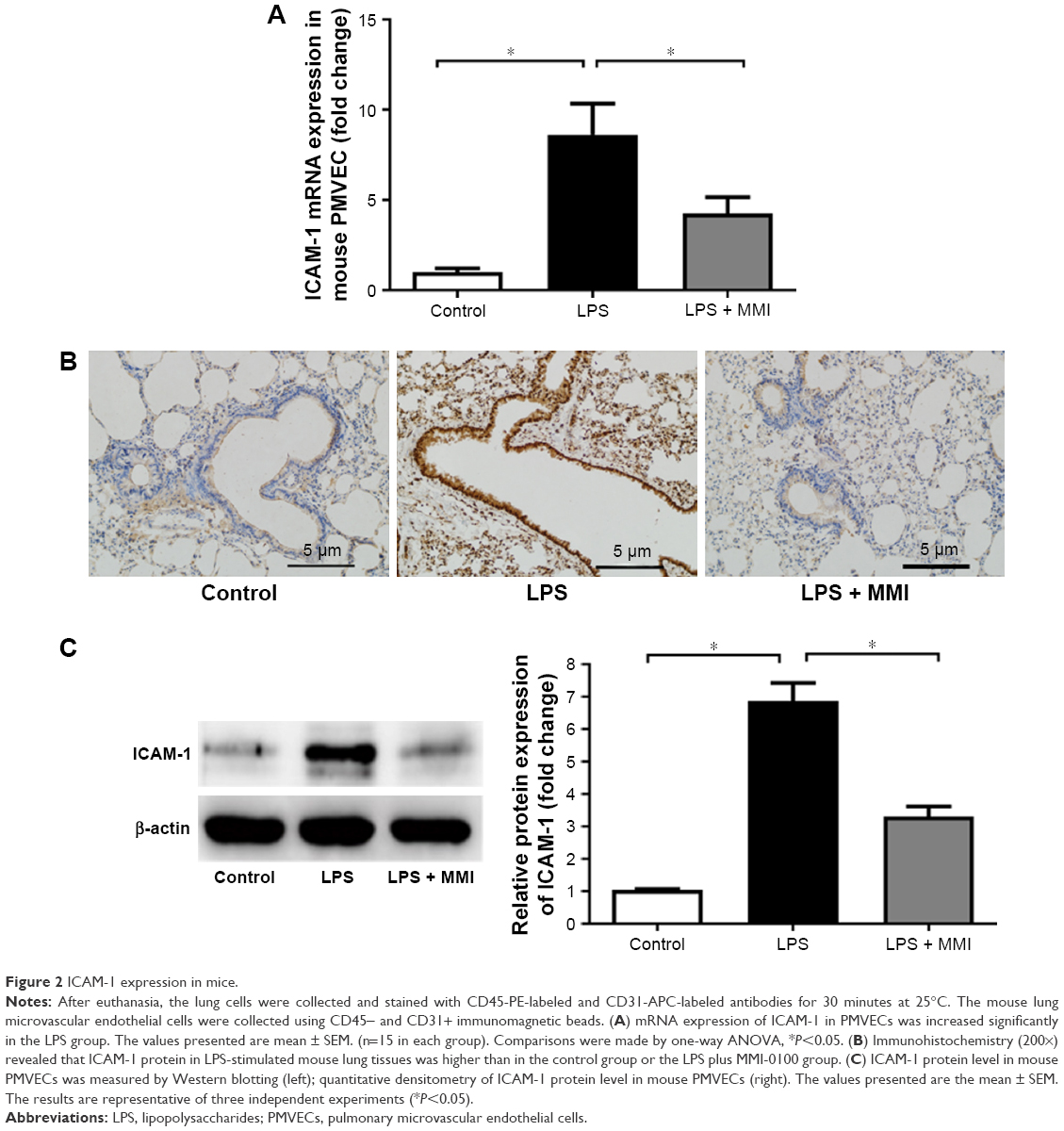 | Figure 2 ICAM-1 expression in mice. |
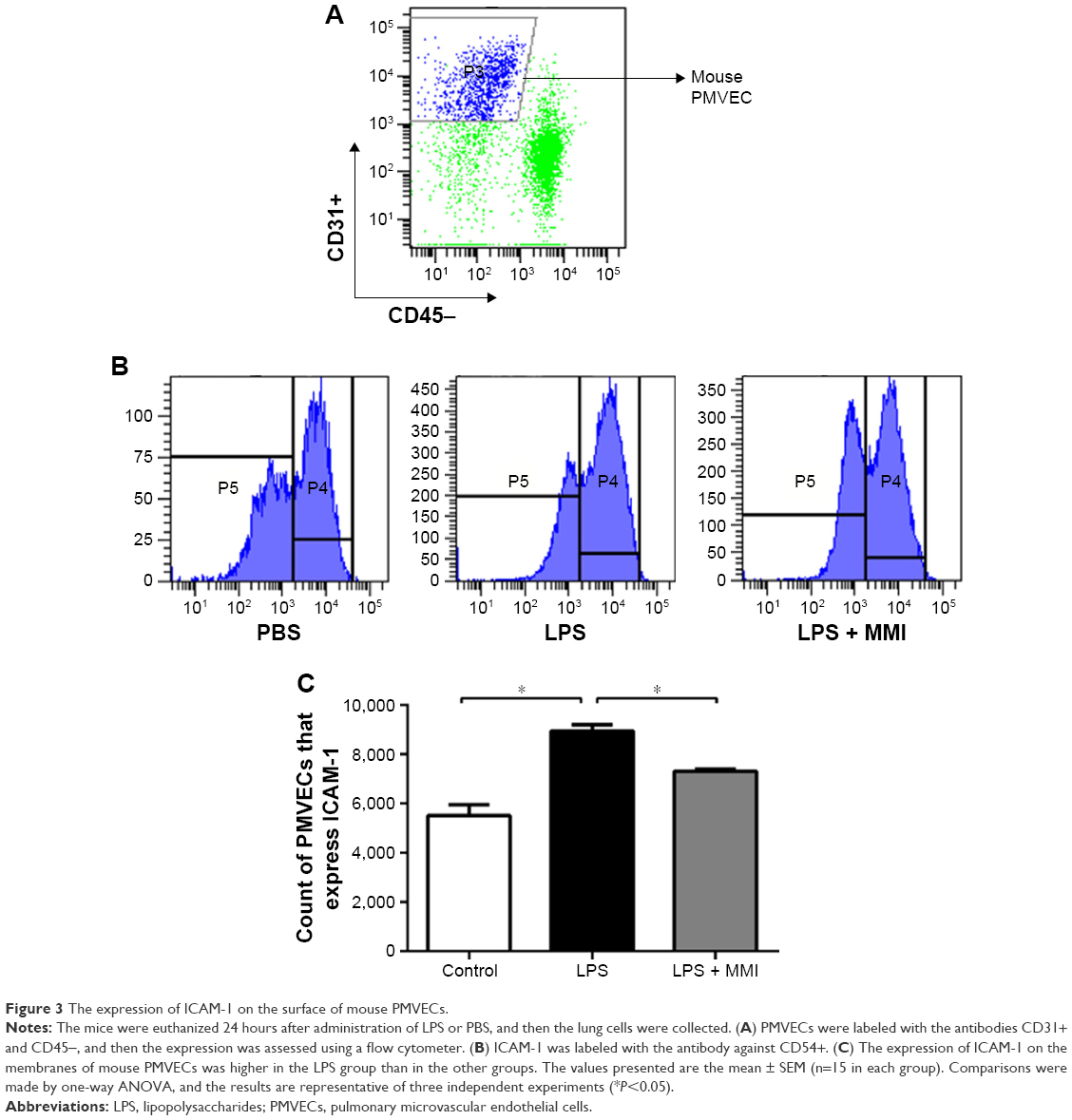 | Figure 3 The expression of ICAM-1 on the surface of mouse PMVECs. |
Administration of MMI-0100 reduced the expression of p-MK2 in mice with LPS-induced ARDS
To examine the effect of MMI-0100, we measured the total MK2 (T-MK2) and p-MK2 levels in mouse PMVECs. The protein expression level of p-MK2/T-MK2 was higher in the LPS group than in the control group and LPS plus MMI-0100 group in mouse PMVECs (Figure 4). Immunohistochemistry revealed that p-MK2 (Figure 5) in LPS group was higher than that of the other two groups.
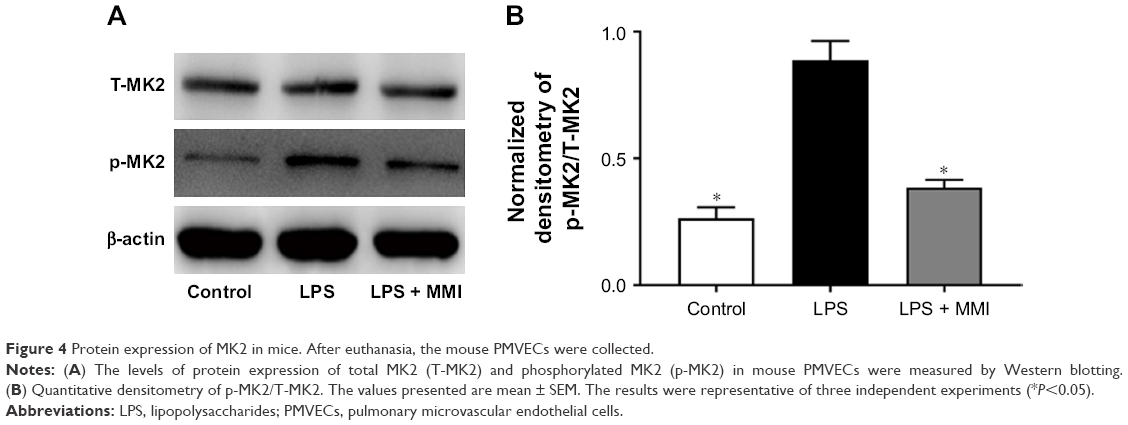 | Figure 4 Protein expression of MK2 in mice. After euthanasia, the mouse PMVECs were collected. |
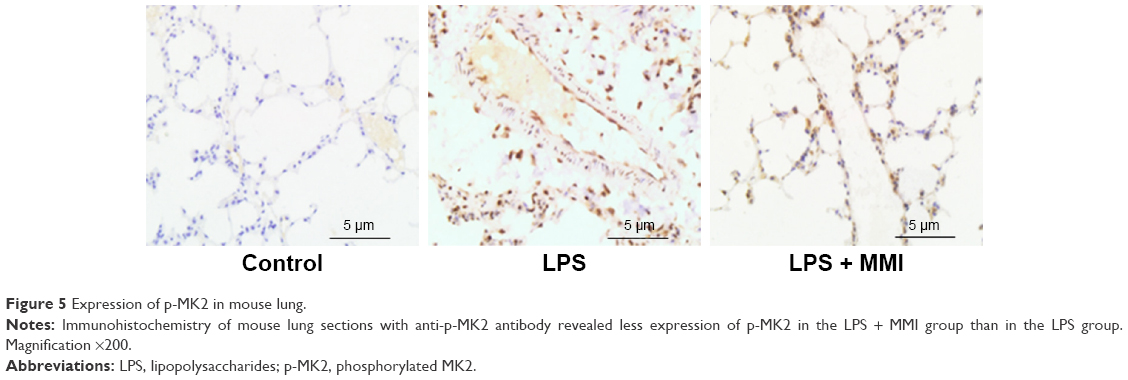 | Figure 5 Expression of p-MK2 in mouse lung. |
Discussion
The endothelial cell barrier plays a major role in ARDS.13 ICAM-1 is known to play a critical role in ARDS by mediating the neutrophil influx into the lung and exacerbating inflammation.3 Consistent with this, our study showed that in mice with ARDS, ICAM-1 is highly expressed in mouse PMVECs.
MK2 can regulate the expression of ICAM-1 and IL-8 post-transcriptionally without influencing NF-κB activation or ICAM-1 and IL-8 mRNA levels in HPMECs.9 We hypothesized that MK2 has a very important and powerful influence on ICAM-1 expression and this regulation may finally affect the core process of ARDS inflammation in vivo. Our results support these hypotheses. Our results showed that after inhibiting the activation of MK2, the expression of ICAM-1 on the membrane of PMVECs in mice with ARDS was reduced. The mRNA and protein expression level of ICAM-1 in mouse PMVECs was reduced as well. The LPS plus MMI-0100 group of mice exhibited less severe inflammation, with a lower W/D and neutrophil count.
MMI-0100, known as an MK2 inhibitor, is a synthetic compound that can inhibit the activation of MK2 rather than the expression of MK2,10 which can lead to a reduction in the expression of inflammatory cytokines, such as IL-6 and TNF-α. Another study has shown that inhibition of MK2 reduced abdominal adhesion proteins.14 These observations verified that inhibition of MK2 results in the suppression of endothelial ICAM-1 expression.
Our study demonstrates, for the first time, that the MK2 inhibitor MMI-0100 was able to regulate ICAM-1 expression in mouse PMVECs and ameliorated lung inflammation in the ARDS mouse model (Figure 6). A limitation of our study is that the mechanism by which MK2 regulates ICAM-1 has not been demonstrated completely. Thus, further studies are needed to detect the mechanism(s) by which MK2 regulates ICAM-1 in cell lines and in animal models.
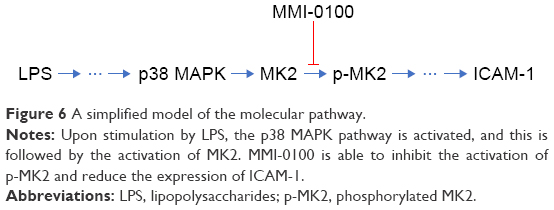 | Figure 6 A simplified model of the molecular pathway. |
Conclusion
Our study showed that MMI-0100 ameliorates lung inflammation in mice with ARDS, possibly by causing a reduction in endothelial expression of ICAM-1. This suggests that MK2 may be a therapeutic target in ARDS patients. MMI-0100 is likely to be particularly efficacious for the treatment of ARDS.
Acknowledgments
We would like to thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript. This study was supported by the National Natural Science Foundation of China (grant no 81670073) and the Natural Science Foundation of Jiangsu Province (BK20161383).
Author contributions
XS designed the experiments. BH, SG, WZ, and YR performed experiments; SG and BH analyzed data; XM and CZ prepared figures; SG, BH, and XS drafted the manuscript; WZ, YR, and XS edited and revised the manuscript; XM, CZ, and XS interpreted the results of the experiments; XS and QY conceived and designed the research project, and XS approved the final version of the manuscript. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Rubenfeld GD, Caldwell E, Peabody E, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353(16):1685–1693. | ||
Chen W, Chen YY, Tsai CF, et al. Incidence and outcomes of acute respiratory distress syndrome: a nationwide registry-based study in Taiwan, 1997 to 2011. Medicine. 2015;94(43):e1849. | ||
Montefort S, Roche WR, Howarth PH, et al. Intercellular adhesion molecule-1 (ICAM-1) and endothelial leucocyte adhesion molecule-1 (ELAM-1) expression in the bronchial mucosa of normal and asthmatic subjects. Eur Respir J. 1992;5(7):815–823. | ||
Sumagin R, Lomakina E, Sarelius IH. Leukocyte-endothelial cell interactions are linked to vascular permeability via ICAM-1-mediated signaling. Am J Physiol Heart Circ Physiol. 2008;295(3):H969–H977. | ||
Bird MD, Morgan MO, Ramirez L, Yong S, Kovacs EJ. Decreased pulmonary inflammation after ethanol exposure and burn injury in intercellular adhesion molecule-1 knockout mice. J Burn Care Res. 2010;31(4):652–660. | ||
Sousa A, Raposo F, Fonseca S, et al. Measurement of cytokines and adhesion molecules in the first 72 hours after severe trauma: association with severity and outcome. Dis Markers. 2015;2015:1–8. | ||
Brook M, Tchen CR, Santalucia T, et al. Posttranslational regulation of tristetraprolin subcellular localization and protein stability by p38 mitogen-activated protein kinase and extracellular signal-regulated kinase pathways. Mol Cell Biol. 2006;26(6):2408–2418. | ||
Ronkina N, Kotlyarov A, Dittrich-Breiholz O, et al. The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol Cell Biol. 2007;27(1):170–181. | ||
Su X, Ao L, Zou N, et al. Post-transcriptional regulation of TNF-induced expression of ICAM-1 and IL-8 in human lung microvascular endothelial cells: an obligatory role for the p38 MAPK-MK2 pathway dissociated with HSP27. Biochim Biophys Acta. 2008;1783(9):1623–1631. | ||
Vittal R, Fisher A, Gu H, et al. Peptide-mediated inhibition of mitogen-activated protein kinase-activated protein kinase-2 ameliorates bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2013;49(1):47–57. | ||
Shi JX, Su X, Xu J, Zhang WY, Shi Y. MK2 posttranscriptionally regulates TNF-α-induced expression of ICAM-1 and IL-8 via tristetraprolin in human pulmonary microvascular endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2012;302(8):L793–L799. | ||
Wu T, Shi JX, Geng S, Zhou W, Shi Y, Su X. The MK2/HuR signaling pathway regulates TNF-α-induced ICAM-1 expression by promoting the stabilization of ICAM-1 mRNA. BMC Pulm Med. 2016;16(1):84. | ||
Teijaro JR, Walsh KB, Cahalan S, et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011;146(6):980–991. | ||
Ward BC, Kavalukas S, Brugnano J, Barbul A, Panitch A. Peptide inhibitors of MK2 show promise for inhibition of abdominal adhesions. J Surg Res. 2011;169(1):e27–e36. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.