")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Mixed nanomicelles as potential carriers for systemic delivery of Z-GP-Dox, an FAPα-based doxorubicin prodrug: formulation and pharmacokinetic evaluation
Authors Zhang Y, Zhang X, Lui H, Cai S, Wu B, Wang H
Received 17 October 2014
Accepted for publication 7 December 2014
Published 26 February 2015 Volume 2015:10(1) Pages 1625—1636
DOI https://doi.org/10.2147/IJN.S75954
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Yuchen Zhang,1,* Xingwang Zhang,2,* Hongming Liu,2 Shaohui Cai,1 Baojian Wu2
1Department of Pharmacology, 2Division of Pharmaceutics, College of Pharmacy, Jinan University, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Abstract: Z-GP-Dox, the FAPα (fibroblast activation protein-α)-based doxorubicin prodrug, demonstrates excellent tumor targeting effects and a favorable toxicokinetic profile. However, the insoluble nature of Z-GP-Dox becomes a significant barrier to drug administration, particularly when it comes to the clinical stage. Here we developed a nanomicelle system to facilitate the systemic delivery of Z-GP-Dox, and evaluated its disposition in rats following administration of the micelles using a physiologically-based pharmacokinetic model. Z-GP-Dox-loaded mixed nanomicelles (ZGD-MNs) were prepared by dispersion of an ethanol solution of Z-GP-Dox, lecithin, and sodium oleate in water. The obtained ZGD-MNs were 86.6 nm in size with a drug loading of 14.03%. ZGD-MNs were fairly stable in phosphate-buffered saline and simultaneously showed satisfactory physical and chemical stability over a 2-week observation period. Accumulative drug release was more than 56% within 24 hours. Further, the physiologically-based pharmacokinetic rat model consisting of various organs (ie, heart, liver, spleen, lung, kidney, and intestine) was fitted to the experimental data following administration of ZGD-loaded cosolvent (control) or micelles. Derived partition coefficient values revealed that the nanomicelles significantly altered the biodistribution of Z-GP-Dox. Of note, drug distribution to the lung, liver, and spleen was greatly enhanced and the fold change ranged from 2.4 to 33. In conclusion, this is the first report of a mixed micelle system being a viable carrier for delivery of Z-GP-Dox. Also, the pharmacokinetic behavior of Z-GP-Dox was satisfactorily described by the physiologically-based pharmacokinetic model.
Keywords: doxorubicin, Z-GP-Dox, micelles, systemic delivery, pharmacokinetics, physiologically-based pharmacokinetic model
Introduction
Fibroblast activation protein-α (FAPα) is a membrane prolyl-specific proteinase. The FAPα enzyme is of great interest in prodrug design for tumor targeting due to its selective and high expression in reactive stromal fibroblasts of epithelial cancers.1 Z-GP-Dox (Figure 1A) is an FAPα-based doxorubicin prodrug that selectively releases doxorubicin in tumors, showing enhanced anticancer effects and an improved toxicity profile.2 However, the insoluble nature of Z-GP-Dox becomes a significant barrier to drug administration, particularly when it comes to clinical trials. Therefore, it would be of great value to solve the solubility problem and achieve systemic delivery of this promising anticancer prodrug.
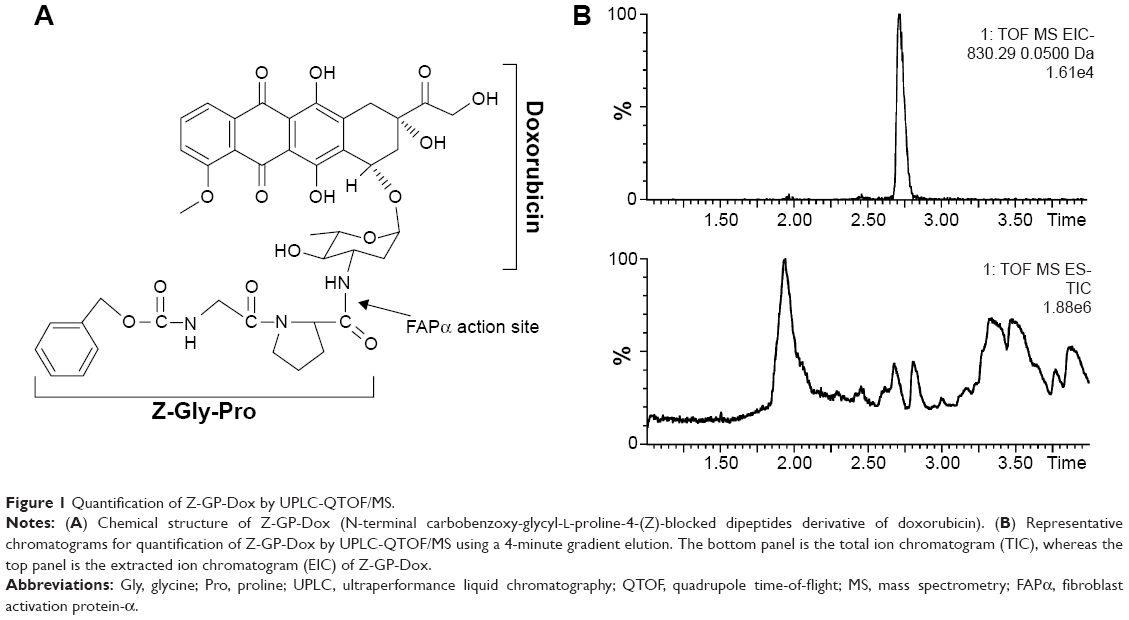 | Figure 1 Quantification of Z-GP-Dox by UPLC-QTOF/MS. |
A variety of nanocarrier systems have shown great potential in drug solubilization, including liposomes,3 microemulsions,4 nanoparticles,5 and micelles.6 Of these, micelles are becoming a powerful tool for systemic delivery of insoluble drugs.7–11 Micelles are colloidal nanoparticulates self-assembled from amphiphilic molecules with a hydrophobic core inside and a hydrophilic shell outside.12 The hydrophobic core serves as a drug reservoir, whereas the hydrophilic shell provides a hydrodynamically stable surface that can be modified for targeting effects. Due to safety concerns, lecithin is the material most frequently used to fabricate the micelles for intravenous delivery. Further, a combination of lecithin and a different type of surfactant (eg, bile salts) is often utilized in developing a micelle system (so-called “mixed micelles”).7,13,14 This is because lecithin (or a phospholipid) alone is generally unable to form stable fine micelles because of a lower hydrophilic-lipophilic balance value and/or inadequate solubilization power.15
Physiologically-based pharmacokinetic (PBPK) modeling has gained increasing attention in drug research and development.16,17 A PBPK model quantitatively describes drug absorption, distribution, metabolism, and elimination in the body, facilitating a deeper understanding of the effects of these intricate processes on drug exposure and how these processes interact with each other.18,19 An important feature of a PBPK model is that it allows extrapolation of data from a preclinical animal species to humans, thereby providing excellent guidance on clinical trials.20 Although PBPK modeling has become a powerful tool in identifying the factors determining drug pharmacokinetics, its ability to evaluate the effects of the formulation factor on drug disposition has not been fully established.21
In this study, we aimed to develop a mixed micelle system that can enhance the solubility of Z-GP-Dox and facilitate intravenous delivery of the drug. A combination of lecithin and sodium oleate (SO) was used to fabricate mixed nanomicelles using the solvent-diffusion technique. SO is a nonendogenous fatty acid salt that is frequently used in injectable lipid emulsions,22 and has been approved as a pharmaceutical excipient for injection purposes in the People’s Republic of China. The potential of nanomicelles self-assembled from lecithin and SO for systemic delivery of Z-GP-Dox was investigated. Further, whole-body PBPK modeling was used to compare the disposition of Z-GP-Dox in rats following administration of the drug micelles or the cosolvent (control).
Materials and methods
Materials
Z-GP-Dox was synthesized in our laboratory as described elsewhere.2 Soybean lecithin (S100) was obtained from Lipoid KG (Ludwigshafen, Germany). SO was purchased from Sigma-Aldrich (Shanghai, People’s Republic of China). Deionized water was prepared using a water purifier (Chengdu, People’s Republic of China). All other chemicals were of analytical grade and used as received.
Preparation of Z-GP-Dox-loaded mixed nanomicelles
Z-GP-Dox-loaded mixed nanomicelles (ZGD-MNs) were prepared using the solvent-diffusion technique with a slight modification.23 In brief, Z-GP-Dox, lecithin, and SO were dissolved in 90% ethanol and then slowly injected into water using a microsyringe. The materials were spontaneously assembled into nanomicelles upon diffusion of the solvent into the aqueous phase. Residual ethanol was removed under reduced pressure by a rotary evaporator, and the nanosuspension was condensed to an appropriate volume (keeping the Z-GP-Dox concentration at around 3.0 mg/mL). Factors affecting the performance of nanomicelles were investigated, including the amounts of SO, drug, and lecithin, as well as water volume upon injection.
Characterization of ZGD-MNs
The particle size of the ZGD-MNs was determined by dynamic light scattering using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) at 25°C. To measure particle size, 1 mL of the sample was transferred into a disposable cuvette and then subjected to laser diffraction after equilibration for 120 seconds. The mean particle size was calculated using the built-in software.
Transmission electron microscopy was used to observe the morphology of the ZGD-MNs. In brief, an aliquot of ZGD-MNs was dropped onto a carbon-coated copper grid and dried using a heat lamp. The anchored nanoparticles were inspected by transmission electron microscopy (Tecnai 10, Philips, Eindhoven, the Netherlands) and photographed at an acceleration voltage of 100 kV.
Z-GP-Dox is almost insoluble in water (less than 25 μg/mL), so the amount of free drug in the micelle system is negligible relative to the micellar drug. Therefore, the drug load parameter is more indicative for evaluation of the formulation than encapsulation efficiency. The drug load of the ZGD-MNs was determined by separating nonentrapped and free Z-GP-Dox from the micelle system. Briefly, the micelle system was centrifuged at 8,000 g for 3 minutes to remove nonentrapped bulk drug. The resulting ZGD-MNs were then subjected to centrifugal filtration to further remove free drug using a centrifugal filter device (Amicon® Ultra-0.5, molecular weight cut-off 50,000, Millipore, Billerica, MA, USA). The concentration of Z-GP-Dox entrapped in the mixed micelles was quantified by ultraperformance liquid chromatography/quadrupole time-of-flight mass spectrometry (UPLC-QTOF/MS). The drug load was estimated according to the following equation:
|
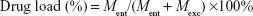
where Ment is the amount of Z-GP-Dox entrapped in the micelles quantified on the basis of the concentration of micellar Z-GP-Dox, calculated from the difference of initial concentration of drug × initial volume of micelle − free concentration of drug × volume of filtrate; Mexc denotes the amount of all excipients used in the micelles quantified based on weighing.
In vitro release study
Drug release from the mixed micelles was performed using a dialysis method. In brief, 2 mL of ZGD-MNs was dialyzed at 37°C against 900 mL of phosphate-buffered saline (pH 7.4) medium containing 0.1% (w/v) sodium dodecyl sulfate as a solubilizer. At predetermined time points, 5 mL of the sample was withdrawn and immediately replaced with the same volume of fresh medium. Z-GP-Dox concentrations were determined by UPLC-QTOF/MS and the percentage of drug release was calculated as the mean ± standard deviation (n=3). The release study was terminated at 24 hours based on assessment on the pharmacokinetic curve previously performed in our laboratory.
Stability studies
For evaluation of stability, the nanomicelle suspensions were stored at room temperature (25°C±2°C) for 2 weeks and the changes in particle size and drug content with time were determined. Moreover, the dilution stability of ZGD-MNs in the blood was simulated in vitro using a physiological buffer solution (0.14 M NaCl, 20 mM phosphates, pH 7.2).24 Typically, 5 mL of ZGD-MNs was added into 500 mL of phosphate-buffered saline. The particle size was monitored real-timely upon dilution against the medium. The stability of the ZGD-MNs was gleaned from particle size, polydispersity index, and the percentage of drug remaining.
Pharmacokinetic study of Z-GP-Dox
The pharmacokinetic study was performed in male Sprague-Dawley rats weighing 180–220 g. All animal experiments were conducted following the Guidelines on the Care and Use of Animals for Scientific Purposes (2004, Singapore) and the protocol was approved by the experimental animal ethical committee of Jinan University (Guangzhou, People’s Republic of China). The rats were randomly divided into two groups (n=5), ie, a control (cosolvent) group and a micelle group. The control group received the Z-GP-Dox cosolvent (45% propylene glycerol) by intravenous injection at a dose of 10 mg/kg body weight, whereas the micelle group received ZGD-MNs at the same dose. At predetermined time points (5, 15, and 30 minutes, and 1, 2, 4, 6, and 8 hours) after drug administration, the rats were rendered unconscious for blood and tissue sampling by injection of phenobarbital. Blood was collected by cardiac puncture. After washout of blood with ice-cold saline, the heart, liver, spleen, lung, kidney, and intestine were rapidly removed, weighed, and stored at −80°C for further analysis.
The blood samples were processed as described previously.25 The concentrations of Z-GP-Dox in these samples were analyzed by UPLC-QTOF/MS. The heart, liver, spleen, lung, kidney, and intestinal tissues were homogenized in saline solution (1:2, w/v). Next, 0.2 mL of tissue homogenate was mixed with 1 mL of acetonitrile containing 2 μM internal standard (SNX-2112). The mixture was then vortexed and centrifuged at 13,000 g for 15 minutes (4°C). The supernatant was collected and dried using an Eppendorf Concentrator Plus (Hamburg, Germany). The residue was reconstituted in 250 μL of water/methanol (50:50, v/v) and centrifuged at 13,000 g for 15 minutes (4°C). A 3 μL aliquot of the supernatant was injected into the UPLC-QTOF/MS system.
Quantification of Z-GP-Dox
Quantification of Z-GP-Dox was performed using UPLC-QTOF/MS. Our instrument configuration and parameter settings have been described in a previous publication.26 In brief, chromatographic separation was performed using an Acquity UPLC BEH column (2.1×50 mm, 1.7 μm; Waters, Milford, MA, USA). A gradient elution was applied using formic acid (0.1%) in water (mobile phase A) versus formic acid (0.1%) in acetonitrile (mobile phase B) at a flow rate of 0.45 mL/min. The gradient elution program was 5% B at 0–1 minutes, 5%–95% B at 1–2 minutes, 95% B at 2–3.5 minutes, and 95%–5% B at 3.5–4 minutes. Quantitation was performed based on the full scan analysis and extracted ion chromatograms using MassLynx version 4.1 as described elsewhere.25,26 A typical chromatogram for analysis of Z-GP-Dox is shown with the drug molecule eluted at 2.73 minutes in Figure 1B.
PBPK modeling and fitting
A whole-body PBPK model consisting of heart, liver, spleen, lung, kidney, intestine, and remaining tissue was constructed to describe Z-GP-Dox disposition in rats following administration of the cosolvent or micelles (Figure 2A). Elimination of Z-GP-Dox primarily occurred via the kidney and liver, which are represented by CLR and CLH, respectively. CLH is the sum of metabolic and biliary clearances. Drug concentration in each tissue was determined by the tissue to plasma partition coefficient (Kpti), plasma flow (Qti), and organ volume (Vti). A single-tissue model and the derived function are illustrated in Figure 2B. The plasma flow and organ volume values used for model fitting were taken from the literature.27 The plasma cardiac output (Qco) for rats was calculated as 2.69 L/hour. The mass balance equations (1–8) for the model are shown as follows:
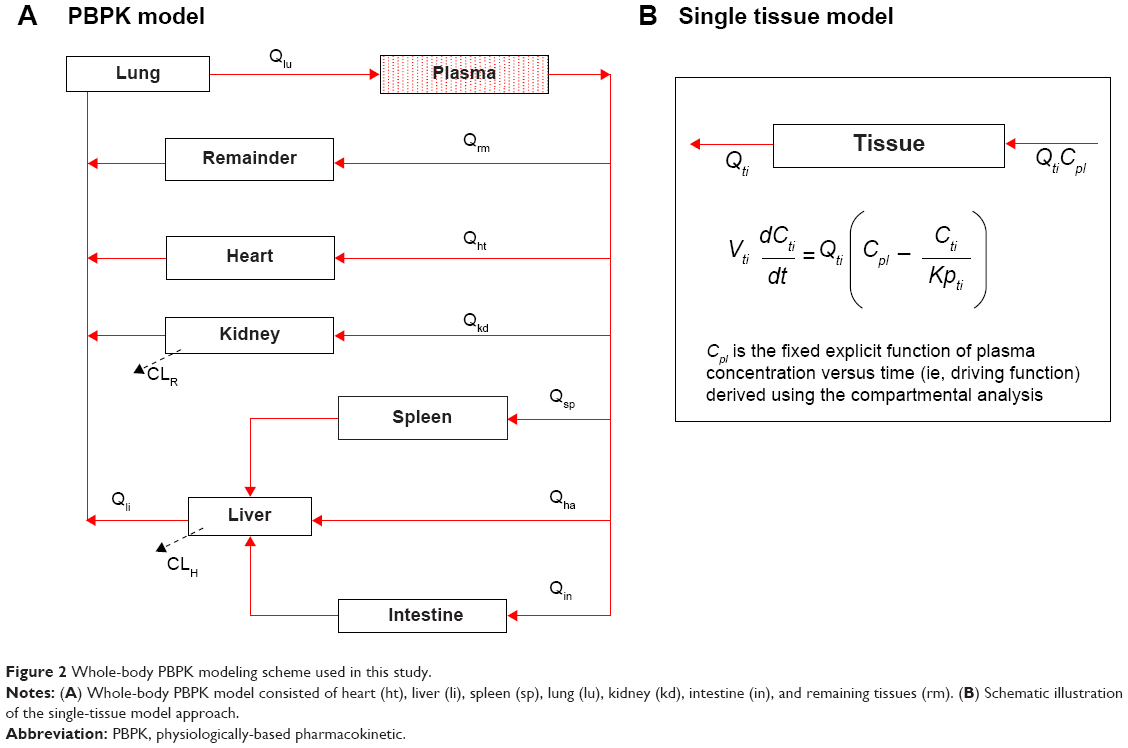 | Figure 2 Whole-body PBPK modeling scheme used in this study. |
| (1) |
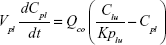
| (2) |
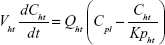
| (3) |
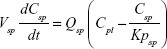
| (4) |
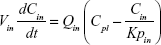
| (5) |
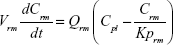
| (6) |
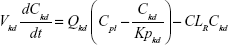
|
|
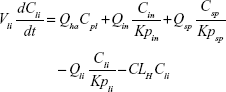
|
|
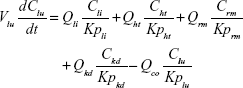
The partition coefficient values (Kp) and the elimination clearance (CLR and CLH) values were obtained by simultaneously fitting the PBPK model to experimental data for plasma/tissue concentrations versus time. PBPK modeling and fitting was performed with MatLab® software (Mathsworks Inc, Natick, MA, USA) using a weighting scheme of 1/y2. Compartmental analysis of the plasma data was performed using WinNonlin® software (Pharmsight, Sunnyvale, CA, USA).
Statistical analysis
The data are expressed as the mean ± standard deviation. The two-tailed Student’s t-test was used to compare the parameter difference between the cosolvent (control) group and the mixed micelle group. The level of statistical significance was set at 5% (P<0.05).
Results and discussion
Preparation and characterization of ZGD-MNs
The solvent diffusion technique was used to prepare ZGD-MNs for this study. The preparation methods were relatively straightforward. All ingredients (ie, drug, lecithin, and SO) were dissolved in 90% ethanol and then slowly injected into water. Factors that may affect the particle size of the ZGD-MNs include the amounts of drug, lecithin, and SO, as well as the volume of water. Various ZGD-MNs were prepared by varying the level of one factor at a time, while keeping the others unchanged (Figure 3). SO had significant effects on the formation of micelles and the drug load (Figure 3A). Less SO (5 mg) resulted in large micelles and a low drug load. Equally, a higher SO content was unfavorable for production of fine micelles. Z-GP-Dox could be solubilized by SO at a suitable level. However, more SO used in the formulation might increase the membrane toughness of the micelles, resulting in development of larger micelles. This can be inferred from the drug load that the increase of SO amount posed less effect on drug load enhancement. In the same way, lecithin affected the performance of micelles that a middle proportion of lecithin would generate micelles having a smaller particle size (Figure 3B). It was intimated that there was a synergistic effect between SO and lecithin in the formation of micelles and drug loading. The effect of drug amount on the formulation was also significant (Figure 3C). The drug load was enhanced with an increase in drug amount below 12 mg, but the particle size of the micelles showed less change. However, the trends went to the opposite direction that the particle size was significantly raised and the drug load declined at a high drug level, indicating a limited drug load capacity provided by the micelle system. The water volume in the organic phase versus aqueous phase upon injection was another factor affecting the properties of the micelles (Figure 3D). A low water-organic phase ratio was not conducive to spontaneous formation of micelles due to the space limitation. A water volume above 6 mL (to 1 mL ethanol solute) was appropriate for production of Z-GP-Dox-loaded micelles.
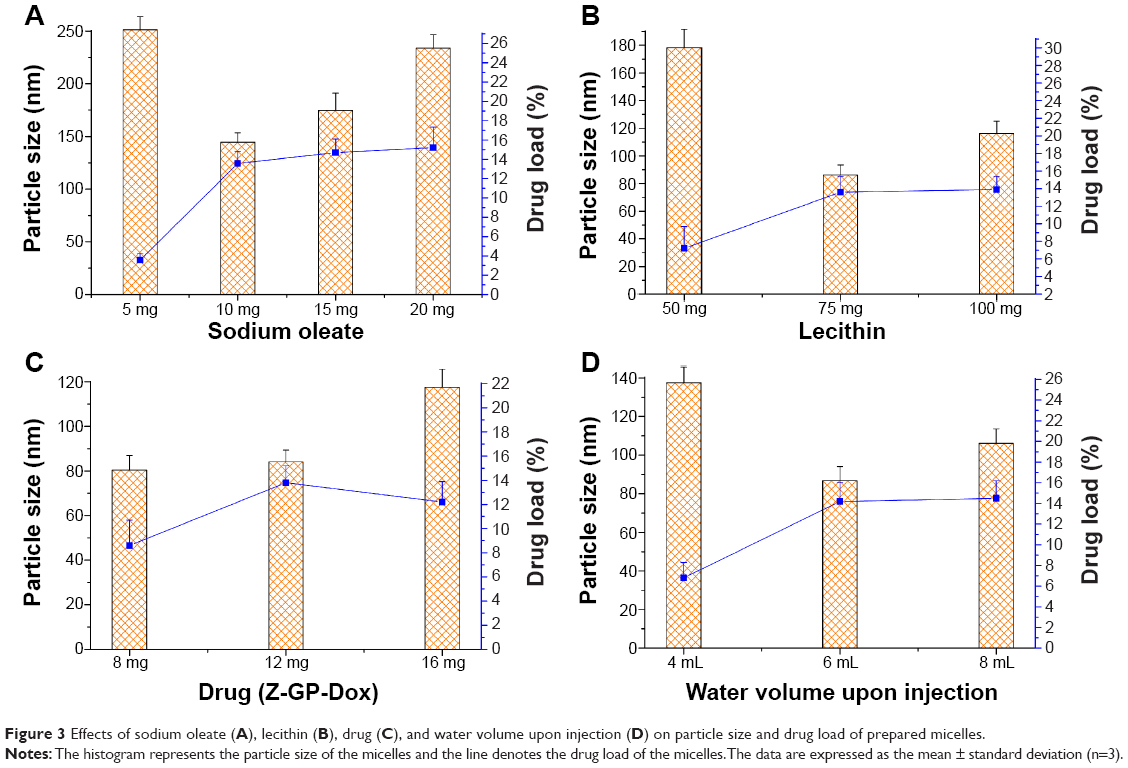 | Figure 3 Effects of sodium oleate (A), lecithin (B), drug (C), and water volume upon injection (D) on particle size and drug load of prepared micelles. |
Based on these results, a typical micelle formulation consisting of 12 mg Z-GP-Dox, 10 mg SO, and 75 mg lecithin was selected to obtain nanoscale micelles (<100 nm). Using the above formula, the obtained ZGD-MNs had a particle size of 86.6 nm with a polydispersity index of 0.269 (Figure 4A). The size at such a level tends to prevent rapid clearance of nanocarriers from the body and reduce nonspecific protein binding and uptake by the reticuloendothelial system.28,29 The zeta potential was determined to be −48.4 mV, a value that is associated with a stable colloidal system. ZGD-MNs showed an acceptable drug load of 14.03%, and the concentration of Z-GP-Dox was 3.0 mg/mL, ie, satisfactory for intravenous delivery. In addition, our prepared MNs were shown to be spherical on transmission electron microscopy (Figure 4B). The particle size estimated from the scale bar (400 nm) was roughly consistent with the hydrodynamic size given by the Nano ZS analyzer.
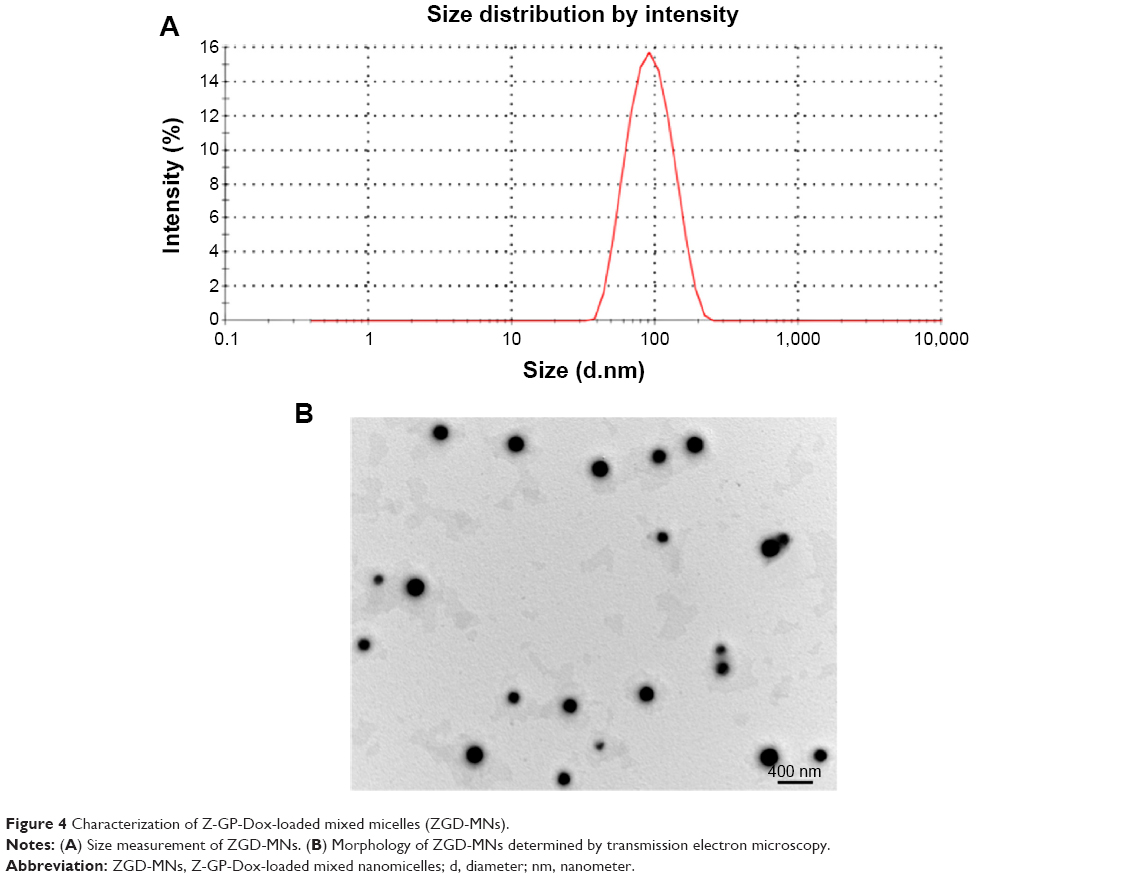 | Figure 4 Characterization of Z-GP-Dox-loaded mixed micelles (ZGD-MNs). |
Drug release
The release profile of Z-GP-Dox from the micelles is shown in Figure 5. The cumulative release increased as a function of time, indicating that drug release into the medium was continuous. This was suggestive of a passive diffusion mechanism for release of Z-GP-Dox. The observed release pattern was consistent with that seen with other micelle systems in the literature.30,31 It was noteworthy that release was fairly slow in the first few hours. With the passage of time, the release was accelerated by degrees. This may be explained by the disassembly of micelles as the result of a dilution effect, since the volume of release medium was far more than that of the micelle sample. The cumulative release of Z-GP-Dox was up to 56.87% within 24 hours, suggesting that Z-GP-Dox could be released from the nanomicelles. The pharmacological function of Z-GP-Dox would not be shielded because of encapsulation into the micelle. From the release kinetics, it can be observed that the major portion of Z-GP-Dox was retained in the micelles in the initial stages. This slow release allowed ZGD-MNs to circulate in the blood in the form of micelles, avoiding drug recrystallization. Meanwhile, the accelerated release seen upon dilution allowed the nanomicelles to exert the pharmacological effects of Z-GP-Dox at the target sites in a timely manner.
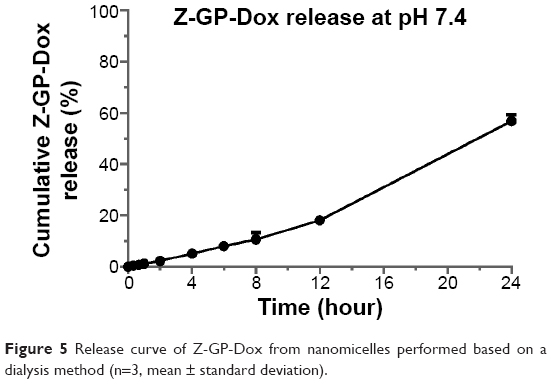 | Figure 5 Release curve of Z-GP-Dox from nanomicelles performed based on a dialysis method (n=3, mean ± standard deviation). |
Stability of ZGD-MNs
Figure 6 shows the changes in particle size and polydispersity index of ZGD-MNs as a function of time. There was no significant difference in particle size between the various time points of storage (Figure 6A) (one-way analysis of variance, P>0.05). Although the polydispersity index fluctuated on a daily basis, the value was between 0.25 and 0.37, demonstrating acceptable physical stability. The Z-GP-Dox content in the micelle suspensions at 14 days was measured to be 99.41%±0.78% of the initial level, indicating satisfactory chemical stability.
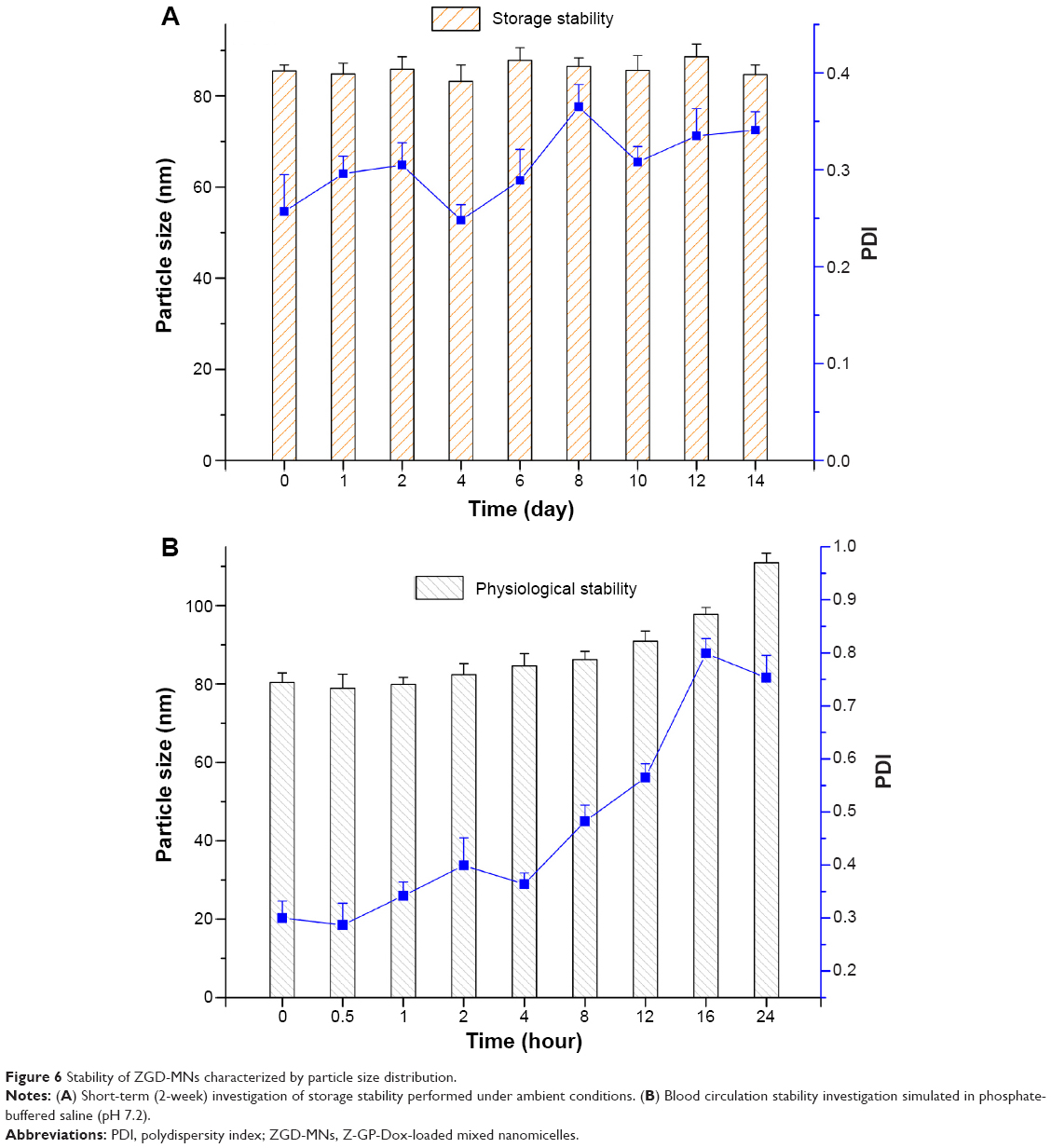 | Figure 6 Stability of ZGD-MNs characterized by particle size distribution. |
The particle size changes of ZGD-MNs upon dilution against an excess of physiological buffer are shown in Figure 6B. The particle size of ZGD-MNs slowly increased with the extension of dilution. The particle size increased by 37.94% (from 80.4 nm to 110.9 nm) after 24 hours. Although the rise in particle size was less rapid, the polydispersity index was significantly broadened with time. This might be ascribable to dissociation of partial micelles due to dilution. From the point of nanoarchitecture being kept, it can be predicted that the nanomicelles would not break up immediately and abruptly release Z-GP-Dox into the blood. Overall, the ZGD-MNs showed satisfactory physical, chemical, and physiological stability.
Pharmacokinetic profiles and tissue distribution of Z-GP-Dox
The plasma concentration-time profiles displayed two distinct phases following intravenous administration of cosolvent (control) or nanomicelles (Figure 7). The data were analyzed using the compartmental approach and found to be well described by the two-compartment model (Figure 7, Table 1). Drug elimination was significantly faster in the nanomicelle group than in the cosolvent group (P<0.05). The elimination half-life for the control group was almost double that for the micelle group (2.97±0.408 hours versus 1.51±0.172 hours, respectively). As already known, the liver and kidney are the main organs responsible for drug elimination. Micelles are colloidal nanoparticles that are easily sequestered into the organs of the reticuloendothelial system,32 ie, the liver, spleen, and lymph. The rapid elimination of micellar Z-GP-Dox could be attributable to sequestration in the liver and its subsequent elimination. It was consistent with the outcomes of tissue distribution.
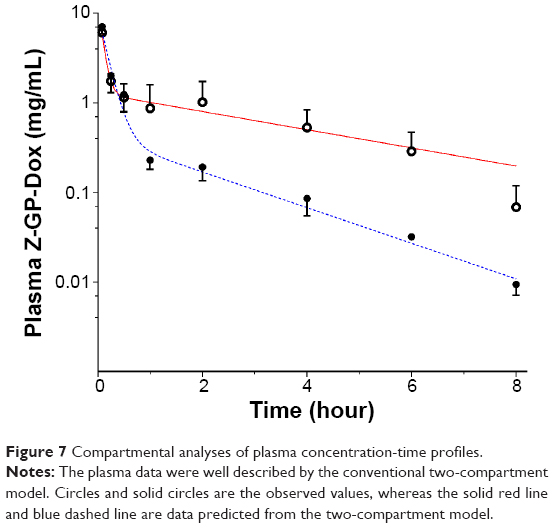 | Figure 7 Compartmental analyses of plasma concentration-time profiles. |
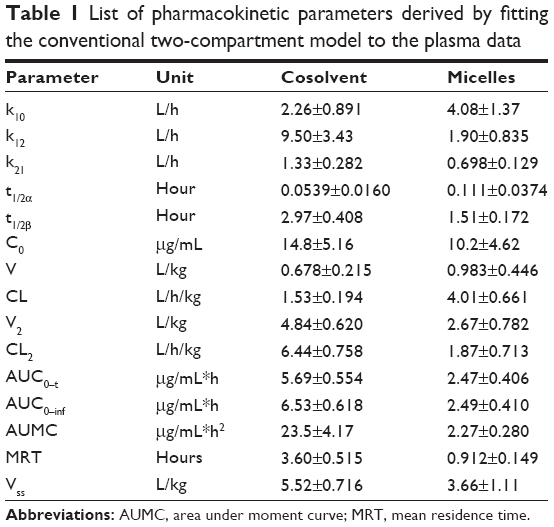 | Table 1 List of pharmacokinetic parameters derived by fitting the conventional two-compartment model to the plasma data |
Following administration of the cosolvent, the organ exposures (AUC0–t, area under the curve) of Z-GP-Dox were 1.49, 14.1, 28.3, 51.5, 21.6, and 156 μg/mL*hour for the heart, intestine, lung, kidney, spleen, and liver, respectively. The results indicate that drug distribution to the liver and kidney was more extensive compared with the other four organs (ie, heart, intestine, lung, and spleen, Figure 8). After administration of the nanomicelles, the organ exposures of Z-GP-Dox were 5.52, 2.42, 37.0, 7.03, 147, and 277 μg/mL*hour for the heart, intestine, lung, kidney, spleen, and liver, respectively. Drug distribution was most extensive in the liver, followed by the spleen, and then the lung, kidney, heart, and intestine. It was noted that organ exposures were significantly elevated in the liver and spleen, but were reduced in the lung, intestine, and kidney. The organ exposure for the heart was not significantly different between the control and micelle groups (Figure 8A).
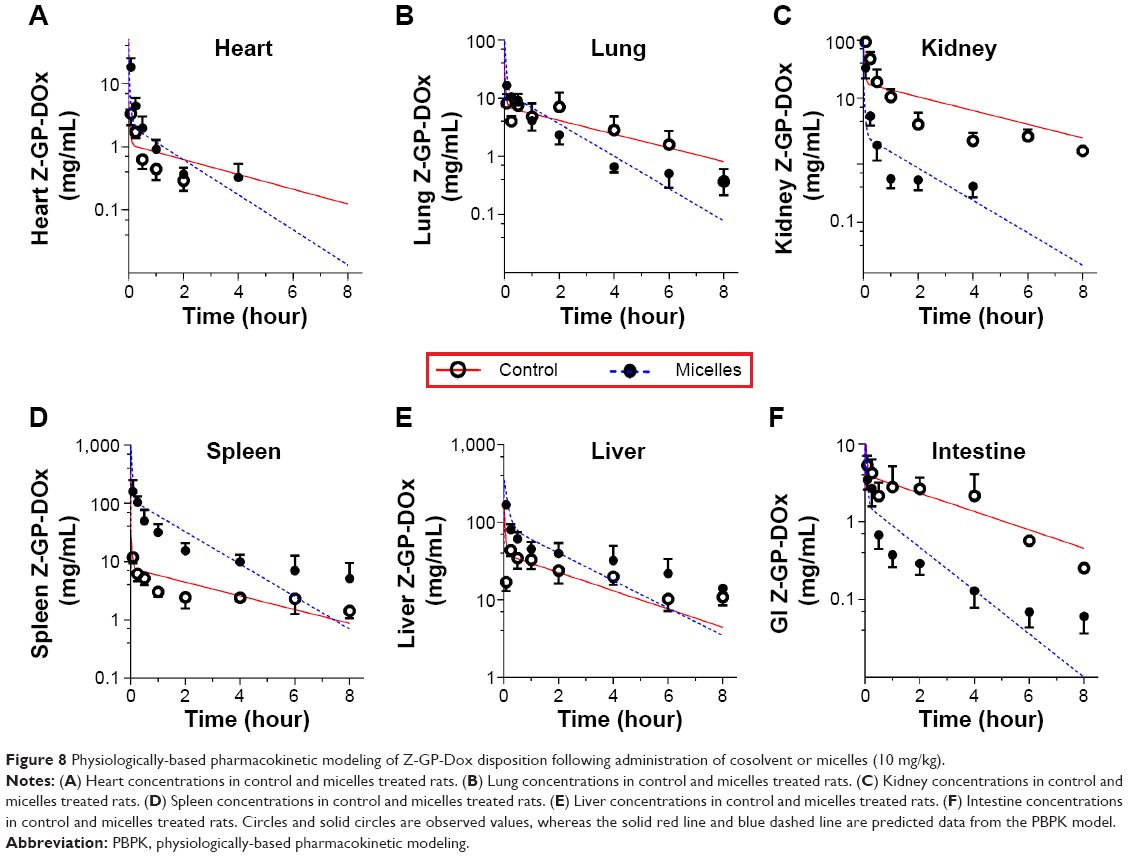 | Figure 8 Physiologically-based pharmacokinetic modeling of Z-GP-Dox disposition following administration of cosolvent or micelles (10 mg/kg). |
Besides cardiotoxicity, doxorubicin-induced renal toxicity also disappoints its anticancer acceptance.33 Reduced kidney exposure to Z-GP-Dox is advantageous when the drug is delivered via nanomicelles. Z-GP-Dox is a prodrug that is much less cytotoxic if it is unable to be cleaved to doxorubicin, the parent drug. Z-GP-Dox is highly stable in blood and various tissue homogenates such as the heart and liver. However, Z-GP-Dox can release doxorubicin in an opportune manner upon hydrolysis of FAPα or incubation with an FAPα-positive tumor homogenate.2 In a previous study, we found that Z-GP-Dox shows significantly lower accumulation than doxorubicin in the heart at all time points, but showed higher accumulation in tumor tissue. Generally, nanocarriers achieve passive tumor targeting via the enhanced permeation and retention effect.34 In our study, the engineered nanomicelles could competently solubilize, load, and intravenously deliver Z-GP-Dox to target tissues. Use of micelles is a promising approach to reducing the injection risk as well as limiting adverse effects and enhancing tumor targeting.
PBPK modeling
The mechanistic PBPK model was fitted to the experimental data and used to derive the pharmacokinetic parameters (Table 2). Comparison of tissue-plasma partition coefficient values (Kp) for control-treated and micelle-treated rats showed that drug distribution to the liver and spleen was markedly enhanced by using the nanomicelles (Figure 8D and E). The Kp,li value was increased by 8.2-fold, whereas the Kp,sp was increased by 32-fold (Table 2). This is in accordance with the notion that nanosized micelles have the ability to target the macrophage system and accumulate in the liver and spleen.7 Enhancement of drug distribution was also noted in the lung (Figure 8B), where the Kp,lu value was increased by 1.4-fold. In contrast, drug distribution to the kidney was significantly reduced (Figure 8C), ie, the Kp,kd value was 2.6-fold lower in micelle-treated rats when compared with control rats (2.96±0.587 versus 10.6±1.34, respectively).
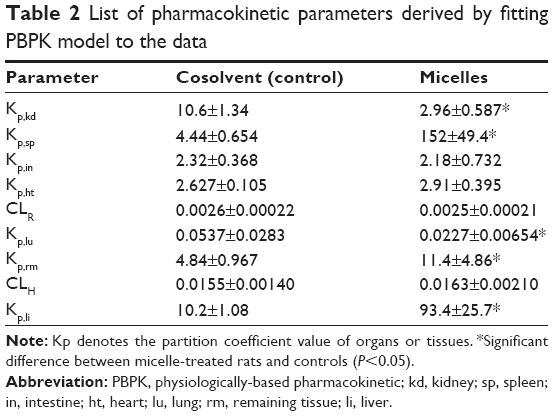 | Table 2 List of pharmacokinetic parameters derived by fitting PBPK model to the data |
Further, no significant difference in Kp,in was observed between the control and micelle groups, indicating that the micelles did not alter the extent of drug distribution to the intestine (Table 2). This appears to be in conflict with the significantly higher intestine concentrations (at the later phase) in control-treated rats when compared with micelle-treated rats (Figure 8F). However, this may not be the case. It is noteworthy that the partition coefficient (Kp) is the ratio of tissue to plasma concentration at steady state. The Kp value rather than the absolute tissue concentration is a direct measure of the extent of tissue distribution. Although absolute tissue concentrations were higher in control-treated rats than in micelle-treated rats, the Kp values for the intestine could be similar because the plasma concentrations were also higher in control rats than in micelle-treated rats.
The CLH (and CLR) values showed no difference between the control-treated and micelle-treated rats, suggesting that the micelle system did not alter the elimination of the drug (Table 2). However, as discussed previously, terminal elimination of the drug was much faster in micelle-treated rats than in the controls (Figure 7). This was because the elimination rate is influenced by both clearance and drug concentration (rate = clearance × drug concentration). Drug elimination was faster because drug concentrations in the liver were markedly increased regardless of hepatic clearance (Figure 8E).
The micelle system showed great potential for enhancement of Z-GP-Dox solubility and facilitating intravenous delivery of the drug. However, it also significantly altered the pharmacokinetic behavior of Z-GP-Dox as compared with the cosolvent. It is possible to exploit the biodistribution pattern in cancer treatment using micelles. For example, micelles may be advantageous in the treatment of spleen and liver cancers because drug molecules tend to accumulate in these organs (Figure 8). However, whether the micelles do have such advantages requires further investigation.
Conclusion
Z-GP-Dox is a promising doxorubicin prodrug with a significantly improved toxicity profile. To overcome the administration difficulties caused by poor solubility, we developed a mixed micelle system allowing systemic delivery of Z-GP-Dox. ZGD-MNs were prepared by dispersion of an ethanol solution of Z-GP-Dox, lecithin, and SO in water. The obtained ZGD-MNs were 86.6 nm in size with a drug load of 14.03%. They were stable in phosphate-buffered saline and drug release was more than 56% within 24 hours. Further, we evaluated the disposition of Z-GP-Dox in rats following administration of the cosolvent alone or micelles using whole-body PBPK models. The Kp values showed that the micelles significantly altered the biodistribution of Z-GP-Dox. Of note, drug distribution to the liver and spleen was markedly enhanced. In conclusion, this is the first report of a mixed micelle system being a viable carrier for delivery of Z-GP-Dox. The pharmacokinetic behavior of Z-GP-Dox was satisfactorily characterized by the established PBPK model.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (81373496).
Disclosure
The authors report no conflicts of interest in this work.
References
Christiansen VJ, Jackson KW, Lee KN, Downs TD, McKee PA. Targeting inhibition of fibroblast activation protein-alpha and prolyl oligopeptidase activities on cells common to metastatic tumor microenvironments. Neoplasia. 2013;15(4):348–358. | ||
Huang S, Fang R, Xu J, et al. Evaluation of the tumor targeting of a FAPα-based doxorubicin prodrug. J Drug Target. 2011;19(7):487–496. | ||
Liu XL, Lynn BC, Zhang JH, et al. A versatile prodrug approach for liposomal core-loading of water-insoluble camptothecin anticancer drugs. J Am Chem Soc. 2002;124(26):7650–7651. | ||
Gupta S. Biocompatible microemulsion systems for drug encapsulation and delivery. Curr Sci. 2011;101(2):174–188. | ||
Dutta P, Dey J. Drug solubilization by amino acid based polymeric nanoparticles: characterization and biocompatibility studies. Int J Pharm. 2011;421(2):353–363. | ||
Zhang X, Zhang X, Yu P, Han Y, Li Y, Li C. Hydrotropic polymeric mixed micelles based on functional hyperbranched polyglycerol copolymers as hepatoma-targeting drug delivery system. J Pharm Sci. 2013;102(1):145–153. | ||
Gill KK, Kaddoumi A, Nazzal S. Mixed micelles of PEG(2000)-DSPE and vitamin-E TPGS for concurrent delivery of paclitaxel and parthenolide: enhanced chemosenstization and antitumor efficacy against non-small cell lung cancer (NSCLC) cell lines. Eur J Pharm Sci. 2012;46(1–2):64–71. | ||
Li J, Huo MR, Wang J, et al. Redox-sensitive micelles self-assembled from amphiphilic hyaluronic acid-deoxycholic acid conjugates for targeted intracellular delivery of paclitaxel. Biomaterials. 2012;33(7):2310–2320. | ||
Saxena V, Hussain MD. Formulation and in vitro evaluation of 17-allyamino-17-demethoxygeldanamycin (17-AAG) loaded polymeric mixed micelles for glioblastoma multiforme. Colloids Surf B Biointerfaces. 2013;112:350–355. | ||
Saxena V, Hussain MD. Polymeric mixed micelles for delivery of curcumin to multidrug resistant ovarian cancer. J Biomed Nanotechnol. 2013;9(7):1146–1154. | ||
Chen Y, Zhang W, Gu J, et al. Enhanced antitumor efficacy by methotrexate conjugated Pluronic mixed micelles against KBv multidrug resistant cancer. Int J Pharm. 2013;452(1–2):421–433. | ||
Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Deliv Rev. 2001;47(1):113–131. | ||
Song X, Jiang Y, Ren CJ, et al. Nimodipine-loaded mixed micelles: formulation, compatibility, pharmacokinetics, and vascular irritability study. Int J Nanomedicine. 2012;7:3689–3699. | ||
Mehta SK, Jindal N. Mixed micelles of lecithin-Tyloxapol as pharmaceutical nanocarriers for anti-tubercular drug delivery. Colloids Surf B Biointerfaces. 2013;110:419–4425. | ||
Hammad MA, Muller BW. Increasing drug solubility by means of bile salt-phosphatidylcholine-based mixed micelles. Eur J Pharm Biopharm. 1998;46(3):361–367. | ||
Rowland M, Peck C, Tucker G. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol. 2011;51:45–73. | ||
Atkinson AJ, Smith BP. Models of physiology and physiologically based models in clinical pharmacology. Clin Pharmacol Ther. 2012;92(1):3–6. | ||
Chow EC, Pang KS. Why we need proper PBPK models to examine intestine and liver oral drug absorption. Curr Drug Metab. 2013;14(1):57–79. | ||
Pang KS, Durk MR. Physiologically-based pharmacokinetic modeling for absorption, transport, metabolism and excretion. J Pharmacokinet Pharmacodyn. 2010;37(6):591–615. | ||
Rostami-Hodjegan A. Physiologically based pharmacokinetics joined with in vitro-in vivo extrapolation of ADME: a marriage under the arch of systems pharmacology. Clin Pharmacol Ther. 2012;92(1):50–61. | ||
Kagan L, Gershkovich P, Wasan KM, Mager DE. Dual physiologically based pharmacokinetic model of liposomal and nonliposomal amphotericin B disposition. Pharm Res. 2014;31(1):35–45. | ||
Ren TY, Cong L, Wang, et al. Lipid emulsions in parenteral nutrition: current applications and future developments. Expert Opin Drug Deliv. 2013;10(11):1533–1549. | ||
Zhang X, Zhang T, Zhou X, et al. Enhancement of oral bioavailability of tripterine through lipid nanospheres: preparation, characterization, and absorption evaluation. J Pharm Sci. 2014;103(6):1711–1719. | ||
Mano N, Mao F, Heller A. A miniature biofuel cell operating in a physiological buffer. J Am Chem Soc. 2002;124(44):12962–12963. | ||
Liu H, Sun H, Lu D, et al. Identification of glucuronidation and biliary excretion as the main mechanisms for gossypol clearance: in vivo and in vitro evidence. Xenobiotica. 2014;44(8):696–707. | ||
Liu W, Liu H, Sun H, et al. Metabolite elucidation of the Hsp90 inhibitor SNX-2112 using ultraperformance liquid chromatography/quadrupole time-of-flight mass spectrometry (UPLC-QTOF/MS). Xenobiotica. 2014;44(5):455–464. | ||
Kagan L, Gershkovich P, Wasan KM, Mager DE. Physiologically based pharmacokinetic model of amphotericin B disposition in rats following administration of deoxycholate formulation (Fungizone®): pooled analysis of published data. AAPS J. 2011;13(2):255–264. | ||
Mayer LD, Tai LC, Ko DS, et al. Influence of vesicle size, lipid composition, and drug-to-lipid ratio on the biological activity of liposomal doxorubicin in mice. Cancer Res. 1989;49(21):5922–5930. | ||
Nie S. Understanding and overcoming major barriers in cancer nanomedicine. Nanomedicine (Lond). 2010;5(4):523–528. | ||
Cho YW, Lee J, Lee SC, Huh KM, Park K. Hydrotropic agents for study of in vitro paclitaxel release from polymeric micelles. J Control Release. 2004;97(2):249–257. | ||
Agrawal SK, Sanabria-DeLong N, Coburn JM, Tew GN, Bhatia SR. Novel drug release profiles from micellar solutions of PLA-PEO-PLA triblock copolymers. J Control Release. 2006;112(1):64–71. | ||
Guo S, Huang L. Nanoparticles escaping RES and endosome: challenges for siRNA delivery for cancer therapy. J Nanomater. 2011;2011:1–12. | ||
Hassan MH, Ghobara M, Abd-Allah GM. Modulator effects of meloxicam against doxorubicin-induced nephrotoxicity in mice. J Biochem Mol Toxicol. 2014;28(8):337–346. | ||
Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65(1):71–79. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.