")
Back to Journals » International Journal of Nanomedicine » Volume 15
Mitigation of Rheumatic Arthritis in a Rat Model via Transdermal Delivery of Dapoxetine HCl Amalgamated as a Nanoplatform: In vitro and in vivo Assessment
Authors Salem HF, Nafady MM, Kharshoum RM, Abd el-Ghafar OA , Farouk HO
Received 15 November 2019
Accepted for publication 1 February 2020
Published 6 March 2020 Volume 2020:15 Pages 1517—1535
DOI https://doi.org/10.2147/IJN.S238709
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Heba Farouk Salem,1 Mohamed Mahmoud Nafady,2 Rasha Mostafa Kharshoum,1 Omnia Ahmed Abd el-Ghafar,3 Hanan Osman Farouk2
1Pharmaceutics and Industrial Pharmacy Department, Faculty of Pharmacy, Beni- Suef University, Beni Suef, Egypt; 2Pharmaceutics and Clinical Pharmacy Department, Faculty of Pharmacy, Nahda University, Beni Suef, Egypt; 3Pharmacology Department, Faculty of Pharmacy, Nahda University, Beni Suef, Egypt
Correspondence: Heba Farouk Salem
Pharmaceutics and Industrial Pharmacy Department, Faculty of Pharmacy, Beni- Suef University, Beni Suef city, Egypt
Tel +2 01001944381
Email [email protected]
Purpose: Dapoxetine HCl (DH), a selective serotonin reuptake inhibitor, may be useful for the treatment of rheumatic arthritis (RA). The purpose of this study was to investigate the therapeutic efficacy of transdermal delivery of DH in transethosome nanovesicles (TENVs). This novel delivery of DH may overcome the drawbacks associated with orally administered DH and improve patient compliance.
Methods: DH-TENV formulations were prepared using an injection- sonication method and optimized using a 33 Box-Behnken-design with Design Expert® software. The TENV formulations were assessed for entrapment efficiency (EE-%), vesicle size, zeta potential, in vitro DH release, and skin permeation. The tolerability of the optimized DH-TENV gel was investigated using a rat skin irritation test. A pharmacokinetic analysis of the optimized DH-TENV gel was also conducted in rats. Moreover, the anti-RA activity of the optimized DH-TENV gel was assessed based on the RA-specific marker anti-cyclic cirtullinated peptide antibody (anti-CCP), the cartilage destruction marker cartilage oligomeric matrix protein (COMP) and the inflammatory marker interleukin-6 (IL-6). Level of tissue receptor activator of nuclear factor kappa-&Bgr; ligand (RANKL) were also assessed.
Results: The optimized DH-TENV formulation involved spherical nanovesicles that had an appropriate EE- % and skin permeation characteristic. The DH-TENV gel was well tolerated by rats. The pharmacokinetics analysis showed that the optimized DH-TENV gel boosted the bioavailability of the DH by 2.42- and 4.16-fold compared to the oral DH solution and the control DH gel, respectively. Moreover, it significantly reduced the serum anti-CCP, COMP and IL-6 levels and decreased the RANKL levels. Furthermore, the DH-TENV gel attenuated histopathological changes by almost normalizing the articular surface and synovial fluid.
Conclusion: The results indicate that DH-TENVs can improve transdermal delivery of DH and thereby alleviate RA.
Keywords: SSRIs, nano carrier via skin, pharmacokinetic parameters, autoimmune disease, RANKL
Introduction
Rheumatoid arthritis (RA) is a progressive inflammatory autoimmune disease. The pathogenesis of RA involves numerous factors, including environmental and genetic factors.1 Studies have shown that bias with RA is seen mainly in developed countries, with a prevalence of 1-2%, and it affects more women than men because of hormonal effects on immune cells.2–4 RA is mostly associated with bone erosion, bone destruction, cartilage degradation, synovial inflammation, and joint rigidity.5 Long-term RA therapy, including glucocorticoids, non-steroidal anti–inflammatory drugs, biological therapies, and disease-modifying anti-RA drugs such as methotrexate (MTX) and leflunomide, have detrimental effects on the kidneys, bones, stomach, and other organs. However, new drug classes with less harmful effects are being studied.6
Selective serotonin reuptake inhibitors (SSRIs) may be suitable candidates for the treatment of RA as they down regulate the pro-inflammatory cytokines interleukin (IL-6), and tumor necrosis factor (TNF-α) and suppress cyclooxygenase-2 (COX-2) protein expression.7
Dapoxetine hydrochloride (DH) is an SSRI that is commonly used to treat depression.8 DH is also the first and only drug approved in men aged 18–64 years for the treatment of premature ejaculation.9 Due to its anti–inflammatory and immune modulatory effects,10 DH is a promising drug for treating RA. Therefore, we investigated the anti–inflammatory effects of DH on Complete Freund’s Adjuvant (CFA)-induced RA in rats. Unfortunately, DH has several limitations. It has a very short elimination half-life of 1.31 h.11 In addition, after rapid absorption from the gastrointestinal tract it is extensively metabolized by the liver, resulting in poor bioavailability (42- %).11 These factors collectively cause DH to have poor efficacy. However, transdermal delivery of DH is a promising administration route.
Transdermal delivery has numerous benefits as it avoids factors that affect gastrointestinal drug absorption, such as pH, enzymatic activity, and drug–food interactions, and bypasses liver metabolism.12,13 Additionally, transdermal delivery reduces the required dose and treatment frequency, and ensures sustained release, with minimal fluctuations in the plasma level of the drug, which is beneficial during long-term RA therapy. Nevertheless, the stratum corneum, the most drug-resistant barrier of the skin, limits the bioavailability of transdermal drugs. Many approaches have been explored to combat the skin barrier to ensure successful transdermal drug delivery, including the use of nanovesicular drug carriers.14
Cevc and Blume introduced deformable (elastic) liposomes known as “transfersomes”15 that can act as nanovesicular drug delivery system. Subsequently, Touitou et al proposed a nanovesicular system known as ethosomes16 that diverge from liposomes as they contain a relatively high ethanol concentration. “Transethosomes” were first reported by Song et al17 and they include the basic components of classical ethosomes with additional compounds such as penetration enhancers and/or surfactants. Research has shown that various penetration enhancers and surfactants improved physicochemical properties of ethosomes.18 Nanovesicular drug delivery systems are extremely biocompatible and have tremendous drug delivery potential.
The aim of this study was to develop a transdermal delivery system for DH by constructing DH-loaded ultra-deformable transethosome nanovesicles (TENVs) embedded in gel in order to boost the systemic absorption of DH and to treat RA. The physicochemical characteristics of the DH-TENV formulations were assessed. The pharmacokinetic parameter of DH in rats after oral and transdermal DH administration were compared. In vivo assessment of efficacy was conducted in a rat model of CFA-induced RA.
Materials and Methods
Materials
DH was kindly supplied as a gift by Marcyrl Pharmaceutical Industries (El-Obour, Egypt). Phosphatidylcholine (PC) and sodium deoxycholate (SDC) were purchased from Acros Organics (Cairo, Egypt). Propylene glycol (PG) was provided as a gift by CID Pharmaceutical (Asuit, Egypt). Carbopol 971 was purchased from El-Nasr Pharmaceutical Chemical Company (Cairo, Egypt). High-performance liquid chromatography-grade ethanol, CFA (from dried Mycobacteria), and MTX were acquired from Sigma-Aldrich (St. Louis, MO, USA). Dialysis bags with a molecular weight cut-off of 12,000 Da were purchased from Sigma-Aldrich (St. Louis, MO, USA). Quantikine enzyme-linked immunosorbent assay (ELISA) kits for serum anti-cyclic citrullinated peptide antibody (anti-CCP), cartilage oligomeric matrix protein (COMP), and IL-6 were obtained from MyBiosource (San Diego, CA, USA). Western blotting antibodies and chemicals were obtained from Thermo Fisher Scientific (-Rockford, IL, USA). All other materials and solvents used in the experiments were of high analytical grade.
Box–Behnken Design
A33 Box–Behnken design was used, with formulations (F1-F17) to investigate the effects of three independent variables on the characteristics of TENV formulations using Design-Expert software® (version 11.0.6.0, Stat-Ease Inc. Minneapolis, MN, USA). The independent variables were the concentration of PC (A), ethanol (B), and SDC (C). The dependent variables were entrapment efficiency (EE %), vesicle size (VS), zeta potential (ZP), cumulative % of DH released from TENVs after 8h (Q8h), and the cumulative amount of DH that permeated a rat skin specimen after 24h (Q24) (Table 1).
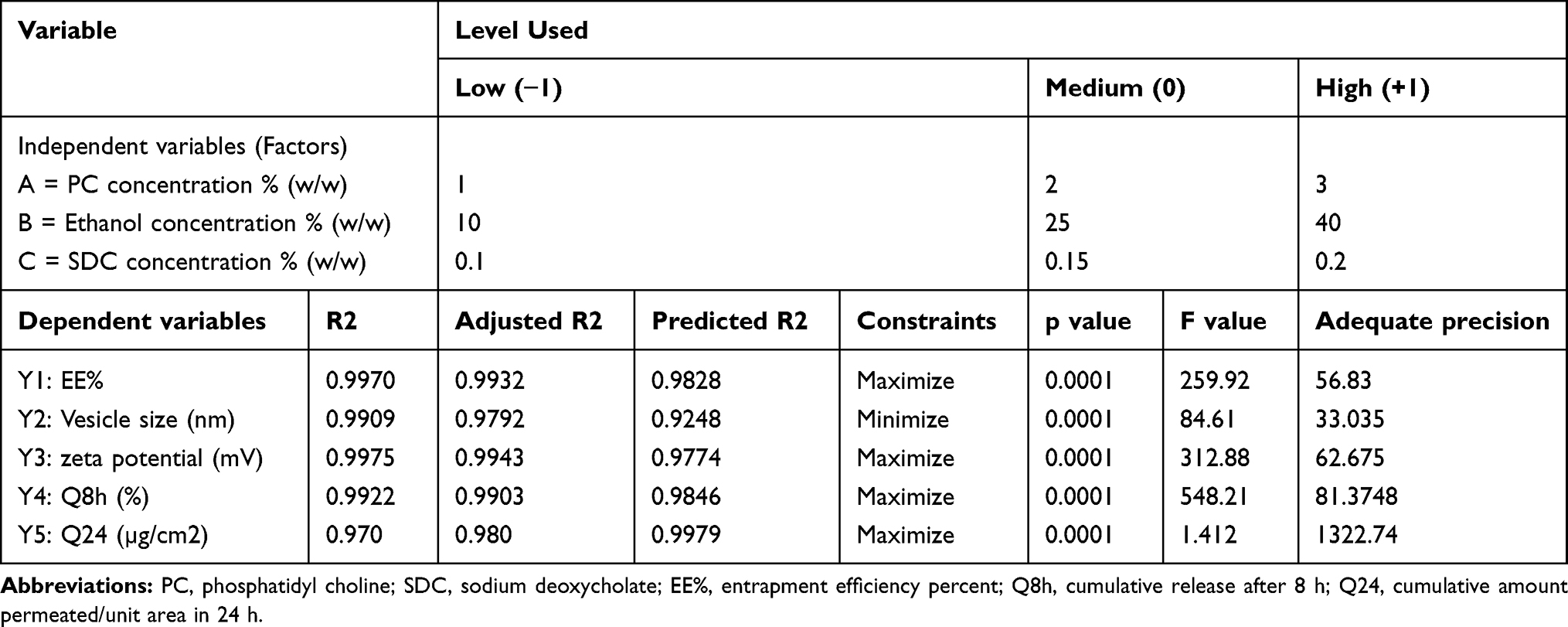 |
Table 1 The Independent Variables, Their Respective Levels, and the Summarize Statistics Model of Box–Behnken Design Used for Optimization of DH Trans- Ethosomes |
Preparation of DH- TENVs
DH-TENVs were constructed using an injection sonication method.19 Various concentrations of PC, and the edge activator (surfactant) SDC were dissolved along with a 0.5 mL of propylene glycol (PG) in various concentrations of ethanol (the alcohol phase). Each mixture was continuously agitated at 40 °C in a sealed container. Next, 10 mg DH was dispersed in distilled water. This was slowly added to the alcohol phase drop by drop and stirred using a magnetic stirrer at 13,000 rpm for four 5-min cycles with a 3-min rest between cycles. The dispersed vesicles were left at ambient temperature (25±1°c) for 45 min and then preserved at 4°c until use.
Characterization of DH -TENVs
The EE % of DH
TENVs were estimated using an ultracentrifugation method. The samples were kept at 4 °C overnight and then centrifuged at 14,000 rpm for 2 h using a refrigerated centrifuge (SIGMA 3–30K, Steinheim Germany). The supernatant (containing free DH) was diluted with distilled water and then analyzed using a “UV spectrophotometer” (Jasco V-530, USA) at a λmax of 292 nm. The EE% was computed using Equation (1):20
(1)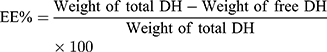
VS, Polydispersity Index (PDI) and ZP
The VS, PDI and ZP of the DH-TENVs (diluted 50 times with deionized water) were determined using “ a Malvern Zetasizer” (Malvern Instruments, Worcestershire, UK)21 at 25 ± 1°C. The measurements were performed in triplicate.
In vitro DH Release Analysis
A cellophane based dialysis bag diffusion technique (molecular weight cut off 12,000; Sigma-Aldrich) was used to compare the in vitro DH release of DH-TENVs and free DH solution.22 The DH-TENV formulations (equivalent to 3 mg DH) and free DH solution were each incorporated into a dialysis bag, which was subsequently wrapped around a cylinder. The bag was then placed in a container with 30 mL phosphate-buffered saline (pH 5.5)23 at 37 ± 0.5°C, with stirring at 50 rpm using a magnetic stirrer to diminish the impact of the non-stirred solution layer.24 At (1, 2, 3, 5, 6, 7, and 8 h) time points, a 3 mL aliquot was withdrawn and immediately replenished with an equivalent volume of phosphate-buffered saline to maintain the sink condition. The DH content of the aliquots was analyzed using a “UV spectrophotometer” at a λmax of 292 nm. The analysis was conducted in triplicate and the % DH released was expressed as mean (±SD). The cumulative % of DH released from DH-TENVs was plotted against time. To identify the DH release pattern, the data were used with various models of release kinetics such as zero order, first order, Higuchi’s matrix,25 and Korsmeyer- Peppas model.26
Ex vivo Skin Permeation Analysis
To assess skin permeation, the DH-TENV formulations and DH solution (control) were evaluated. A Franz diffusion cell with an effective permeation area of 5.0 cm2 and 100 mL receiver compartment was used. A shaved rat skin specimen was prepared according to a previous study27 and placed in the receiver compartment with the stratum corneum side facing upwards. The receiver compartment was full of distilled water,28 and the donor compartment contained a DH-TENV formulation (equivalent to 3 mg DH) or DH solution. The analysis was conducted at 37 ± 2 °C with continuous stirring at 100 rpm for 24 h. At (1, 2, 3, 4, 5, 6, 8, 12, and 24 h) time points, samples were removed, and the DH content was determined using a spectrophotometer at λmax of 292. After each withdrawal, the receiver compartment was refilled with fresh medium of equal quantity to preserve the sink condition.
The cumulative amount of DH that permeated the rat skin specimen per unit area (µg/cm2) was plotted against time (h) for each formulation. The lag time was determined from the x-axis intercept (the linear portion of the graph). Permeation parameters, including the steady state flux (Jss) in µg/cm2/h and permeability coefficient (Kp) in cm/h, were calculated for each DH-TENV formulation to assess the improvement in the DH permeation in comparison to the DH solution (control).29
Optimization of DH-TENVs
Using the desirability function, Design-Expert® software was used to select the optimized DH-TENV formulation based on maximizing EE-%, ZP, Q8h and Q24h and minimizing VS (Table 1). All parameters were optimized simultaneously by assessing the overall desirability.30 The optimized DH-TENV formulation was selected based on having an overall desirability close to 1. To confirm the prediction ability, the selected DH-TENV formulation was prepared, characterized, and the actual values of EE%, ZP, Q8h, Q24h, and VS were compared with the predicted values.
Transmission Electron Microscopy (TEM) of the Optimized DH-TENV
The surface structure of the optimized DH-TENV formulation was imaged via TEM (JEM-1400, Jeol, Tokyo, Japan) with an 80 kV accelerating voltage of 80 kV.31 For visualization by TEM, a drop of diluted formulation was deposited on a carbon-coated grid and then negatively stained with 2% phosphotungstic acid.
Differential Scanning Calorimetry (DSC) of the Optimized DH-TENV
The thermal properties of pure DH, PC, SDC and the optimized DH- TENV formulation were assessed using DSC (DSC 50 Shimadzu, Kyoto, Japan). First, 5 mg samples were placed in a standard aluminum pan and they were then heated at 25 to 300 °C at a scanning rate of 5 °C/min under inert nitrogen flow at 25 mL/min32
Physical Stability of the Optimized DH-TENV
The optimized DH-TENV was stored for 30, 60 and 90 days in a glass vial at 4 °C and then the VS and EE% were assessed.33
Skin Irritation Analysis of the Optimized DH-TENV
The optimized DH-TENV formulation was converted to gel using 1% (w/w) carbopol 971 aqueous solution for topical application. Its safety was assessed in female Wistar albino rats randomly grouped into DH-TENV gel treatment group and a normal control group (10 per group). The rats weighed 150–200 g, had free access to food and tap water, and were kept under standard controlled conditions in polyacrylic boxes. The hair on each rat’s dorsal side was shaved and gel was applied for 4 weeks. The relevant site was monitored for any adverse effects during treatment. Each rat was euthanized by cervical decapitation and the treated site was removed along with an untreated control skin specimen. These skin specimens were washed with normal saline and conserved in 10% neutral buffered formalin for histopathological assessment. The protocol was approved by the institutional animal ethics committee of Nahda University. All experimental procedures followed the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animal (NIH Publications no. 8023, revised 1978).
Skin specimens from four random rats in each group were rinsed with normal saline, buffered with 10% neutral buffered formalin (phosphate buffer), and coated in paraffin. Based on the Bancroft and Gamble process,34 5-μm sections were stained with hematoxylin and eosin (H&E). A digital camera attached to a light microscope was used to photograph the sections.
Pharmacokinetic Analysis of the Optimized DH-TENV
The pharmacokinetics of DH in rats after administration of the optimized DH-TENV formulation, oral DH solution, and control DH gel were compared. The control DH gel was prepared by adding DH to a 1% (w/w) carbopol 971 aqueous solution, stirring to completely dissolve the DH, and then incubating the gel at 4°C overnight to allow complete polymerization.
First, 18 Wister albino rats weighing 200–250 g were obtained from the animal facility of Nahda University. The protocol was approved by the institutional animal ethics committee of Nahda University. All experimental procedures followed the National Institutes of Health (NIH) Guide (NIH Publications No. 8023, revised 1978).
The rats initially had free access to food and water but were fasted for 10 h prior to treatment. The 18 rats were randomly divided into three groups (six rats per group). Each rat received 10 mg/kg/day DH.35 The first group received DH solution by oral gavage, the second group received the optimized DH-TENV gel, and the third group received the control DH gel. Gel (1 g) was spread over 4 cm2 of the dorsal surface of the relevant rats after shaving. Blood samples (0.5 mL) were obtained from the retro-orbital plexus at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, and 24 h. To avoid coagulation, the blood samples were collected in heparinized tubes. The samples were centrifuged for 15 min at 4000 rpm to extract the plasma, which was stored at –20°C until assay.
For the Liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis, 100 μL reboxetine (500 ng/mL) was added to 500 μL plasma as the internal standard (IS). The samples were vortexed for 30 seconds and then precipitation was induced by adding 5 mL tertiary butyl methyl ether, followed by vortexing for 1 min, centrifugation for 10 min at 3000 rpm, and evaporation of the supernatant in a vacuum concentrator. The dry residue was reconstituted with 250 µL acetonitrile: 0.1% formic acid (80:20% v/v), which acted as the isocratic mobile phase, before injection into the LC system.
DH in the plasma samples was analyzed using a sensitive, selective, and accurate validated LC-MS/MS method.36 An 8-µL aliquot of the samples was injected into a Shimadzu Prominence LC system (Shimadzu, Kyoto, Japan) equipped with a SunFire C18 column (4.6 × 50 mm with 5 µm particle size (Waters). Analysis was carried out at room temperature. The mobile phase was delivered to the electrospray ionization chamber of the API-3200 mass spectrometer (fitted with a 400°C Turbolonspray TM interface SCIEX) at a flow rate of 0.550 mL/min. Quantification of both DH and the reboxetine IS was accomplished by MS/MS in positive ion mode. The voltage of the ion spray was set at 5500 V. The nebulizer gas, curtain gas, auxiliary gas, and collision gas were set at 30, 20, 40, and 5 psi, respectively. The declustering potential (DP), collision energy (CE), entrance potential (EP), and collision exit potential (CXP) were 31, 33, 6, and 4 V for DH, and 8, 26, 2, and 1 V for the reboxetine IS, respectively. Ion detection was performed in the multiple reactions monitoring (MRM) mode, monitoring the transition of the m/z 306.209 precursor ion to m/z 157.200 for DH and the m/z 314.251 precursor ion to m/z 176.159 for the reboxetine IS. The Q1 and Q3 quadrupoles were set to unit resolution. The analytical data were processed using Analyst software (version 1.USA).
Pharmacokinetic parameters were estimated based on the plasma data using the Excel add-in PKSolver.37 Non-compartmental analysis was applied to assess the maximum DH concentration (Cmax; ng/mL) and the time needed to reach this concentration (tmax; h). The area under the plasma DH concentration–time curve (AUC) was determined using the trapezoidal rule from 0 to 24 h (AUC0-24, ng.h/mL) and from 0 to infinity (AUC0-∞, ng.h/mL).
In vivo Analysis of Optimized DH-TENVs in CFA-Induced RA in Rats
Adult female Wister albino rats weighing 180–200 g was acquired from the animal house of Nahda University. They were kept at 25±2°C and 60±10% humidity with 12/12h light/dark cycles, with free access to standard food and water. The protocol was approved by the Ethics Committee of Nahda University
The rats were divided into seven groups (eight rats per group) as follows: normal control group (rats received normal saline), DH-TENV gel group (rats received 1 g/day optimized DH-TENV gel), RA group (rats received 0.4 mL/limb/3 day subcutaneous CFA for RA induction;38), MTX+RA group (rats received CFA as in the RA group and 0.1 mg/kg/day oral methotrexate),39 DH-TENV gel+RA group (rats received CFA as in the RA group and 1 g/day optimized DH-TENV gel), DH gel+RA group (rats received CFA as in the RA group and 1 g/day DH gel), and oral DH+RA group (rats received CFA as in the RA group and 10 mg/kg/day oral DH solution).40 To induce RA, 0.4 mL CFA was subcutaneously injected into the planter surface of the hind limbs on days 1 (right hind limb), 4 (left hind limb), and 7 (right hind limb), with a 3-day interval between each dose.38 The gel was applied on the dorsal surface of each rat to a 4-cm2 shaved area. All treatments were administered from day 13 until day 27.
On day 28 (24 h after the last treatment dose), serum was obtained to assess serum anti-CCP, COMP, and IL-6 levels. Additionally, three knee joints from three rats in each group was used for Western blot analysis of receptor activator of nuclear factor kappa-Β ligand (RANKL). Furthermore, three knee joints per group were X-rayed and changes were examined by a radiologist. Finally, three knee joints per group (with adjacent capsules, cartilage, and bones) together with the spleen and liver underwent histopathological examination.
On Day 28, the rats were anesthetized with diethyl ether and blood was collected from the retro-orbital plexus. Serum was obtained by centrifugation at 3000 rpm for 30 min and stored at −20°C until use. Serum anti-CCP, COMP, and IL-6 were subsequently assessed using ELISA kits according to the manufacturer guidelines based on principles described in previous studies.2,41,42
Receptor activator of nuclear factor kappa-Β ligand (RANKL) tissue levels were measured using a previously described Western blot technique43,44 using a Mini PROTEIN electrophoresis system (Bio-Rad). Briefly, a portion of the synovial tissue was exposed to radioimmunoprecipitation assay (RIPA) lysis buffer. The lysate sample was kept on ice for 30 min and cell debris was then removed by centrifugation at ~16,000g for 30 min at 4°C. The supernatants were collected in tubes for protein concentration determination. Proteins were then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Next, primary antibodies against RANKL and B-actin were added and incubated at 4°C overnight. The blot was then dipped in tris-buffered saline plus Tween 20 (TBST) 3–5 times for 5 min. Horseradish peroxidase (HRP)-conjugated secondary antibody was then added for 1 h at room temperature. The blot was then dipped in TBST again 3–5 times for 5 min. Chemiluminescent substrate was added following the manufacturer’s recommendations. Chemiluminescent images were then obtained using a charge-coupled device (CCD) camera in the ChemiDoc MP System (Bio-Rad). After standardization using β-actin, the band intensity associated with RANKL was assessed in each group and compared to the untreated RA group.
On day 28, the rats were anaesthetized by intraperitoneal injection of 50 mg/kg pentobarbitone sodium. Radiographs were taken with X-ray apparatus. The knee joints were inspected by a radiologist based on joint space narrowing and deformity.45
Lastly, knee joints, spleen, and liver were embedded in 10% formalin saline for 24 h for histopathological examination. The knee Joints, spleen and liver were cut into -μm sections, stained with H&E, and viewed with a microscope (Nikon) by a pathologist.
Statistical Analysis
Statistical analysis was performed using one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test using GraphPad (Prism 6, USA). A p value <0.05 was considered statistically significant. Results are expressed as mean±SD.
Results and Discussion
Box–Behnken Design
The data obtained using a Box–Behnken design were statistically analyzed using Design-Expert® software. Based on preliminary experiments on the feasibility of preparing DH-TENVs, the three-factor was chosen and the three level of each of the three factors (PC, SDC, and ethanol) were set (Table S1). The signal-to-noise ratio was accurately measured to ensure that the model could be used to navigate the design space.46,47 All parameters (EE-%, VS, ZP, Q8h, and Q24h) had a ratio >4 (Table 1), which is desirable. To be in fair agreement, the adjusted and predicted R2 values should be similar to each other,48 which were accomplished for all parameters (Table 1). The experimental run and the measured parameters are listed and computed in (Table 2).
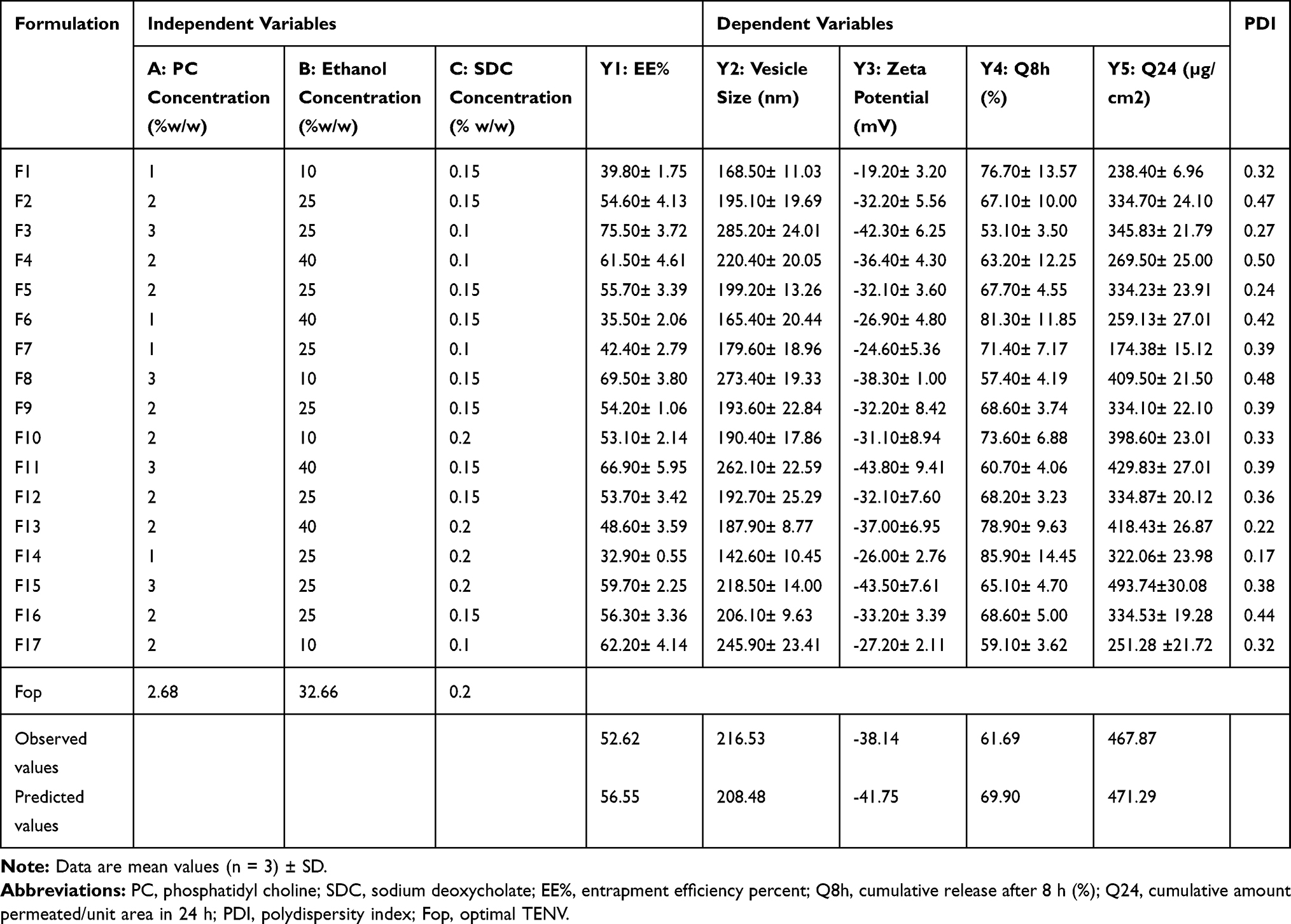 |
Table 2 Observed Response in Box–Behnken Design for Optimization of DH Loaded TENVs |
EE% of DH-TENVs
The EE% for the DH-TENV formulations was 32.9–75.5% (Table 2). Thus, DH was successfully entrapped in the TENVs, indicating that lipid based TENVs can be used as a delivery system for water-soluble drugs.
ANOVA showed that the sequential model proposed for assessing the EE% values was a quadratic model. Equation (2) shows the quantitative effects of the independent variables on the EE% of the TENV formulations.
(2)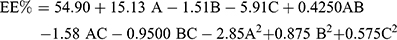
Where A is PC, B is ethanol, and C is SDC
The EE% values were significantly affected by all three independent variables (p<0.05). The regression coefficients indicate that the PC concentration had a noticeable positive effect on EE%, while the ethanol and SDC concentrations had negative effects on EE%. The effects of PC and ethanol concentrations on EE% at the middle level of the third independent variable (SDC concentration) are shown in Figure 1A. Increasing the PC concentration from 1% to 3% significantly increased the EE%, possibly because there was increased space to accommodate the DH.49 This finding is in agreement with the study by Singh et al.50
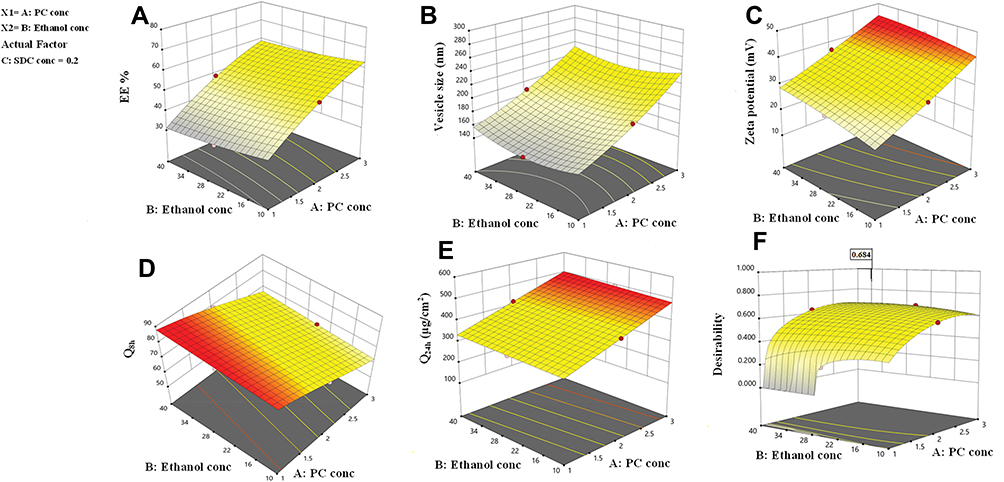 |
Figure 1 Response surface plot for the effect of PC concentration (A), ethanol concentration (B) at the middle levels of the 3rd (SDC concentration) on (A) EE%, (B) vesicle size, (C) zeta potential, (D) Q8h, (e) Q24 and (F) desirability of the developed TENVs dispersions. |
The EE% values for 10% ethanol were superior to those for 40% ethanol, as ethanol may increase the fluidity of the vesicle membrane, leading to drug leakage.51,52 These findings are similar to previous studies on the effect of ethanol concentration on the EE% of water-soluble drug-loaded vesicles.53 However, research on water-insoluble drugs is not in agreement.54
The decrease in EE% with increasing SDC concentration may be attributable to the coexistence of micelles with vesicles.55 Moreover, the high hydrophilicity of SDC, which interacts with the hydrophobic domain of the vesicles, leads to less rigid vesicles and increases drug solubilization in the aqueous medium during preparation, which lowers the EE%.56 Furthermore, edge activators such as SDC are single-chain components that destabilize the vesicles and enhance the deformability of the phospholipid bilayer by decreasing the interfacial tension.57
VS and PDI of DH-TENVs
One of the greatest challenges regarding improving the transdermal steady-state flux (Jss) of DH was the development of vesicles with optimized VS. The VS of the DH-TENV formulations was 142.6–285.2 nm (Table 2), indicating a nanometer range. The PDI values of the DH-TENV formulations, which could be indicative of the VS distribution,58 were 0.17–0.5.
VS values were subjected to polynomial analysis and quadratic model was selected. Equation (3) shows the quantitative effects of the independent variables on the VS of the TENV formulations.
(3)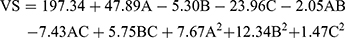
where A is PC, B is ethanol, and C is SDC
The concentrations of PC, ethanol, and SDC significantly influenced the mean VS of TENVs (p<0.05). Increasing the concentration of PC from 1% to 3% significantly increased the mean VS (Figure 1B). This may be attributable to the increased amount of entrapped DH, which resulted in swollen transethosomes. These findings are similar to those of a previous study.59
Increasing the ethanol concentration significantly reduced the VS. This could be due to the interaction between the ethanol and the lipid bilayer, contributing to a decrease in the vesicle membrane thickness.52,60–62 Also, ethanol changes the net charge of the transethosome and leads to steric stabilization, ultimately reducing the VS.63 Verma and Pathak64 reported similar findings regarding ethanol-containing ethosomes.
The VS decreased with increasing SDC concentration from 0.1 to 0.2%. This may have resulted from the decrease in the interfacial tension between the aqueous and lipid phase, leading to the formation of smaller droplets. Additionally, surfactants such as SDC stabilize the particles by creating a steric barrier on the surface, thereby preventing aggregation into larger particles.65,66 Accordingly, the negative relationship between surfactant concentration and VS was unsurprising.
ZP of DH-TENVs
the ZP has a pivotal impact on the storage stability of particle dispersions. If the ZP of particles is too low to offer an adequate steric barrier or electric repulsion between the particles, aggregation will occur. Muller67 reported that particles are considered stable when the ZP value is around ±35 mV. The ZP of the TENV formulations ranged between −19.2±3.2 and −43.8±9.4 mV (Table 2). Equation (4) shows the quantitative effects of the independent variables on the ZP of the TENV formulations
(4)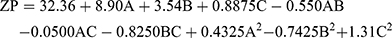
where A is PC, B is ethanol, and C is SDC
The three had significant positive impacts on the ZP value (p<0.05) (Figure 1C). The equation shows that vesicles with the greatest PC concentration had the highest ZP. Although PC heads are zwitterionic and therefore theoretically uncharged at a neutral pH, they produce a negative ZP value in water68 because of the head group orientation and the development of hydration layers around its surface.69 Furthermore, Makino et al70 argued that the charge of a PC bilayer is due to the orientation of the dipole, involving the negative charge of the phosphatidyl group and the positive charge of the choline group in the head group. It has been reported that, in a medium with low ionic strength, the head group is oriented so that the negative phosphatidyl group is outside and the positive choline group is inside resulting in a negative surface charge.69
Increased ethanol concentration significantly increased the ZP values of DH-TENVs. Similar findings have been reported previously, demonstrating that the concentration-dependent negative surface charge of ethanol results in electrostatic repulsions that prevent vesicle aggregation, increasing the ZP value.71
Similarly, increasing SDC concentrations significantly increased the ZP values. A plausible explanation is that SDC results in negative surface charges, leading to strong electrostatic repulsions between the particles.72
These results indicate the good stability of the vesicles. However, negatively charged TENVs lead to strongly decreased drug permeation during transdermal delivery.23,73,74
DH Release of DH-TENVs
The release behavior of DH from DH-TENVs was investigated to confirm whether the DH-TENVs had the ability to release DH in a sustained manner. Rapid DH release from free DH solution in the dialysis bag was observed, with approximately 95% of the DH being released in the first 3 h (Figure S1A–C). In contrast, the DH in DH-TENVs demonstrated a slow and controlled release, with about 53–85% of the DH being released within 8 h (Table 2).
Cumulative % of DH released from TENVs after 8 h (Q8h) was subjected to polynomial analysis and a linear model was selected; the statistical analysis indicated the ability of the linear model to demonstrate the significant impact of three independent variables on the DH release. The Box–Cox plot of Q8h indicated the need for logarithmic transformation in order to fulfill the ANOVA assumptions. Equation (5) shows the quantitative effects of the independent variables on ln(Q8h).
(5)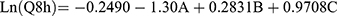
where A is PC, B is ethanol, and C is SDC.
The independent variables significantly influenced the cumulative % of DH released from TENVs after 8 h (Q8h) (p<0.05) (Figure 1D). As the PC concentration increased, Q8h was significantly reduced. This may be due to the presence of firm lipid membranes, which prevents DH release. This result is in accordance with a study by Aboud et al.75
Notably, increasing ethanol concentrations yielded a significantly higher DH release rate. This indicates that ethanol may provide the vesicles with soft flexible characteristics that allow easy drug diffusion through membranes. Furthermore, the inclusion of ethanol may minimize the vesicle hydration layer, which encourages drug release.76
Increasing SDC concentrations significantly increased Q8h. This could be due to the fact that increased SDC concentrations led to decreased VS, increasing the total surface area and thus accelerating the release rate.77 Furthermore, increased SDC concentration may lead to the development of micelles in the bilayer, which may enhance the membrane permeability and thus increase drug release.78
Linear analysis of the DH release data showed that, for all the DH-TENV formulations, the Higuchi kinetics equation had the best R2 value, indicating slow and sustained drug release,54 while, for the free DH solution, the first-order kinetics equation had a good R2 value, indicating that the DH release rate was dependent on the DH concentration.
For further elucidation of DH release kinetics, the Korsmeyer–Peppas model was employed to shed light on other mechanisms of DH release. In this model, the diffusional exponent (n) values for Fickian (diffusional), zero-order, and non-Fickian (anomalous) release kinetics are 0.5, 1, 0.5<n<1, respectively. The DH release profiles of most formulations were fitted to the Korsmeyer–Peppas model. The n values (between ≤ 0.5 and 0.96) indicated non-Fickian (anomalous) diffusion for the majority of the formulations. This indicates that the DH release rate is controlled by more than one process, i.e., erosion and diffusion.79,80 In contrast, formulations F8, F17 exhibited zero order release kinetic (n=1) (Table S2).
Ex-Vivo Permeation of DH-TENVs
Ex vivo permeation analyses provide valuable insights into in vivo performance.81 The ex vivo skin permeation analysis (Figure S2A–C) showed that the permeation of the DH-TENV formulations ranged from 174.3±15.12 to 493.8±30.08 µg/cm2 (Table 2), which indicated increased permeation compared to the DH solution (135.6±17.0 µg/cm2). This may have been due to the DH-TENVs composition involving both phospholipids and edge activators,55,82 their small vS creating a large surface area for increased contact of released DH with the skin surface, and ethanol-induced fluidization of the skin’s intercellular lipid layer. The DH-TENV formulations were superior to the DH solution regarding all permeation parameters (Table 3). The transdermal steady state flux (Jss) of the DH-TENV formulations ranged from 7.2±0.6 to 20.5±1.2 µg/cm2/h compared to only 5.6±0.7 µg/cm2/h for the DH solution. The enhancement ratios of the DH-TENV formulations compared to the DH solution ranged from 1.8 to 7.5. Equations (6) shows the quantitative effects of the independent variables on the cumulative amount of DH that permeated the rat skin specimen after 24 h (Q24).
(6)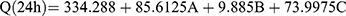
where A is PC, B is ethanol, and C is SDC
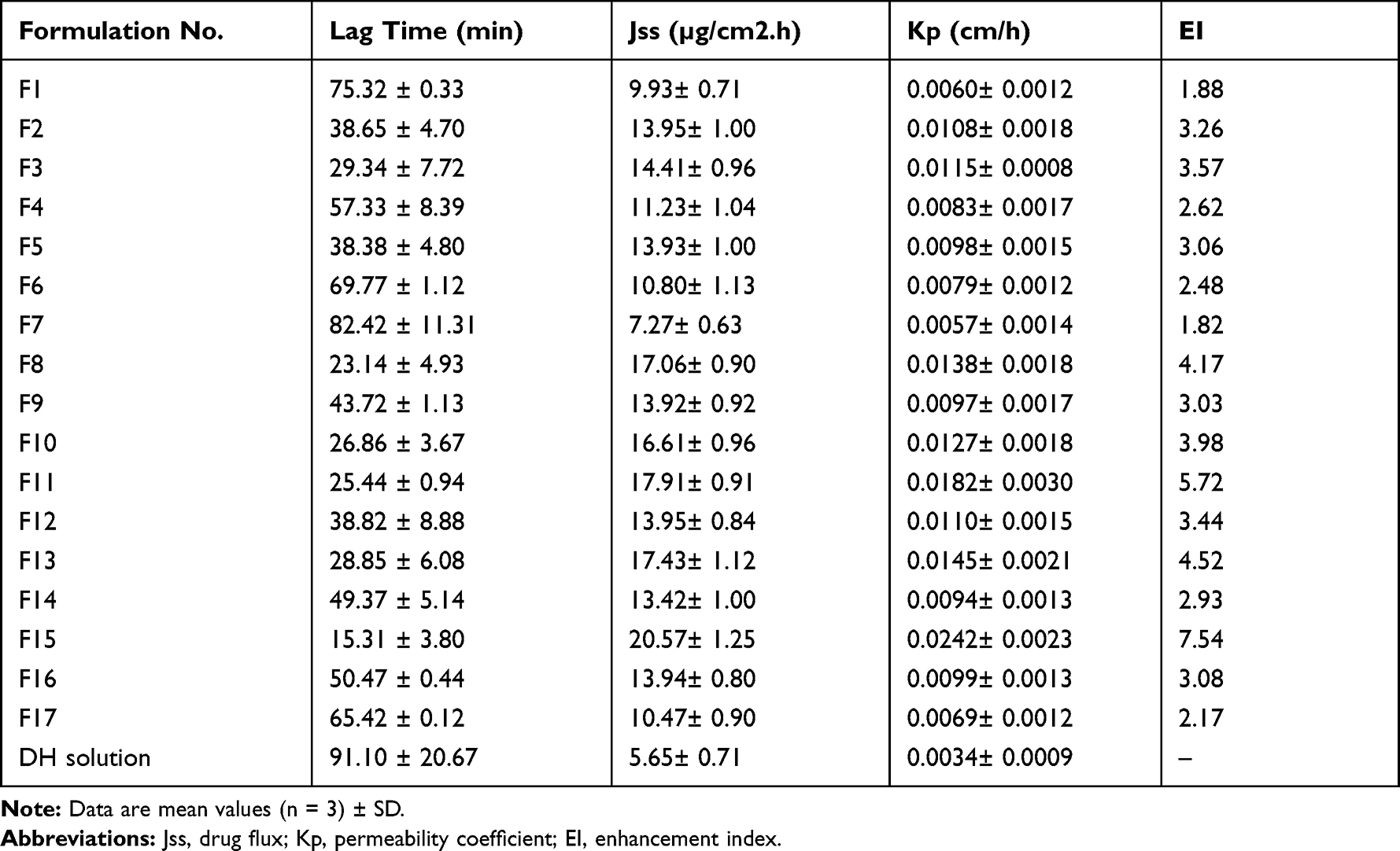 |
Table 3 Ex vivo Permeation Parameters of DH-TENVs Formulation versus DH Solution |
The independent variables significantly influenced the Q24 of DH-TENVs (p<0.05) (Figure 1E). All terms had a positive regression coefficient, indicating their positive impacts on diffusion. The positive effect of PC concentration on Q24 could be due to the presence of lipids helping the vesicles to fuse with the membrane lipid layers deep in the skin, supporting previous speculation.83
Additionally, 40% ethanol led to a significantly higher Q24 than 10% ethanol. A plausible explanation is that ethanol has a penetration-enhancing effect that lowers the lipid transition temperature, making the vesicles more flexible and thereby allowing them to penetrate the skin.16 Additionally, ethanol might interact with the polar head groups of the lipids in the intercellular lipid multilayers of the stratum corneum and induce stratum corneum fluidization, thereby increasing the penetration of elastic vesicles.84
The increase in Q24 with the increase in SDC concentration could be due to the ability of SDC to boost the vesicles’ flexibility by altering the bilayer packing characteristics. This would help the vesicles to squeeze through the skin pores.
Selection of the Optimized DH-TENV
After satisfactory selection of materials, we identified the DH-TENV formulations with the most suitable physicochemical properties. For optimization, Design Expert® software suggested several solutions representing diverse combinations of the levels of the independent variables that best satisfied the predetermined constraints. The combination of a PC concentration of 2.68% w/w, an ethanol concentration of 32.67% w/w, and a SDC concentration of 2% w/w led to an EE% of 52.62%, a VS of 216.53 nm, a ZP of −38.14 mV, a Q8h of 61.69%, and a Q24h of 467.87 µg/cm2, with an overall desirability of 0.684 (Figure 1F). Table 2 shows that the observed and predicted values of the optimized DH-TENV formulation are highly similar, with no significant differences (p>0.05). This confirmed the validity of Equations (2–6) and confirmed their prediction capability within data uncertainty limits.85
Morphology of the Optimized DH-TENV
TEM analysis was useful for investigating the morphology of the optimized DH-TENV formulation. The DH-TENV were approximately spherical with a smooth surface, appearing as black dots, well dispersed and well separated from each other. (Figure 2). Furthermore, the TEM image indicates the presence of a very thin layer around the particles, suggesting that they have a DH-enriched core.
 |
Figure 2 Transmission electron micrograph of the optimized DH-TENV formulation. |
DSC Analysis of the Optimized DH-TENV
DSC thermograms of pure DH, PC, SDC, and the optimized DH-TENV formulation are shown in Figure 3. The thermogram of DH exhibited an endothermic peak at 182.31°C, as reported previously for DH.86 The SDC thermograms of PC had an endothermic 147°C.87 The thermograph of SDC had a broad endothermic peak that started at 72.22°C and 117.40°C probably due to the loss of water molecules followed by an exothermic recrystallization peak at 196.90°C88. The thermogram of the optimized DH-TENV formulation indicated DH melting endotherm subsidence. The distinguishing endothermic peak of DH was no longer present, and the endothermic peaks of the surfactant/lipid bilayers were broadened and/or shifted. This suggests that DH interacts with the bilayer components, causing numerous lattice defects, with DH residing in amorphous regions; this may account for the enhanced entrapment of DH in the TENVs in an amorphous state.89
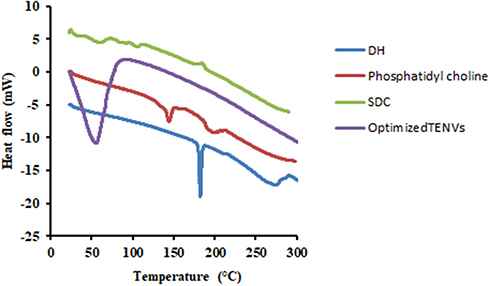 |
Figure 3 DSC thermograms of DH, Phosphatidyl choline, SDC and the optimized TENVs. |
Physical Stability of the Optimized DH-TENV
The stability, based on EE% and VS, of the optimized DH-TENV formulation after 90 days of storage at 4°C was assessed. There were minimal changes in EE% (decreased from 52.62±5.63% to 47.58±6.33%) and VS (increased from 216.53±6.65 to 224.57±14.05 nm) after 90 days (Figure 4). These changes were non-significant (p>0.05) according to one-way ANOVA. The stability of the optimized DH-TENV formulation may be ascribed to the rigidity of the vesicles and/or the ability of the vesicles to retain DH, as suggested previously.90
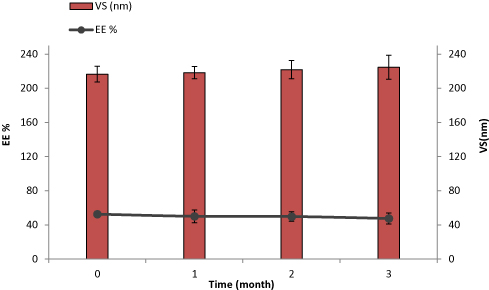 |
Figure 4 Effect of storage at 4°C for 3 months on entrapment efficiency (EE) and vesicle size (VS) of the optimized DH-TENV formulation. |
Skin Irritation Analysis of the Optimized DH-TENV
There was no evidence of adverse effects (erythema, edema, or irritation) at the DH-TENV gel application site. This indicates that the transdermal DH-TENV gel was safe and non-irritating. Histopathological analysis showed that the DH-TENV gel-treated skin had a normal structure compared to the untreated skin (Figure 5), suggesting good tolerance.
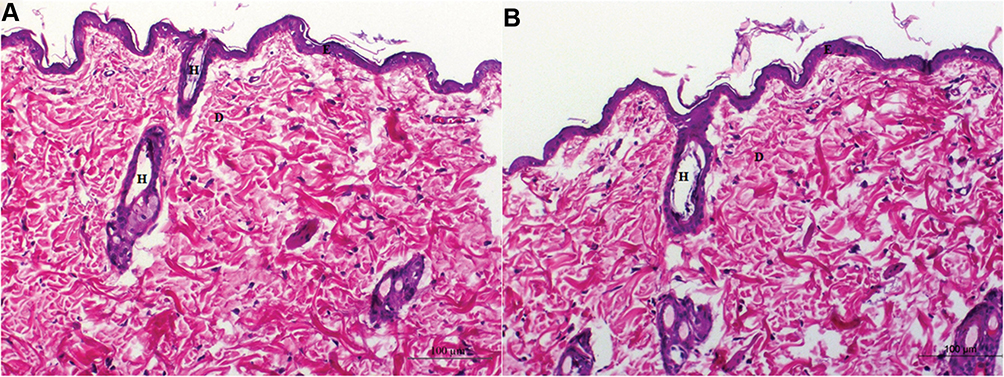 |
Figure 5 Light photomicrographs showing histopathological sections of (A) Normal untreated rat skin and (B) Rat skin treated with DH- TENV gel (X200 H&E stain). |
Pharmacokinetic Analysis of the Optimized DH-TENV
The pharmacokinetic parameters of all the DH-TENV formulations are provided in Table 4. The DH Cmax values of the oral DH solution, DH-TENV gel, and control DH gel were 197.6, 120.7 and 32.2 ng/mL, respectively, the tmax values were 1.50±0.06, 4.00±0.55, and 3.00±0.34 h, respectively, and the AUC0-∞ values were 733.6, 1773.3, and 426.3 ng·h/mL, respectively. Compared to the oral DH solution and control DH- gel, the Cmax value was significantly lower for the DH-TENV gel. For transdermal DH administration, the tmax values were significantly higher relative to oral administration and the differences were significant (p<0.05). This disparity may be due to the stratum corneum, which most likely slows the permeation of DH. In comparison, the oral DH solution results reflect the fact that it is an immediate-release administration form. The DH-TENV gel had a significantly higher tmax value than the control gel (4 h vs. 3 h; P ˂ 0.05). This could be because, unlike the control DH gel, the DH-TENV gel acts as a DH depot, providing a means of sustained release.13
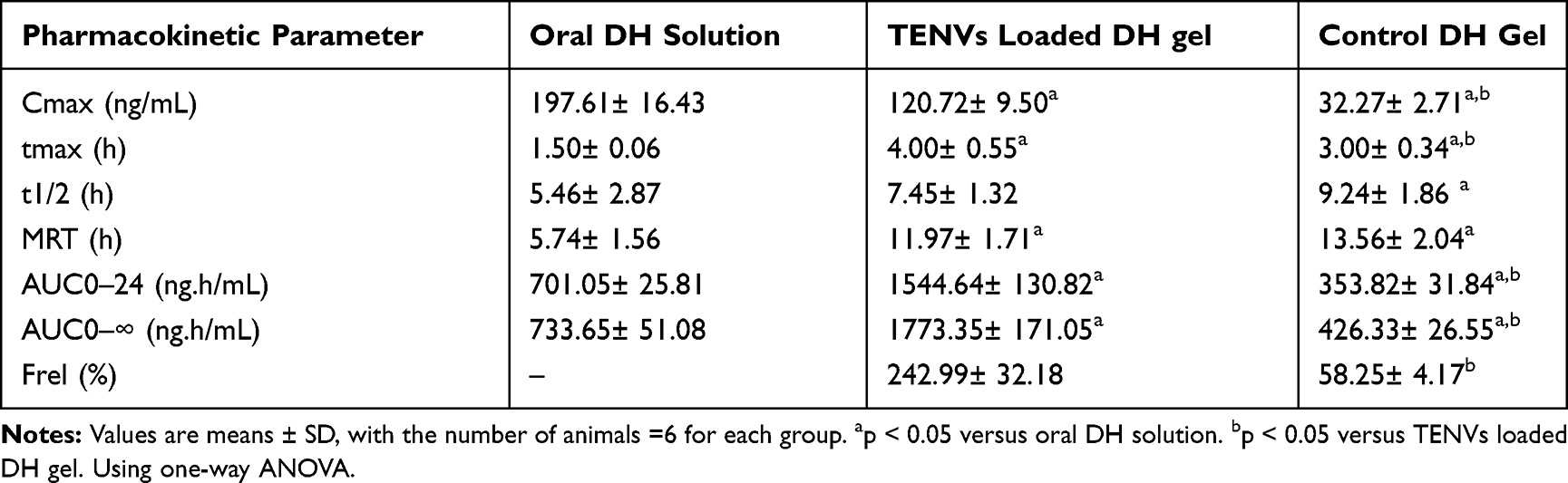 |
Table 4 Pharmacokinetic Parameters of DH in Rat Plasma After Administration of an Oral Solution, TENVs Loaded Gel, and Control Gel |
In addition, the mean AUC0-∞ value was 2.4-fold higher for the DH-TENV gel than the oral solution and the difference was significant (p<0.05). Compared with both the oral DH solution and the control DH gel, the DH-TENV gel had a significantly higher AUC0-24 value P˂ 0.05, meaning that it was more efficient in delivering DH to the systemic circulation. Compared to the oral DH solution, the improved bioavailability of the DH-TENV gel was most likely due to the avoidance of first-pass hepatic metabolism. In comparison, the improved bioavailability of the DH-TENV gel compared to the control DH gel may have been due to the capacity of the former gel to boost the permeation of DH. The relative bioavailability of DH in the DH-TENV gel and control DH gel was 242.9% and 58.25% respectively, compared to the oral DH solution.
In vivo Analysis of the Optimized DH-TENV in CFA-Induced RA in Rats
For the first time, a gel formulation of transdermal DH-TENV carrying the SSRI DH was studied. Its anti-RA activity was shown to involve alleviating the inflammatory cascade. Transdermal formulations are promising and innovative materials, as they maintain the drug concentration within the therapeutic window for a prolonged period, they are easy to prepare, and they are responsive to stimuli.91
Compared to the normal control group and the DH-TENV gel group, the untreated RA group had increased serum anti-CCP, COMP, and IL-6 levels (Table 5) and increased tissue expression of RANKL. This is in agreement with previous studies,92–94 which showed that RA is associated with significant increases in these biomarkers.
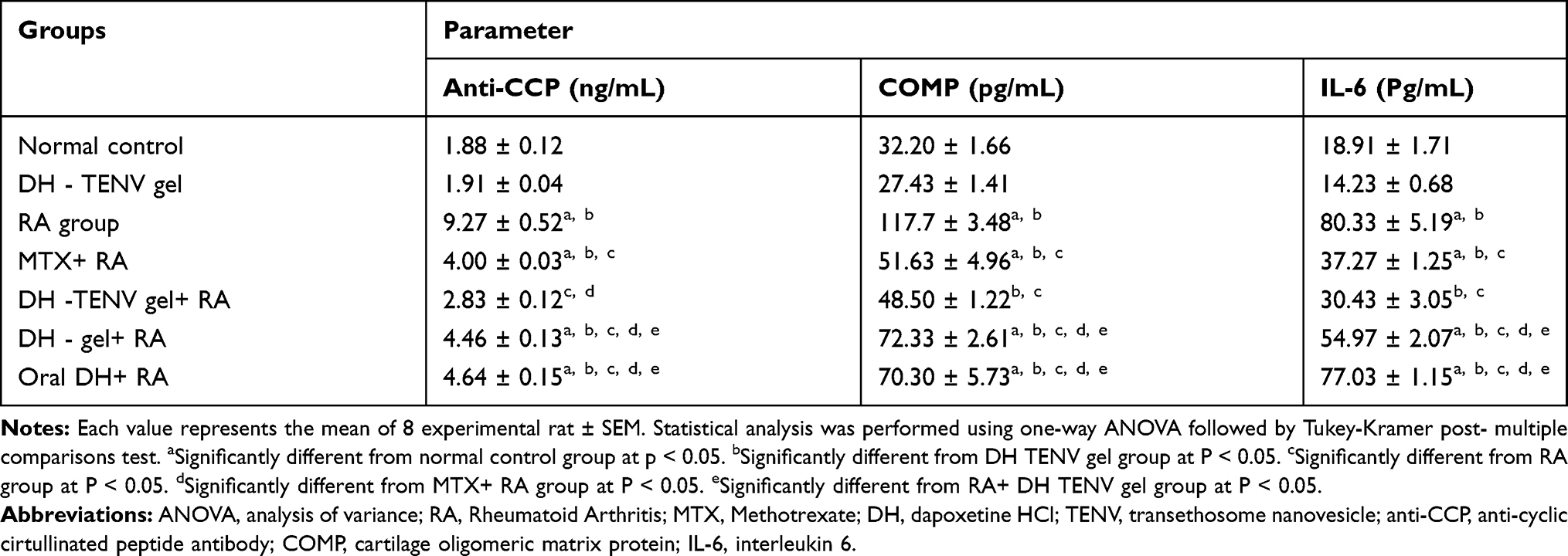 |
Table 5 Biochemical Analysis in Different Studied Groups |
All treated groups exhibited a significant decrease in anti-CCP level compared to the untreated RA group (Table 5). These findings are in agreement with a study by Sacre et al,95 which highlighted the potential anti-RA effect of SSRIs. Notably, the DH-TENV gel+RA group had a larger decrease in anti-CCP than the MTX+RA, DH gel+RA, and oral DH+RA groups. These findings are supported by previous research96 on the therapeutic advantages of nano-gels compared to other formulations.
Increased serum COMP level is indicative of RA (Table 5). The MTX+RA and DH-TENV gel+RA groups had nearly normalized COMP levels. The DH gel+RA and oral DH+RA groups had a significantly smaller effect on COMP compared to the MTX+RA and DH-TENV gel+RA groups. No previous studies have assessed the effect of SSRIs related drugs on the COMP level.
Research shows that IL-6 may be an inflammatory mediator that can lead to joint inflammation in RA. Moreover, anti–IL-6 monoclonal antibodies may represent a new RA treatment.97 The MTX+RA and DH-TENV gel+RA groups had significantly decreased IL-6 levels compared to the untreated RA group. Many studies have shown the anti–inflammatory effects of SSRIs. For example, Durairaj et al98 showed that the SSRI paroxetine has critical but differential effects on IL-6 and TNF-α production in macrophages, which likely involves distinct mechanisms. Furthermore, Fonseka et al99 reported that an SSRI can modulate the elevated IL-6- levels associated with major depressive disorder. The DH gel+RA group had a significantly smaller effect on IL-6 compared to the MTX+RA and DH-TENV gel+RA groups. The oral DH+RA group had no significant effect on IL-6 compared to the untreated RA group.
Administration of DH-TENV gel to RA rats significantly (P < 0.05) decreased tissue RANKL expression compared to the untreated RA group, with a significantly greater decrease compared to MTX, DH gel, or oral DH (Figure 6). These data agree with a previous study40 that showed that the SSRI paroxetine had an anti-RA effect on CFA-induced RA by modulating RANKL expression.
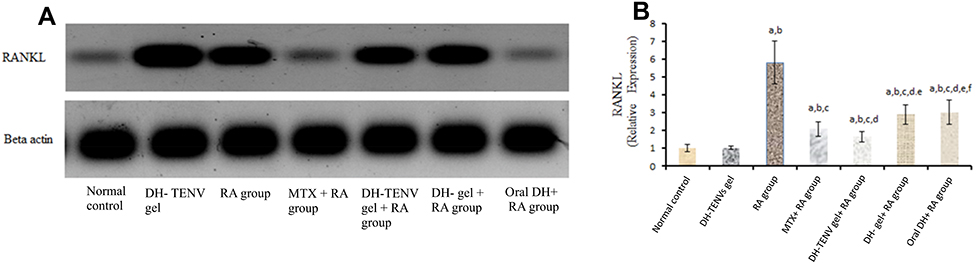 |
Figure 6 Effect of transdermal application of DH- TENV gel on RANKL relative expression as compared to arthritic group using Western blot analysis. Each value represents the mean of 8 experimental rat ± SEM. Statistical analysis was performed using one-way ANOVA followed by Tukey-Kramer post- multiple comparisons test. aSignificantly different from normal control group at p < 0.05. bSignificantly different from DH -TENV gel group at P < 0.05. cSignificantly different from RA group at P < 0.05. dSignificantly different from MTX+ RA group at P < 0.05. eSignificantly different from DH-TENV gel+ RA group at P < 0.05. fSignificantly different from oral DH+ RAgroup at P < 0.05. Abbreviations: ANOVA, analysis of variance; RA, Rheumatoid Arthritis; MTX, Methotrexate; DH, dapoxetine HCl; TENV, transethosome nanovesicle; RANKL, Receptor activator of nuclear factor kappa-Β ligand; B-Actin, Beta-Actin. |
The radiological changes in normal control, RA, and treated RA rats are shown in Figure 7. The untreated RA group had excessive soft tissue volume, joint space, narrowing, subchondral erosion, and degenerative joint changes with signs of inflammation on the radiographs. Treatment of RA rats with DH-TENV gel significantly inhibited soft tissue swelling (surrounding the foot bones), and bone enlargement in (Figure 7). The DH-TENV gel was more effective than the other treatments, as DH was targeted to the site of inflammation, reducing the inflammation and halting disease progression. DH oral solution and DH gel were associated with milder bone erosion and inflammation compared to DH-TENV.
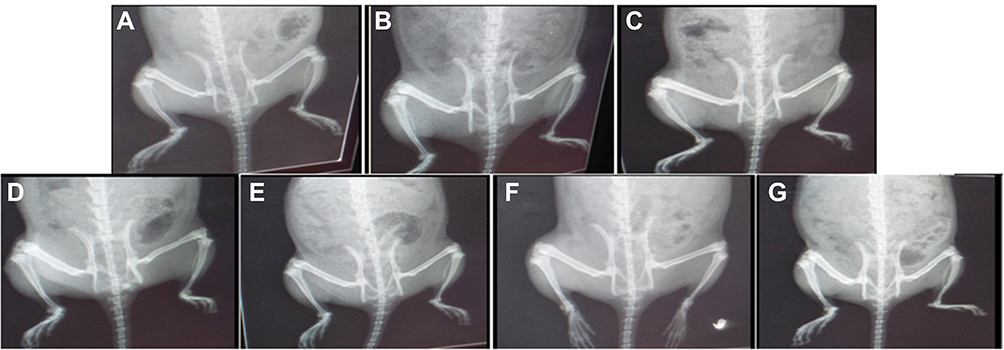 |
Figure 7 Radiographic analysis of rat hind paw from different groups. (A) Normal control group and (B) DH - TENV gel group showed normal tissue with no signs of inflammation, bone enlargement or bone damage. (C) Complete Freund’s adjuvant (CFA) arthritic group showed observable excess soft tissue volume, joint space, sub-chondral erosion, periostitis, osteolysis, subluxation, degenerative joint changes with signs of inflammation at the metatarsal-phalangeal joint and the regions in-between the bones of the phalanges and the metatarsals. (D) Rheumatoid arthritis group treated with methotrexate showed almost normal soft tissue with disappearance of inflammatory signs and no bone enlargement was observed. (E) Rats treated with DH - gel, (F) DH - TENV gel and (G) Oral DH exhibited significant and comparable inhibition of soft tissues swelling that surrounded the bones of the foot and bone enlargement. |
The biochemical and radiographic results were confirmed by histopathological findings regarding the knee joints, spleen, and liver. The DH-TENV gel-treated RA rat knee joint sections showed considerable amelioration of joint tissue damage compared to sections from untreated RA rats, and the former rats had normal red and white pulp in the spleen and improved hepatocytes in hepatic cords (Figure 8).
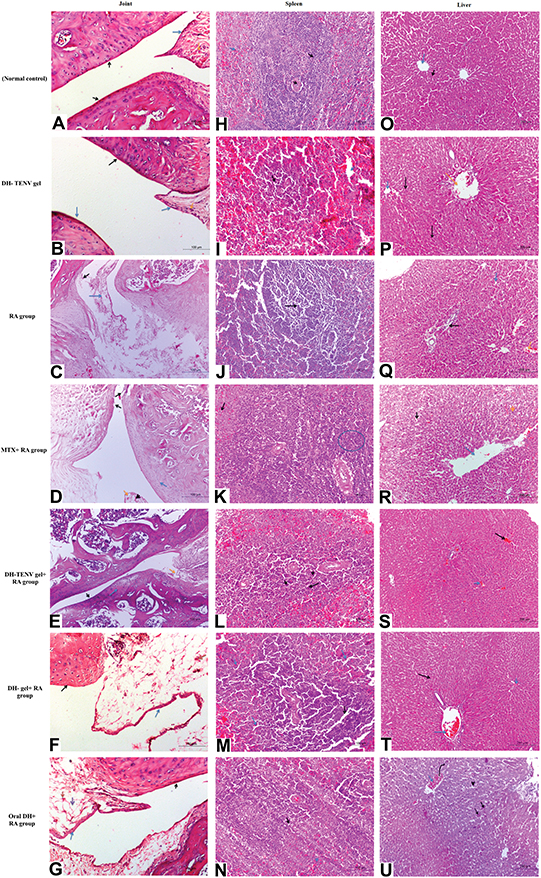 |
Figure 8 Photomicrographs of joint sections, stained with routine H&E stain (×100) and obtained from different groups. (A) Sections from normal control rats showing articulating ends of two bones that covered by articular surface (black arrow) and normal synovial membrane (blue arrow) lining the inner surface of capsule with normal meniscus (orange arrow). (B) Sections from DH - TENV gel group illustrated normal articular surface (black arrow) with normal synovial membrane (blue arrow) and normal meniscus (orange arrow). (C) RA- arthritic rats showing bone in the focal manner in the articular surface (black arrow). Also resorption of meniscus is appeared, as well as the resorbed area is reached with collaged and fibroblast cell together with a few chondroblast (blue arrow). (D) RA group that treated with MIX showed slight smooth articular surface (black arrow) with thickened articular cartilage with proliferating chondrocytes (blue arrow) and synovial membrane showing congestion (arrow head) and edema (orange arrow). (E) Articular surface of rheumatoid arthritis group that treated with DH - TENV gel showed smooth articular surface (black arrow). The proliferating chondrocytes showing hypercellularity, cloning and slightly ordered distribution (blue arrow) and nearly normal synovial membrane (orange arrow). (F) The joint capsule and most of the synovial membrane of RA- treated gel group showed slight normal articular surface (black arrow) and synovial membrane showed focal inflammatory cell aggregation, moderate edema, capillary are increased in number, thickness and hypercellularity (blue arrow). (G) Rheumatoid arthritis group that treated with oral DH showed rough cerrated articular surface (black arrow), smooth intact synovial membrane (blue arrow) and the associated connective tissue showing multiple capillaries (red arrow) and moderate edema (purple arrow). Photomicrographs sections of spleen that obtained from different groups and stained with routine H&E stain (×100) were are followed: (H) Normal control rats showing Normal white pulb containing follicular artery (black arrow) and normal red pulb containing numerous blood sinusoids (blue arrow). (I) Sections from DH- TENV gel group illustrated normal white pulb (black arrow) with dilated in sinusoid in red pulb (blue arrow). (J) RA- arthritic rats showing marked atrophy of white pulp (black arrow). (K) RA group that treated with MIX showed hyperplagia in the white pulb (blue circle), congestion and dilation in sinusoid of the red pulp (black arrow). (L) Sections from DH - TENV gel group illustrated hyperplasia in white pulb with multiple macrophages indicated immunoreaction (black arrow). (M) spleen sections of RA- treated gel group showed Hyperplasia in white pulb (black arrow) and dilation in sinusoid in the red pulb (blue arrow). (N) Rheumatoid arthritis group that treated with oral DH showed marked activation in the macrophage in the white pulb (black arrow) and nearly normal red pulb (blue arrow). Liver photomicrographs sections obtained from different groups and stained with routine H&E stain (×200) were are followed: (O) Normal control rats showing normal liver with normal central vein (blue arrow) with normal hepatic cords (black arrow). (P) Sections from DH - TENV gel group illustrated nearly normal liver with normal central vein (blue arrow) and portal area (orange arrow). (Q) RA- arthritic rats showing slight congestion (orange arrow) and edema (black arrow) together with sever vascular degeneration in the hepatocyte (blue arrow). (R) RA group that treated with MIX showed dilated central vein (blue arrow) and dilation in hepatic sinusoid (black arrow) with some with vascular degeneration in the hepatocyte (arrow head). (S) Sections from DH - TENV gel group illustrated nearly normal liver except slight congestion in central (black arrow) and portal vein (blue arrow). (T) liver sections of RA- treated gel group showed slight congestion in both central vein and portal vein (blue arrow) and hepatic cord and hepatocyte appear normally (black arrow). (U) Rheumatoid arthritis group that treated with DH oral solution showed marked congestion of central veins (blue arrow) with dilated engorged sinusoid (arrow head) and focal coagulative necrosis of sore hepatocyte (black arrow). |
Together, the findings strongly suggest that DH-TENV gel has an anti-RA effect that can control RA progression, at least partly by inhibiting serum anti-CCP, COMP, and IL-6 levels and tissue RANKL gene expression. Additionally, this anti-RA effect of DH-TENV gel is greater than the anti-RA effects of DH in oral or simple gel formulations
Conclusion
Novel ultra-deformable vesicular carriers (TENVs) were developed for the transdermal delivery of DH to treat RA. The DH-TENVs were prepared by an injection sonication method and optimized using a Box–Behnken design. The optimized DH-TENVs contained 2.68% (w/w) of PC, 32.67% (w/w) of ethanol, and 2% (w/w) of SDC. This led to an EE% of 52.62%, a VS of 216.53 nm, a ZP of −38.14 mV, a Q8h of 61.69%, and a Q24h of 467.87µg/cm2. Pharmacokinetic analysis showed that DH-TENV gel had significantly higher AUC0-t values and slower elimination rate compare to oral DH solution, indicating sustained DH release from the DH-TENVs. Moreover, the bioavailability of DH after DH-TENV gel administration was approximately 242.99% greater than that of oral DH solution. The DH-TENV gel clearly had a superior anti-RA effect compared to MTX, DH gel, and DH oral solution. Our experiments showed that the DH-TENV gel formulation effectively prevented the aggressive progression of RA and could be used rather than MTX (to minimize side effects). The future research required includes equipping the optimized DH-TENV formulation with factors that allow drug targeting and improved RA management.
Acknowledgments
We express our gratitude to Dr. Omnia Ahmed Abdel El-Gaphar, Department of Pharmacology, Faculty of Pharmacy, Nahda University for her help in pharmacological studies.
Author Contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Choy E. Understanding the dynamics: pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology. 2012;51(suppl_5):3–11. doi:10.1093/rheumatology/kes113
2. Ahmed YM, Messiha BAS, Abo-Saif AA. Granisetron and carvedilol can protect experimental rats against adjuvant-induced arthritis. Immunopharmacol Immunotoxicol. 2017;39(2):97–104. doi:10.1080/08923973.2017.1286502
3. Riemekasten G, Siegert E. Geschlechtsspezifische Unterschiede des Immunsystems. Zeitschrift für Rheumatologie. 2014;73(7):600–606. doi:10.1007/s00393-014-1357-4
4. Cutolo M, Sulli A, Capellino S, et al. Sex hormones influence on the immune system: basic and clinical aspects in autoimmunity. Lupus. 2004;13(9):635–638. doi:10.1191/0961203304lu1094oa
5. Kumar LD, Karthik R, Gayathri N, et al. Advancement in contemporary diagnostic and therapeutic approaches for rheumatoid arthritis. Biomed Pharmacother. 2016;79:52–61. doi:10.1016/j.biopha.2016.02.001
6. Lampropoulos C, Orfanos P, Bournia V-K, et al. Adverse events and infections in patients with rheumatoid arthritis treated with conventional drugs or biologic agents: a real world study. Clin Exp Rheumatol. 2015;33(2):216–224.
7. Xia Z, Depierre JW, Nässberger L. Tricyclic antidepressants inhibit IL-6, IL-1β and TNF-α release in human blood monocytes and IL-2 and interferon-γ in T cells. Immunopharmacology. 1996;34(1):27–37. doi:10.1016/0162-3109(96)00111-7
8. Bowen PD. Use of selective serotonin reuptake inhibitors in the treatment of depression in older adults: identifying and managing potential risk for hyponatremia. Geriatr Nurs (Minneap). 2009;30(2):85–89. doi:10.1016/j.gerinurse.2008.04.007
9. McCarty E, Dinsmore W. Dapoxetine: an evidence-based review of its effectiveness in treatment of premature ejaculation. Core Evid. 2012;7:1. doi:10.2147/CE.S13841
10. Taler M, Gil-Ad I, Lomnitski L, et al. Immunomodulatory effect of selective serotonin reuptake inhibitors (SSRIs) on human T lymphocyte function and gene expression. Eur Neuropsychopharmacol. 2007;17(12):774–780. doi:10.1016/j.euroneuro.2007.03.010
11. McMahon CG. Dapoxetine: a new option in the medical management of premature ejaculation. Ther Adv Urol. 2012;4(5):233–251. doi:10.1177/1756287212453866
12. Burger C, Shahzad Y, Brummer A, et al. Traversing the skin barrier with nano-emulsions. Curr Drug Deliv. 2017;14(4):458–472. doi:10.2174/1567201813666160824125444
13. Vogt A, Wischke C, Neffe AT, et al. Nanocarriers for drug delivery into and through the skin—do existing technologies match clinical challenges? J Controlled Release. 2016;242:3–15. doi:10.1016/j.jconrel.2016.07.027
14. Ashtikar M, Nagarsekar K, Fahr A. Transdermal delivery from liposomal formulations–Evolution of the technology over the last three decades. J Controlled Release. 2016;242:126–140. doi:10.1016/j.jconrel.2016.09.008
15. Cevc G, Blume G. Lipid vesicles penetrate into intact skin owing to the transdermal osmotic gradients and hydration force. Biochim Biophys Acta Bioenerg. 1992;1104(1):226–232. doi:10.1016/0005-2736(92)90154-E
16. Touitou E, Dayan N, Bergelson L, et al. Ethosomes—novel vesicular carriers for enhanced delivery: characterization and skin penetration properties. J Controlled Release. 2000;65(3):403–418. doi:10.1016/S0168-3659(99)00222-9
17. Song CK, Balakrishnan P, Shim C-K, et al. A novel vesicular carrier, transethosome, for enhanced skin delivery of voriconazole: characterization and in vitro/in vivo evaluation. Colloids and Surf B. 2012;92:299–304. doi:10.1016/j.colsurfb.2011.12.004
18. Abdulbaqi IM, Darwis Y, Abdul Karim Khan N, et al. Ethosomal nanocarriers: the impact of constituents and formulation techniques on ethosomal properties, in vivo studies, and clinical trials. Int J Nanomedicine. 2016;11:2279. doi:10.2147/IJN
19. Meng M, Chen Z, Yang L, et al. Enhanced transdermal bioavailability of testosterone propionate via surfactant-modified ethosomes. Int J Nanomedicine. 2013;8:3051. doi:10.2147/IJN
20. Thapa C, Ahad A, Aqil M, et al. Formulation and optimization of nanostructured lipid carriers to enhance oral bioavailability of telmisartan using Box–Behnken design. J Drug Deliv Sci Technol. 2018;44:431–439. doi:10.1016/j.jddst.2018.02.003
21. Mahmood S, Taher M, Mandal UK. Experimental design and optimization of raloxifene hydrochloride loaded nanotransfersomes for transdermal application. Int J Nanomedicine. 2014;9:4331.
22. Ahmad J, Kohli K, Mir SR, et al. Formulation of self-nanoemulsifying drug delivery system for telmisartan with improved dissolution and oral bioavailability. J Dispers Sci Technol. 2011;32(7):958–968. doi:10.1080/01932691.2010.488511
23. Ali MF, Salem HF, Abdelmohsen HF, Attia SK. Preparation and clinical evaluation of nano-transferosomes for treatment of erectile dysfunction. Drug Des Devel Ther. 2015;9:2431.
24. Joshi M, Pathak S, Sharma S, et al. Design and in vivo pharmacodynamic evaluation of nanostructured lipid carriers for parenteral delivery of artemether: nanoject. Int J Pharm. 2008;364(1):119–126. doi:10.1016/j.ijpharm.2008.07.032
25. Higuchi T. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 1963;52(12):1145–1149. doi:10.1002/jps.2600521210
26. Korsmeyer RW, Gurny R, Doelker E, et al. Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15(1):25–35. doi:10.1016/0378-5173(83)90064-9
27. Alam S, Aslam M, Khan A, et al. Nanostructured lipid carriers of pioglitazone for transdermal application: from experimental design to bioactivity detail. Drug Deliv. 2016;23(2):601–609. doi:10.3109/10717544.2014.923958
28. Fouad SA, Shamma RN, Basalious EB, et al. Novel instantly-soluble transmucosal matrix (ISTM) using dual mechanism solubilizer for sublingual and nasal delivery of dapoxetine hydrochloride: in-vitro/in-vivo evaluation. Int J Pharm. 2016;505(1–2):212–222. doi:10.1016/j.ijpharm.2016.04.006
29. Al-mahallawi AM, Khowessah OM, Shoukri RA. Nano-transfersomal ciprofloxacin loaded vesicles for non-invasive trans-tympanic ototopical delivery: in-vitro optimization, ex-vivo permeation studies, and in-vivo assessment. Int J Pharm. 2014;472(1–2):304–314. doi:10.1016/j.ijpharm.2014.06.041
30. Salem HF, Kharshoum RM, Abou-Taleb HA, AbouTaleb HA, AbouElhassan KM. Progesterone-loaded nanosized transethosomes for vaginal permeation enhancement: formulation, statistical optimization, and clinical evaluation in anovulatory polycystic ovary syndrome. J Liposome Res. 2018;29:1–12.
31. Manconi M, Mura S, Sinico C, et al. Development and characterization of liposomes containing glycols as carriers for diclofenac. Colloids Surf a Physicochem Eng Asp. 2009;342(1–3):53–58. doi:10.1016/j.colsurfa.2009.04.006
32. Albash R, Abdelbary A, Refai H, et al. Use of transethosomes for enhancing the transdermal delivery of olmesartan medoxomil: in vitro, ex vivo, and in vivo evaluation. Int J Nanomedicine. 2019;14:1953. doi:10.2147/IJN
33. Nasr M, Mansour S, Mortada ND, et al. Vesicular aceclofenac systems: a comparative study between liposomes and niosomes. J Microencapsul. 2008;25(7):499–512. doi:10.1080/02652040802055411
34. Bancroft JD, Gamble M. Theory and Practice of Histological Techniques. Elsevier health sciences; 2008.
35. Nadeem RI, Ahmed HI, Ezz-El-Din S. Effect of imipramine, paroxetine, and lithium carbonate on neurobehavioral changes of streptozotocin in rats: impact on glycogen synthase kinase-3 and blood glucose level. Neurochem Res. 2015;40(9):1810–1818. doi:10.1007/s11064-015-1670-6
36. Modi NB, Dresser MJ, Simon M, et al. Single-and multiple-dose pharmacokinetics of dapoxetine hydrochloride, a novel agent for the treatment of premature ejaculation. J Clin Pharmacol. 2006;46(3):301–309. doi:10.1177/0091270005284850
37. Zhang Y, Huo M, Zhou J, et al. PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in microsoft excel. Comput Methods Programs Biomed. 2010;99(3):306–314. doi:10.1016/j.cmpb.2010.01.007
38. Wahba MGF, Messiha BAS, Abo-Saif AA. Ramipril and haloperidol as promising approaches in managing rheumatoid arthritis in rats. Eur J Pharmacol. 2015;765:307–315. doi:10.1016/j.ejphar.2015.08.026
39. Wang K, Zhang D, Liu Y, et al. Traditional Chinese medicine formula Bi-Qi capsule alleviates rheumatoid arthritis-induced inflammation, synovial hyperplasia, and cartilage destruction in rats. Arthritis Res Ther. 2018;20(1):43. doi:10.1186/s13075-018-1547-6
40. Shafiey SI, Mohamed WR, Abo-Saif AA. Paroxetine and rivastigmine mitigates adjuvant-induced rheumatoid arthritis in rats: impact on oxidative stress, apoptosis and RANKL/OPG signals. Life Sci. 2018;212:109–118. doi:10.1016/j.lfs.2018.09.046
41. Coenen D, Verschueren P, Westhovens R, et al. Technical and diagnostic performance of 6 assays for the measurement of citrullinated protein/peptide antibodies in the diagnosis of rheumatoid arthritis. Clin Chem. 2007;53(3):498–504. doi:10.1373/clinchem.2006.078063
42. Hibi M, Nakajima K, Hirano T. IL-6 cytokine family and signal transduction: a model of the cytokine system. J Mol Med. 1996;74(1):1–12. doi:10.1007/BF00202068
43. Williams NE. Immunoprecipitation procedures. Method Cell Biol. 1999;2000(62);449–453. Elsevier.
44. Seevaratnam R, Patel BP, Hamadeh MJ. Comparison of total protein concentration in skeletal muscle as measured by the Bradford and Lowry assays. J Biochem. 2009;145(6):791–797. doi:10.1093/jb/mvp037
45. Bhalekar MR, Upadhaya PG, Madgulkar AR. Fabrication and efficacy evaluation of chloroquine nanoparticles in CFA-induced arthritic rats using TNF-α ELISA. Eur J Pharm Sci. 2016;84:1–8. doi:10.1016/j.ejps.2016.01.009
46. Ahmad A, Alkharfy KM, Wani TA, et al. Application of Box–Behnken design for ultrasonic-assisted extraction of polysaccharides from Paeonia emodi. Int J Biol Macromol. 2015;72:990–997. doi:10.1016/j.ijbiomac.2014.10.011
47. Li JC, Zhu N, Zhu JX, et al. Self-assembled cubic liquid crystalline nanoparticles for transdermal delivery of paeonol. Medical science monitor. Int Med j Exp Clin Res. 2015;21:3298.
48. Annadurai G, Ling LY, Lee JF. Statistical optimization of medium components and growth conditions by response surface methodology to enhance phenol degradation by Pseudomonas putida. J Hazard Mater. 2008;151(1):171–178. doi:10.1016/j.jhazmat.2007.05.061
49. Ezzat SM, Salama MM, ElMeshad AN, et al. HPLC–DAD–MS/MS profiling of standardized rosemary extract and enhancement of its anti-wrinkle activity by encapsulation in elastic nanovesicles. Arch Pharm Res. 2016;39(7):912–925. doi:10.1007/s12272-016-0744-6
50. Singh B, Mehta G, Kumar R, et al. Design, development and optimization of nimesulide-loaded liposomal systems for topical application. Curr Drug Deliv. 2005;2(2):143–153. doi:10.2174/1567201053585985
51. Lopez-Pinto J, Gonzalez-Rodriguez M, Rabasco A. Effect of cholesterol and ethanol on dermal delivery from DPPC liposomes. Int J Pharm. 2005;298(1):1–12. doi:10.1016/j.ijpharm.2005.02.021
52. Ahad A, Raish M, Al-Mohizea AM, et al. Enhanced anti-inflammatory activity of carbopol loaded meloxicam nanoethosomes gel. Int J Biol Macromol. 2014;67:99–104. doi:10.1016/j.ijbiomac.2014.03.011
53. El-Menshawe SF, Ali AA, Halawa AA, et al. A novel transdermal nanoethosomal gel of betahistine dihydrochloride for weight gain control: in-vitro and in-vivo characterization. Drug Des Devel Ther. 2017;11:3377. doi:10.2147/DDDT
54. Salem HF, Kharshoum RM, Abou-Taleb HA, et al. Progesterone-loaded nanosized transethosomes for vaginal permeation enhancement: formulation, statistical optimization, and clinical evaluation in anovulatory polycystic ovary syndrome. J Liposome Res. 2019;29(2):183–194. doi:10.1080/08982104.2018.1524483
55. El Zaafarany GM, Awad GAS, Holayel SM, et al. Role of edge activators and surface charge in developing ultradeformable vesicles with enhanced skin delivery. Int J Pharm. 2010;397(1–2):164–172. doi:10.1016/j.ijpharm.2010.06.034
56. El-Say KM, Abd-Allah FI, Lila AE, et al. Diacerein niosomal gel for topical delivery: development, in vitro and in vivo assessment. J Liposome Res. 2016;26(1):57–68. doi:10.3109/08982104.2015.1029495
57. Shaji J, Lal M. Preparation, optimization and evaluation of transferosomal formulation for enhanced transdermal delivery of a COX-2 inhibitor. Int J Pharm Pharm Sci. 2014;6(1):467–477.
58. Cho HJ, Park JW, Yoon IS, Kim DD. Surface-modified solid lipid nanoparticles for oral delivery of docetaxel: enhanced intestinal absorption and lymphatic uptake. Int J Nanomedicine. 2014;9:495.
59. Ansari MD, Ahmed S, Imam SS, et al. CCD based development and characterization of nano-transethosome to augment the antidepressant effect of agomelatine on Swiss albino mice. J Drug Deliv Sci Technol. 2019;54:101234. doi:10.1016/j.jddst.2019.101234
60. Ahad A, Aqil M, Kohli K, et al. Formulation and optimization of nanotransfersomes using experimental design technique for accentuated transdermal delivery of valsartan. Nanomedicine. 2012;8(2):237–249. doi:10.1016/j.nano.2011.06.004
61. Chourasia MK, Kang L, Chan SY. Nanosized ethosomes bearing ketoprofen for improved transdermal delivery. Results Pharma Sci. 2011;1(1):60–67. doi:10.1016/j.rinphs.2011.10.002
62. Dubey V, Mishra D, Dutta T, et al. Dermal and transdermal delivery of an anti-psoriatic agent via ethanolic liposomes. J Controlled Release. 2007;123(2):148–154. doi:10.1016/j.jconrel.2007.08.005
63. Ahad A, Aqil M, Kohli K, et al. Enhanced transdermal delivery of an anti-hypertensive agent via nanoethosomes: statistical optimization, characterization and pharmacokinetic assessment. Int J Pharm. 2013;443(1–2):26–38. doi:10.1016/j.ijpharm.2013.01.011
64. Verma P, Pathak K. Nanosized ethanolic vesicles loaded with econazole nitrate for the treatment of deep fungal infections through topical gel formulation. Nanomedicine. 2012;8(4):489–496. doi:10.1016/j.nano.2011.07.004
65. Dora CP, Singh SK, Kumar S, et al. Development and characterization of nanoparticles of glibenclamide by solvent displacement method. Acta Pol Pharm. 2010;67(3):283–290.
66. Aboud HM, El Komy MH, Ali AA, et al. Development, optimization, and evaluation of carvedilol-loaded solid lipid nanoparticles for intranasal drug delivery. AAPS PharmSciTech. 2016;17(6):1353–1365. doi:10.1208/s12249-015-0440-8
67. Muller R, Jacobs C, Kayse O. Nanosuspension as particulate drug formulations in therapy: rationale for development and what we expect in future. Adv Drug Deliv Rev. 2001;47:3–19. doi:10.1016/S0169-409X(00)00118-6
68. Garcia-Manyes S, Oncins G, Sanz F. Effect of pH and ionic strength on phospholipid nanomechanics and on deposition process onto hydrophilic surfaces measured by AFM. Electrochim Acta. 2006;51(24):5029–5036. doi:10.1016/j.electacta.2006.03.062
69. Egawa H, Furusawa K. Liposome adhesion on mica surface studied by atomic force microscopy. Langmuir. 1999;15(5):1660–1666. doi:10.1021/la980923w
70. Makino K, Yamada T, Kimura M, et al. Temperature-and ionic strength-induced conformational changes in the lipid head group region of liposomes as suggested by zeta potential data. Biophys Chem. 1991;41(2):175–183. doi:10.1016/0301-4622(91)80017-L
71. Shah SM, Ashtikar M, Jain AS, et al. LeciPlex, invasomes, and liposomes: a skin penetration study. Int J Pharm. 2015;490(1–2):391–403. doi:10.1016/j.ijpharm.2015.05.042
72. Shatalebi M, Mostafavi S, Moghaddas A. Niosome as a drug carrier for topical delivery of N-acetyl glucosamine. Res Pharm Sci. 2010;5(2):107.
73. Mitkari B, Korde SA, Mahadik KR, Kokare CR. Formulation and evaluation of topical liposomal gel for fluconazole. Indian J Pharm Educ Res. 2010;44(4):324–333.
74. Shaji J, Lal M. Novel double loaded transferosomes: evidence of superior anti-inflammatory efficacy-a comparative study. Int J Curr Pharm Res. 2014;6(2):16–25.
75. Aboud HM, Ali AA, El-Menshawe SF, et al. Nanotransfersomes of carvedilol for intranasal delivery: formulation, characterization and in vivo evaluation. Drug Deliv. 2016;23(7):2471–2481. doi:10.3109/10717544.2015.1013587
76. Song H, Wen J, Li H, et al. Enhanced transdermal permeability and drug deposition of rheumatoid arthritis via sinomenine hydrochloride-loaded antioxidant surface transethosome. Int J Nanomedicine. 2019;14:3177. doi:10.2147/IJN
77. Moawad FA, Ali AA, Salem HF. Nanotransfersomes-loaded thermosensitive in situ gel as a rectal delivery system of tizanidine HCl: preparation, in vitro and in vivo performance. Drug Deliv. 2017;24(1):252–260. doi:10.1080/10717544.2016.1245369
78. Hinze WL, Pramauro E. A critical review of surfactant-mediated phase separations (cloud-point extractions): theory and applications. Crit Rev Anal Chem. 1993;24(2):133–177. doi:10.1080/10408349308048821
79. Saindane NS, Pagar KP, Vavia PR. Nanosuspension based in situ gelling nasal spray of carvedilol: development, in vitro and in vivo characterization. AAPS PharmSciTech. 2013;14(1):189–199. doi:10.1208/s12249-012-9896-y
80. Gad HA, El-Nabarawi MA, El-Hady SSA. Formulation and evaluation of PLA and PLGA in situ implants containing secnidazole and/or doxycycline for treatment of periodontitis. AAPS PharmSciTech. 2008;9(3):878. doi:10.1208/s12249-008-9126-9
81. Ammar HO, Ghorab M, El-Nahhas SA, et al. Polymeric matrix system for prolonged delivery of tramadol hydrochloride, part II: biological evaluation. AAPS PharmSciTech. 2009;10(3):1065. doi:10.1208/s12249-009-9294-2
82. González-Rodríguez M, Arroyo CM, Cózar-Bernal MJ, et al. Deformability properties of timolol-loaded transfersomes based on the extrusion mechanism. Statistical optimization of the process. Drug Dev Ind Pharm. 2016;42(10):1683–1694. doi:10.3109/03639045.2016.1165691
83. Pal HC, Sharma S, Strickland LR, et al. Fisetin inhibits human melanoma cell invasion through promotion of mesenchymal to epithelial transition and by targeting MAPK and NFκB signaling pathways. PLoS One. 2014;9(1):e86338. doi:10.1371/journal.pone.0086338
84. Kirjavainen M, Mönkkönen J, Saukkosaari M, et al. Phospholipids affect stratum corneum lipid bilayer fluidity and drug partitioning into the bilayers. J Controlled Release. 1999;58(2):207–214. doi:10.1016/S0168-3659(98)00152-7
85. Fouad SA, Shamma RN, Basalious EB, et al. Novel instantly-dispersible nanocarrier powder system (IDNPs) for intranasal delivery of dapoxetine hydrochloride: in-vitro optimization, ex-vivo permeation studies, and in-vivo evaluation. Drug Dev Ind Pharm. 2018;44(9):1443–1450. doi:10.1080/03639045.2018.1459675
86. Attia AK, Souaya ER, Soliman EA. Thermal analysis investigation of dapoxetine and vardenafil hydrochlorides using molecular orbital calculations. Adv Pharm Bull. 2015;5(4):523. doi:10.15171/apb.2015.071
87. Collins JJ, Phillips MC. The stability and structure of cholesterol-rich codispersions of cholesterol and phosphatidylcholine. J Lipid Res. 1982;23(2):291–298.
88. Suzuki H, Ogawa M, Hironaka K, et al. A nifedipine coground mixture with sodium deoxycholate. II. Dissolution characteristics and stability. Drug Dev Ind Pharm. 2001;27(9):951–958. doi:10.1081/DDC-100107676
89. ElMeshad AN, Mohsen AM. Enhanced corneal permeation and antimycotic activity of itraconazole against Candida albicans via a novel nanosystem vesicle. Drug Deliv. 2016;23(7):2115–2123. doi:10.3109/10717544.2014.942811
90. Abdulbaqi IM, Darwis Y, Abou Assi R, et al. Transethosomal gels as carriers for the transdermal delivery of colchicine: statistical optimization, characterization, and ex vivo evaluation. Drug Des Devel Ther. 2018;12:795. doi:10.2147/DDDT
91. Eckmann D, Composto RJ, Tsourkas A, et al. Nanogel carrier design for targeted drug delivery. J Mater Chem B. 2014;2(46):8085–8097. doi:10.1039/C4TB01141D
92. Guerreiro-Cacais A, Norin U, Gyllenberg A, et al. VAV1 regulates experimental autoimmune arthritis and is associated with anti-CCP negative rheumatoid arthritis. Genes Immun. 2017;18(1):48. doi:10.1038/gene.2016.49
93. Schinnerling K, Aguillón JC, Catalán D, et al. The role of interleukin-6 signalling and its therapeutic blockage in skewing the T cell balance in rheumatoid arthritis. Clin Exp Immunol. 2017;189(1):12–20. doi:10.1111/cei.2017.189.issue-1
94. Tanaka S, Tanaka Y, Ishiguro N, et al. RANKL: a therapeutic target for bone destruction in rheumatoid arthritis. Mod Rheumatol. 2018;28(1):9–16. doi:10.1080/14397595.2017.1369491
95. Sacre S, Medghalchi M, Gregory B, et al. Fluoxetine and citalopram exhibit potent antiinflammatory activity in human and murine models of rheumatoid arthritis and inhibit toll‐like receptors. Arthritis Rheum. 2010;62(3):683–693. doi:10.1002/art.27304
96. Prasad LK, O’Mary H, Cui Z. Nanomedicine delivers promising treatments for rheumatoid arthritis. Nanomedicine. 2015;10(13):2063–2074. doi:10.2217/nnm.15.45
97. Wendling D, Racadot E, Wijdenes J. Treatment of severe rheumatoid arthritis by anti-interleukin 6 monoclonal antibody. J Rheumatol. 1993;20(2):259–262.
98. Durairaj H, Steury MD, Parameswaran N. Paroxetine differentially modulates LPS-induced TNFα and IL-6 production in mouse macrophages. Int Immunopharmacol. 2015;25(2):485–492. doi:10.1016/j.intimp.2015.02.029
99. Fonsek TM, McIntyre RS, Soczynska JK, et al. Novel investigational drugs targeting IL-6 signaling for the treatment of depression. Expert Opin Investig Drugs. 2015;24(4):459–475. doi:10.1517/13543784.2014.998334
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.