")
Back to Journals » Cancer Management and Research » Volume 11
miR-625 reverses multidrug resistance in gastric cancer cells by directly targeting ALDH1A1
Authors Gong X, Xu B, Zi L, Chen X
Received 14 March 2019
Accepted for publication 28 May 2019
Published 15 July 2019 Volume 2019:11 Pages 6615—6624
DOI https://doi.org/10.2147/CMAR.S208708
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Harikrishna Nakshatri
Xufei Gong, Baoli Xu, Li Zi, Xinrui Chen
Department of General Surgery, Linyi People’s Hospital, Linyi, Shandong 276002, People’s Republic of China
Background: microRNAs (miRNAs) are emerging as critical regulators of multidrug resistance (MDR) in gastric cancer, a major cause of chemotherapy failure. miR-625 is downregulated in gastric cancer and negatively associated with metastasis. In the current study, we aimed to investigate whether miR-625 regulates MDR in gastric cancer.
Methods: The level of miR-625 in gastric cancer cells with or without MDR was quantified by quantitative reverse transcription PCR (qRT-PCR) analysis. The sensitivity of gastric cancer cells to chemotherapeutic agents was assessed by MTT assay. The protein expression was determined by Western blot analysis, and the luciferase reporter assay was applied to confirm miR-625 regulation of the potential target.
Results: miR-625 is downregulated in MDR gastric cancer cells compared with chemosensitive counterparts. In addition, miR-625 increases the sensitivity and promotes apoptosis of gastric cancer cells when treated with different chemotherapeutic agents. Moreover, miR-625 directly targets the aldehyde dehydrogenase 1A1 (ALDH1A1), and importantly, the restoration of ALDH1A1 expression rescues miR-625 effects on MDR in gastric cancer cells.
Conclusion: miR-625 reverses MDR in gastric cancer cells by targeting ALDH1A1. Hence, our study identifies miR-625 as a novel regulator of MDR in gastric cancer cells, and implicates its potential application for overcoming MDR in gastric cancer chemotherapy.
Keywords: miR-625, multidrug resistance, gastric cancer, ALDH1A1, apoptosis
Introduction
The gastric cancer is the fifth most common malignancies and the second leading cause of cancer-related death in the world.1 Most patients are diagnosed with an advanced stage or relapse after surgical resection, and the systemic chemotherapy is currently the mainstay treatment for advanced gastric cancer.2 However, in many cases, patients show a poor initial response or develop intrinsic or acquired resistance to chemotherapy, known as multidrug resistance (MDR), which becomes a huge obstacle for effective chemotherapy and leads to a poor prognosis of gastric cancer patients.3 The development of gastric cancer MDR is a very complicated process and a large number of drug-resistant molecules have been shown to play an important role, such as P-glycoprotein/ABCB1 and MRP1/ABCC1.4 However, the mechanisms underlying gastric cancer MDR are still not well understood.
microRNAs (miRNAs) are a group of small noncoding RNAs that posttranscriptionally regulate gene expression through translational inhibition and mRNA destabilization.5 Until now, the roles of several miRNAs in gastric cancer MDR have been investigated. For example, miR-15b and miR-16 modulate gastric cancer MDR by modulating apoptosis through targeting BCL2.6 PTEN is a target of miR-19a/b, which mediates the promotive effect on MDR in gastric cancer.7 Additionally, miR-106a induces MDR in gastric cancer by targeting RUNX3.8 Moreover, miR-508-5p regulates MDR in gastric cancer by targeting ABCB1 and ZNRD1.9 These studies suggest that miRNAs can regulate gastric cancer MDR by targeting different genes.
In a recent study, miR-625 was found significantly downregulated and negatively correlated with lymph node metastasis in gastric cancer, and miR-625 was also shown to inhibit the invasion and metastasis of gastric cancer cells by targeting ILK.10 However, to our best knowledge, whether miR-625 is associated with MDR in gastric cancer and the molecular mechanisms are not reported. In this study, we investigated the regulation and functional role of miR-625 in gastric cancer MDR by taking the advantage of SGC7901 cells and their MDR variants, including SGC7901/VCR and SGC7901/ADR.
Materials and methods
Cell lines and culture
The cell line SGC7901 was purchased from the Shanghai cell bank of the Chinese Academy of Sciences. The cells were cultured in RPMI-1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), 100 U/ml penicillin sodium and 100 µg/ml streptomycin at 37°C in an incubator with 5% CO2. The MDR variants of SGC7901, including SGC7901/VCR and SGC7901/ADR, were established in our lab as described previously,11 and cultured with the addition of 1 mg/ml VCR and 0.5mg/ml ADR, respectively, to maintain their MDR phenotype. All procedures were conducted in accordance with the protocols approved by the Ethics Committee of Linyi People’s Hospital.
qRT-PCR analysis
SGC7901 cells were harvested and the total RNA was isolated using TRIzol reagent (Invitrogen). The expression of miR-625 was quantified by the stem-loop RT followed by TaqMan PCR analysis as previously described,12 during which an All-in-One miRNA qRT-PCR Detection Kit (GeneCopoeia, Rockville, MD, USA) was utilized based on the manufacturer’s protocols. The results were calculated by the 2−∆∆CT method,13 and normalized to U6 snRNA. ALDH1A1 mRNA expression was quantified by qRT-PCR analysis using the SYBR Green PCR Kit (Takara Bio Inc., Otsu, Japan), and the results were normalized to GAPDH. Each reaction was conducted in triplicate. Primer sequences are listed as follows: miR-625 GSP: 5ʹ-GCGGCAGACTATAGAACTTT-3ʹ; R: 5ʹ-CAGTGCGTGTCGTGGA-3ʹ; ALDH1A1 F 5ʹ-AGGGGCAGCCATTTCTTCTCA3ʹ; R 5ʹ-CACGGGCCTCCTCCACATT-3ʹ.
In vitro drug sensitivity assay
SGC7901 cells were transfected with antagomir-625, and SGC7901/ADR and SGC/7901VCR cells were transfected with mimic-mir625 using the Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. The transfection of antagomir-NC and mimic-NC was used as control. The drug sensitivity was determined as previously described.6 Briefly, at 48 h after transfection, 5×103 cell were seeded into 96-well plates and then treated with different concentrations of chemotherapeutic agents, including vincristine (VCR), adriamycin (ADR), 5-fluorouracil (5-FU), and cisplatin (CDDP). At 48 h after treatment, 3-(4,5-di-methyl-2-thiazolyl)-2,5-diphenyl-2H tetrazolium bromide (MTT) (Sigma, St Louis, MO, USA) assay was performed, and the absorbance at 490 nm was recorded using a spectrophotometer. The concentration at which each drug produced 50% inhibition of growth (IC50) was calculated by the relative survival curve. Each treatment was performed with 5 replicates.
Flow cytometry analysis of apoptosis
SGC7901 cells were harvested and washed with PBS. Then, cell apoptosis was assessed using an Annexin-V-FITC apoptosis detection kit (BD, Franklin Lakes, NJ, USA) in combination of flow cytometry analysis as previously described.14 A FACSCalibur flow cytometer (BD Biosciences) was used and data were analyzed using the FlowJo software (FlowJo, Ashland, OR, USA).
Luciferase activity assay
The 3ʹ-UTR of human ALDH1A1 cDNA containing the putative binding site for miR-625 was amplified by PCR and inserted downstream of the luciferase gene in the pGL3 vector (Promega, Madison, WI). A mutant 3ʹ-UTR ALDH1A1 construct was generated using the QuikChange II site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) according to the manufacturer’s protocols. Both the wild-type (wt) and mutant (mut) constructs were confirmed by DNA sequencing. For determining luciferase activity, SGC7901 cells were plated with 1×105 cells per well in 24-well plates. 200 ng of pGL3-wt-3ʹ-UTR or pGL3-mut-3ʹ-UTR plus 50 ng pRL-TK Renilla luciferase (Promega, Madison WI USA) were cotransfected with 50 pmol of antagomir-NC, antagomir-625, mimic-NC, or mimic-mir625 using the Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. At 48 h following transfection, the luciferase activity was measured using the Dual Luciferase Reporter Assay System (Promega). The firefly luciferase activity was normalized to that of Renilla luciferase. Each treatment was performed in triplicate.
Western blot analysis
Cells were harvested after treatment and homogenized in RIPA lysis buffer (Beyotime, P0013B) supplemented with complete EDTA-free cocktail protease inhibitor (Roche). The extracted protein samples were resoluted by 10% SDS-PAGE gels and then transferred onto nitrocellulose (NC) membranes (Millipore, Bedford, MA). The NC membranes were blocked with 5% nonfat dry milk in TBST for 1 h, and then probed with primary antibodies overnight at 4°C. At the second day, the NC membranes were washed by TBST for 3 times, and further probed with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature. The protein bands were acquired by incubating with the enhanced chemiluminescence (ECL) (Amersham Biosciences, Amersham, UK). The antibodies used in this study were purchased from the following sources: anti-Bax antibody (Novus Biologicals, NBP1-28566); anti-Bcl-2 antibody (Santa Cruz, sc-7382); anti-cleaved caspase-3 (Asp175) (Cell Signaling Technology, 9661); anti-ALDH1A1 antibody (Abcam, EP1933Y); anti-β-Actin (Abcam, ab14128); anti-rabbit IgG, HRP-linked antibody (Cell Signaling Technology, 7074); anti-mouse IgG, HRP-linked antibody (Cell Signaling Technology, 7076).
Statistical analysis
Data are presented as the mean ± SEM. The two-tailed Student’s t-test, one-way or two-way analysis of variance (ANOVA) was applied to compare the data. The statistical analysis was carried out using the SPSS11.0 software (SPSS Inc., Chicago, IL, USA). Differences were considered to be statistically significant if p<0.05.
Results
miR-625 expression is decreased in MDR gastric cancer cells
We established two gastric cancer cell variants from parental SGC7901 cells, including SGC7901/ADR and SGC7901/VCR, which are resistant to the treatment of adriamycin (ADR) and vincristine (VCR), respectively.9 The resistant characteristic of SGC7901/ADR cells to ADR (Figure 1A) and SGC7901/VCR cells to VCR (Figure 1B) was confirmed by their robust cell viability under contentious drug treatment, as compared to sensitive SGC7901 cells. To explore whether miR-625 is associated with MDR development in gastric cancer, we compared its expression in SGC7901 cells with that in SGC7901/ADR and SGC7901/VCR. As analyzed by quantitative reverse transcription PCR (qRT-PCR) assay, in comparison with SGC7901 cells, miR-625 expression was markedly downregulated in both SGC7901/ADR cells (Figure 1C) and SGC7901/VCR cells (Figure 1D). Therefore, these results may imply a reverse correlation between miR-625 expression and MDR development in gastric cancer.
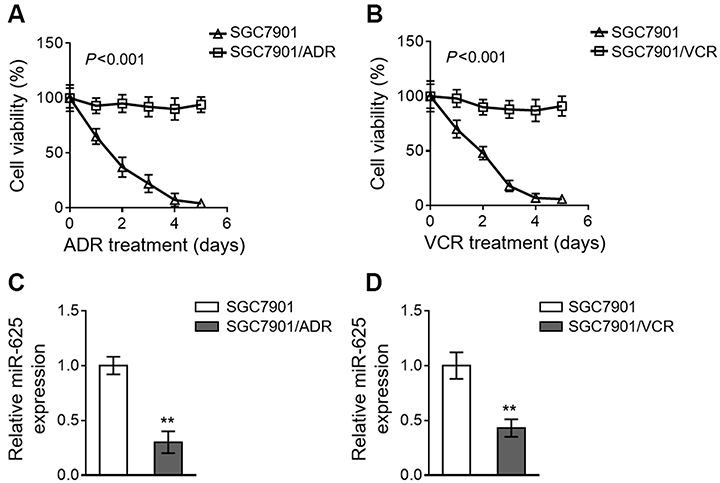 |
Figure 1 miR-625 is downregulated in MDR gastric cancer cells. (A and B) SGC7901/ADR (A) and SGC7901/VCR (B) were obtained from parental SGC7901 cells by stepwise selection of vincristine (VCR) and adriamycin (ADR) treatment, respectively. 1 mg/ml VCR and 0.5 mg/ml ADR were used to maintain their MDR phenotype. SGC7901/ADR cells were treated with 10 µg/ml ADR (A) and SGC7901/VCR cells were treated with 10 µg/ml VCR (B) for consecutive days as indicated. The parental SGC7901 cell line was used as a negative control. The drug sensitivity was determined by MTT assay. The percentage of viable cells is shown (%). Each treatment condition was performed in 5 replicates. (C and D) The expression of miR-625 in SGC7901/ADR cells (C) and SGC7901/VCR cells (D) was determined by qRT-PCR analysis. Results were normalized to U6 snRNA and expressed as relative to SGC7901 cells. Each column represents the mean value from 3 replicates. Data are presented as the mean ± SEM. ANOVA with a post hoc Dunnett’s test (A and B); Two-tailed Student’s t-test (C and D). **P<0.01. |
miR-625 sensitizes MDR gastric cancer cells to chemotherapeutic agents
To establish whether miR-625 is functionally involved in MDR development in gastric cancer, we antagonized miR-625 in SGC7901 cells with the transfection of specific antagomir (antagomir-625). Compared to negative control antagomir (antagomir-NC), the miR-625 level was indeed silenced upon antagomir-625 transfection (Figure 2A). We next treated these cells with commonly-utilized chemotherapeutic agents for gastric cancer, including ADR, VCR, 5-fluorouracil (5-FU), and cis-diamminedichloroplatinum (CDDP).15 An in vitro drug sensitivity assay, which evaluated the concentration at which each drug produced 50% inhibition of growth (IC50), showed that miR-625 inhibition in SGC7901 cells by antagomir-625 increased the IC50 values for all the tested chemotherapeutic agents (Figure 2B), suggesting that miR-625 inhibition increases MDR in SGC7901 cells, which parallels the phenomenon of miR-625 downregulation in MDR SGC7901 cells (Figure 1). Based on these results, we suspected that whether miR-625 restoration decreases MDR in SGC7901/ADR and SGC7901/VCR. To test this possibility, miR-625 was overexpressed in these cells by transfecting with mimic-mir625, which was validated by qRT-PCR analysis (Figure 2C and D). As expected, compared to mimic-NC, miR-625 overexpression mediated by mimic-mir625 transfection leaded to reduced IC50 values for 4 chemotherapeutic agents in both SGC7901/ADR cells (Figure 2E) and SGC7901/VCR cells (Figure 2F). Therefore, these results indicate that miR-625 is able to reverse MDR in gastric cancer.
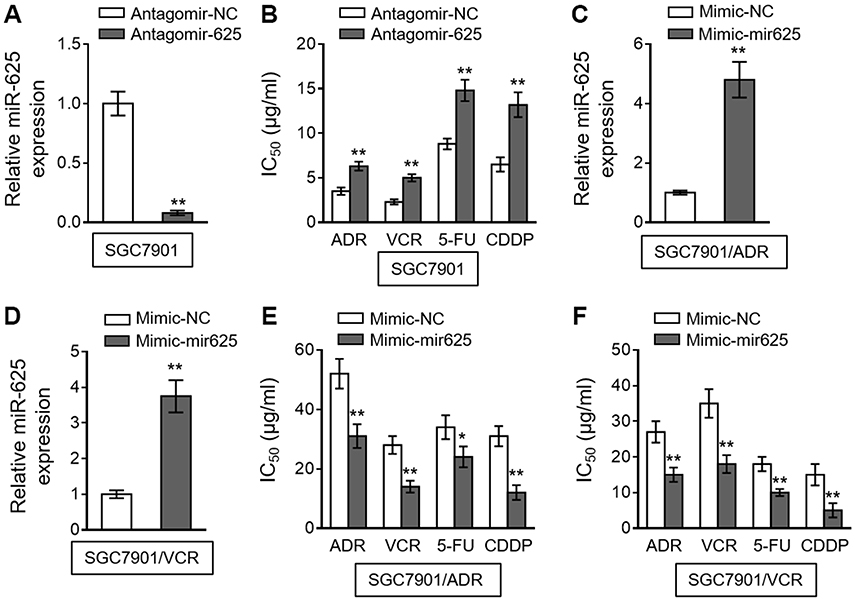 |
Figure 2 miR-625 increases drug sensitivity in MDR gastric cancer cells. (A) SGC7901 cells were transfected with antagomir-625 or negative control antagomir (antagomir-NC). At 2 days after transfection, the miR-625 level was determined by qRT-PCR analysis. Results were normalized to U6 snRNA. The expression relative to that in antagomir-NC group is shown. Each column represents the mean value from 3 replicates. (B) SGC7901 cells were transfected as in (A), and further treated with ADR, VCR, 5FU and CDDP for 2 days. The drug sensitivity was determined by MTT assay. The IC50 is shown. Each concentration treatment was performed in 5 replicates. (C and D) SGC7901/ADR (C) and SGC7901/VCR (D) cells were transfected with mimic-mir625 or negative control (mimic-NC). At 2 days after transfection, the miR-625 level was was determined by qRT-PCR analysis. Results were normalized to U6 snRNA. The expression relative to that in mimic-NC group is shown. Each column represents the mean value from 3 replicates. (E and F) SGC7901/ADR cells (E) and SGC7901/VCR cells (F) were transfected as in (C and D), and further treated with ADR, VCR, 5FU and CDDP for 2 days. The drug sensitivity was determined by MTT assay. The IC50 is shown. Each concentration treatment was performed in 5 replicates. Data are presented as the mean ± SEM. Two-tailed Student’s t-test. **P<0.01; *P<0.05. |
miR-625 promotes apoptosis in MDR gastric cancer cells treated with chemotherapeutic agents
The evasion to apoptosis induced by cytotoxicity of chemotherapeutic agents is an important molecular mechanism for tumor resistance to chemotherapy.16,17 We asked whether miR-625 reverses MDR in gastric cancer cells by promoting apoptosis. To address this issue, we checked the expression of apoptosis-associated markers, including Bax, Bcl-2, and cleaved caspase-3.18,19 Western blot analysis revealed that, miR-625 inhibition decreased the expression of Bax and cleaved caspase-3, and meanwhile, increased Bcl-2 expression in SGC7901 cells treated with 4 chemotherapeutic agents (Figure 3A), which are consistent with the increased survival rate and MDR in these cells (Figure 2B). Moreover, instead, miR-625 overexpression via mimic-mir625 transfection increased the expression of Bax and cleaved caspase-3, and decreased Bcl-2 expression in both SGC7901/ADR cells (Figure 3B) and SGC7901/VCR cells (Figure 3C) under the treatment of 4 chemotherapeutic agents, which are also in concert with results shown in Figure 2E and F. Altogether, these observations suggest that miR-625 reverses MDR in gastric cancer through resensitizing cells to apoptosis induced by chemotherapeutic agents.
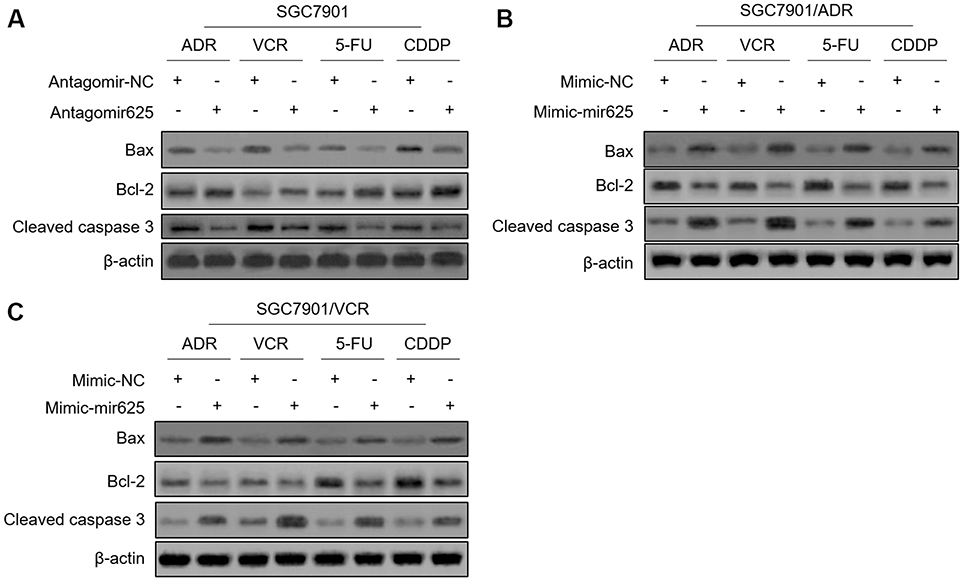 |
Figure 3 miR-625 promotes chemotherapeutic agent-induced apoptosis in MDR gastric cancer cells. (A) SGC7901 cells were transfected with antagomir-625 or antagomir-NC. At 2 days after transfection, cells were further treated with 1 µg/ml ADR, 0.5 µg/ml VCR, 5 µg/ml 5FU and 2 µg/ml CDDP for 2 days. The protein levels of Bax, Bcl-2, and cleaved caspase 3 were determined by Western blot analysis. β-actin was utilized as a loading control. The representative results from 3 independent experiments are shown. (B and C) SGC7901/ADR cells (B) and SGC7901/VCR cells (C) were transfected with mimic-mir625 or mimic-NC. At 2 days after transfection, cells were further treated with 1 µg/ml ADR, 0.5 µg/ml VCR, 5 µg/ml 5FU and 2 µg/ml CDDP for 2 days. The protein levels of Bax, Bcl-2, and cleaved caspase 3 were determined by Western blot analysis. β-actin was utilized as a loading control. The representative results from 3 independent experiments are shown. |
ALDH1A1 is a direct target of miR-625
miRNAs exert their versatile biological activities through regulating gene expression by means of targeting complementary mRNAs.20 To elucidate how miR-625 reverses MDR in gastric cancer, its mRNA targets were predicted by TargetScan tool.21 Among these putative targets, the aldehyde dehydrogenase 1A1 (ALDH1A1) attracted our attention (Figure 4A), since its overexpression is associated with the aggressiveness and poor prognosis of gastric cancer,22 and it also plays a role in cancer drug resistance.23 We confirmed whether ALDH1A1 is a direct target of miR-625 in SGC7901 cells by luciferase reporter assay. As shown, miR-625 silencing increased the luciferase activity of wild-type but not the mutant form of ALDH1A1 construct (Figure 4B). Oppositely, miR-625 overexpression decreased the luciferase activity of wild-type ALDH1A1 construct, but with the mutant form unaffected (Figure 4C). These results prove that miR-625 can directly target ALDH1A1. In addition, consistently, along with the downregulation of miR-625 in SGC7901/ADR cells (Figure 1C) and SGC7901/VCR cells (Figure 1D), both the mRNA level (Figure 4D) and protein level (Figure 4E) of ALDH1A1 were elevated. Furthermore, miR-625 silencing resulted in increased ALDH1A1 expression in SGC7901 cells (Figure 4F), and in agreement with this, miR-625 overexpression leaded to decreased ALDH1A1 expression in SGC7901/ADR cells and SGC7901/VCR cells (Figure 4G). Thus, these data demonstrate that ALDH1A1 could be a direct target of miR-625.
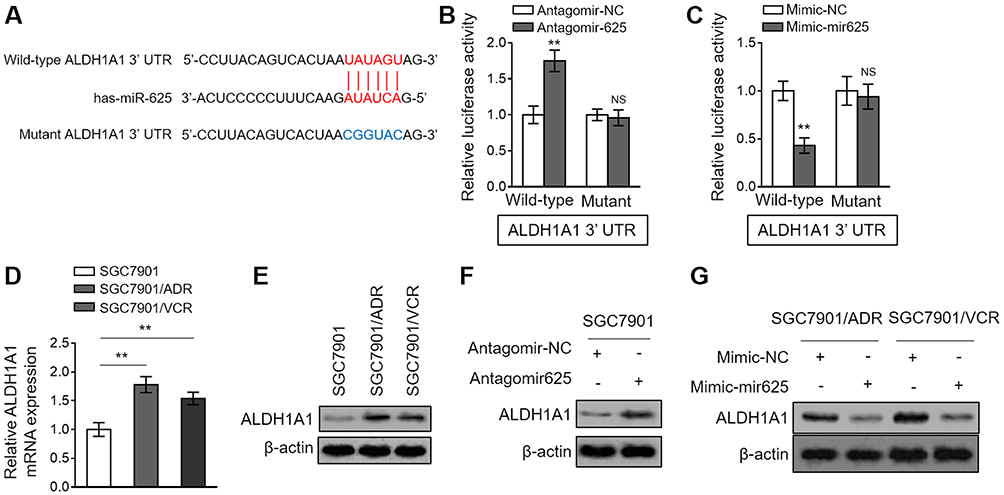 |
Figure 4 miR-625 directly targets ALDH1A1. (A) The schematic representation of complimentary sequences between miR-625 and 3ʹ-UTR of ALDH1A1. (B) SGC7901 cells were transfected with antagomir-625 or antagomir-NC in combination with luciferase reporter construct containing wild-type or mutant 3ʹ-UTR of ALDH1A1. At 2 days after transfection, the luciferase activity was measured. The firefly luciferase activity was normalized to that of Renilla luciferase. Each treatment condition was performed in triplicate. (C) SGC7901 cells were transfected with mimic-mir625 or mimic-NC in combination with luciferase reporter construct containing wild-type or mutant 3ʹ-UTR of ALDH1A1. At 2 days after transfection, the luciferase activity was measured. The firefly luciferase activity was normalized to that of Renilla luciferase. Each treatment condition was performed in triplicate. (D) The mRNA level of ALDH1A1 in SGC7901/ADR and SGC7901/VCR cells was determined by qRT-PCR analysis. Results were normalized to U6 snRNA. The expression relative to that in parental SGC7901 cells is shown. Each column represents the mean value from 3 replicates. (E) The protein level of ALDH1A1 in parental SGC7901 cells, SGC7901/ADR cells and SGC7901/VCR was determined by Western blot analysis. β-actin was utilized as a loading control. The representative results from 3 independent experiments are shown. (F) SGC7901 cells were transfected with antagomir-625 or antagomir-NC. At 2 days after transfection, the protein level of ALDH1A1 was determined by Western blot analysis. (G) SGC7901/ADR cells and SGC7901/VCR cells were transfected with mimic-mir625 or mimic-NC. At 2 days after transfection, the protein level of ALDH1A1 was determined by Western blot analysis. β-actin was utilized as a loading control. The representative results from 3 independent experiments are shown. Data are presented as the mean ± SEM. Two-tailed Student’s t-test. **P<0.01; NS, not significant. |
miR-625 reverses MDR in gastric cancer cells by suppressing ALDH1A1
Finally, we aimed to clarify whether ALDH1A1 contributes to miR-625 function in MDR in gastric cancer. For this purpose, we depleted ALDH1A1 expression in SGC7901 cells through small interfering RNA (siRNA) technique. Indeed, siRNA targeting ALDH1A1 resulted in pronounced reduction of ALDH1A1 expression in SGC7901 cells (Figure 5A). Significantly, the mir-625 silencing-increased IC50 values for 4 tested chemotherapeutic agents were vastly reversed by ALDH1A1 depletion (Figure 5B), proving that ALDH1A1 upregulation mediated by mir-625 silencing plays a critical role in increasing the MDR in gastric cancer. Consistently, in SGC7901/ADR cells, the decreased MDR by mir-625 overexpression was largely recovered when ALDH1A1 expression was restored by overexpression (Figure 5C and D). Furthermore, similar results were obtained in SGC7901/VCR cells (Figure 5E and F). Collectively, these lines of evidence establish ALDH1A1 as a critical target through which miR-625 reverses MDR in gastric cancer cells.
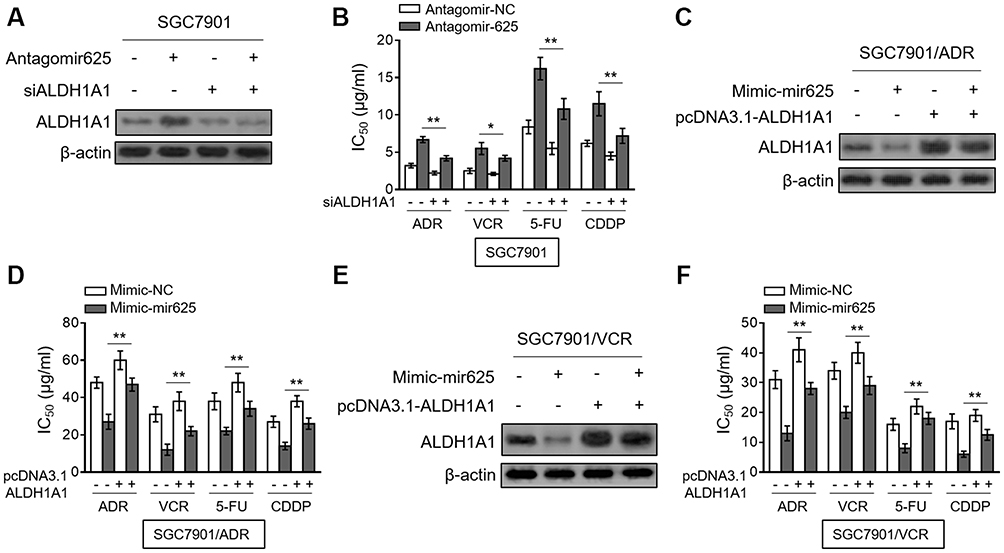 |
Figure 5 Restored ALDH1A1 expression reverses miR-625 effect on MDR in gastric cancer cells. (A) SGC7901 cells were transfected with antagomir-625 or antagomir-NC in combination with control siRNA or ALDH1A1 siRNA (siALDH1A1). At 2 days after transfection, the protein level of ALDH1A1 was determined by Western blot analysis. β-actin was utilized as a loading control. The representative results from 3 independent experiments are shown. (B) SGC7901 cells were transfected as in (A), and further treated with ADR, VCR, 5FU and CDDP for 2 days. The drug sensitivity was determined by MTT assay. The IC50 is shown. Each concentration treatment was performed in 5 replicates. (C) SGC7901/ADR cells were transfected with mimic-625 or mimic-NC in combination with pcDNA3.1 vector or pcDNA3.1 ALDH1A1. At 2 days after transfection, the protein level of ALDH1A1 was determined by Western blot analysis. β-actin was utilized as a loading control. The representative results from 3 independent experiments are shown. (D) SGC7901/ADR cells were transfected as in (C), and further treated with ADR, VCR, 5FU and CDDP for 2 days. The drug sensitivity was determined by MTT assay. The IC50 is shown. Each concentration treatment was performed in 5 replicates. (E) SGC7901/VCR cells were transfected as in (C). The protein level of ALDH1A1 was determined by Western blot analysis. β-actin was utilized as a loading control. The representative results from 3 independent experiments are shown. (F) SGC7901/VCR cells were transfected as in (C), and further treated with ADR, VCR, 5FU and CDDP for 2 days. The drug sensitivity was determined by MTT assay. The IC50 is shown. Each concentration treatment was performed in 5 replicates. Data are presented as the mean ± SEM. Two-tailed Student’s t-test. **P<0.01; *P<0.05. |
Disscussion
The acquired MDR is a major cause of chemotherapy failure during gastric cancer treatment.24 Apart from the well-recognized drug-resistant ATP-binding cassette transporters, such as P-glycoprotein (P-gp) and MDR-associated protein (MRP)1,25 recent studies have revealed that several new molecules and mechanisms are also associated with the development of MDR in gastric cancer, including some miRNAs.4,26,27 For example, miR-15b and miR-16 influence gastric cancer MDR via modulation of apoptosis by targeting BCL2.6 Therefore, the connection between miRNAs and gastric cancer MDR may provide novel therapeutic targets for overcoming gastric cancer MDR. In the present study, we report that miR-625 reverses gastric cancer MDR, in which the targeted ALDH1A1 constitutes the predominant mechanism. Thus, we identify miR-625 as a novel miRNA regulator in gastric cancer MDR and also highlight an important role of ALDH1A1 in mediating miR-625 function.
SGC7901/VCR and SGC7901/ADR cell lines are two MDR variants derived from SGC7901 cells, which are frequently used as experimental models in this research field.28 The aberrant expression of some miRNAs has been observed in gastric cancer, and these dysregulated miRNAs have the potential to be used as biomarkers and therapeutic targets in gastric cancer.29,30 In addition, the abnormal expression of some miRNAs has also been found in clinical gastric cancer tissues with MDR, like miR-30a.31 Moreover, in SGC7901/VCR and/or SGC7901/ADR cells, multiple miRNAs display abnormal expression as compared with sensitive SGC7901, such as miR-19a/b,7 miR-106a,8 and miR-508-5p.9 In the current study, we found that contrary to SGC7901 cells, miR-625 expression was markedly decreased in both SGC7901/VCR and SGC7901/ADR cells. Thus, we in the first time reveal a negative correlation between miR-625 expression and gastric cancer MDR. Notably, in a previous study, miR-625 expression was found significantly downregulated and reversely correlated with the lymph node metastasis in gastric cancer.10 Together with our findings, it appears that miR-625 is not only negatively associated with progression and metastasis, but also with MDR development in gastric cancer, which possibly suggest it as a tumor suppressive miRNA in gastric cancer. In addition to drug resistance and metastasis, miR-625 also influences other activities of cancer, including proliferation of esophageal cancer,32 migration and invasion of hepatocellular carcinoma,33 and metabolism of melanoma.34 To better understand the association between miR-625 and gastric cancer, it would be required to investigate whether miR-625 plays a role in other activates of gastric cancer and how it’s expression is downregulated during tumorigenesis and MDR development of gastric cancer.
As described above, some miRNAs have been shown to reverse gastric cancer MDR. For example, miR-15b and miR-16 were reported to reverse gastric cancer MDR by promoting apoptosis via targeting a common molecule BCL2, a well-known anti-apoptotic regulator.6 Besides, miR-508-5p and miR-129-5p reverse gastric cancer MDR by targeting ABCB1 and ZNRD1,9 and ABC transporters,35 respectively. These previous studies suggest that different miRNAs reverse gastric cancer MDR through distinct mechanisms. We found that miR-625 resensitized MDR gastric cancer cells to four chemotherapeutic agents, including ADR, VCR, 5-FU, and CDDP, and that this was accompanied by the promoted apoptosis of gastric cancer cells, indicating that miR-625 reverses gastric cancer MDR through promoting apoptosis which is induced by chemotherapeutic agents. It has shown that miR-625 induces apoptosis and increases the chemosensitivity of glioma to temozolomide.36 Moreover, miR-625 also promotes apoptosis and increases chemosensitivity of lymphoblastic leukemia cells to vincristine and cytarabine.37 Therefore, we speculate that promoting the cytotoxicity-induced apoptosis derived from chemotherapeutic agents may be a common mechanism by which miR-625 reverses chemoresistance. Given this similarity shared by these findings, it is likely that miR-625 may reverse chemoresistance in other cancer types. Further efforts are needed to validate speculation.
It has been established that the elevated expression and activity of ALDH1A1, which functions as a detoxifying enzyme, are important features of tumor-initiating and/or cancer stem cells in multiple types of cancers.38 In fact, ALDH1A1 is involved in many biological processes, including oxidative stress response, cell differentiation, and drug resistance.39 ALDH1A1 mediates temozolomide resistance in glioblastoma,39 and confers gemcitabine in pancreatic adenocarcinoma cells.38 ALDH1A1 also induces resistance to CHOP in diffuse large B-cell lymphoma.40 Mechanistically, we provide evidence demonstrating that ALDH1A1 is a direct target of miR-625, and that miR-625 reverses gastric cancer MDR by suppressing ALDH1A1 expression. Thus, our study extends ALDH1A1 function to gastric cancer MDR. The limitation of our study is the lacking of molecular evidence elucidating how ALDH1A1 regulates MDR in gastric cancer. Previous studies have proven that the mechanisms that underlie ALDH1A1-conferred chemoresistance are associated with activation and upregulation of drug-transporters and survival proteins, like P-glycoprotein, AKT and BCL2.41,42 Therefore, it is very possible the impairment of these mechanisms may also contribute to miR-625-promoted apoptosis and -reversed MDR in gastric cancer cells when treated with chemotherapeutic agents. It is interesting to test whether this is the case. Nevertheless, other possibilities can not rule out, since we discovered that ALDH1A1 restoration did not completely rescue the reversed gastric cancer MDR by miR-625. Except for ALDH1A1, more study efforts are required to discover other targets which mediate miR-625 effect on reversing gastric cancer MDR. Furthermore, investigating whether miR-625/ALDH1A1 axis reverses MDR in gastric cancer in vivo, such as in xenografted tumor model, and whether miR-625 and ALDH1A1 have possible association with MDR by examining clinical gastric cancer samples would not only strengthen our in vitro findings but also bring more profound significance in therapeutically exploiting it in the future.
In conclusion, we reveal a novel role of miR-625 and ALDH1A1 in the modulation of gastric cancer MDR. Our findings hint that upregulating miR-625 level or directly targeting ALDH1A1 might provide clinical benefit for reversing gastric cancer MDR, thereby improving the effectiveness of chemotherapy for gastric cancer patients.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet. 2016;388(10060):2654–2664. doi:10.1016/S0140-6736(16)30354-3
2. Wagner AD, Syn NLX, Moehler M, et al. Chemotherapy for advanced gastric cancer. Cochrane Database Syst Rev. 2017;8:CD004064.
3. Fodale V, Pierobon M, Liotta L, Petricoin E. Mechanism of cell adaptation: when and how do cancer cells develop chemoresistance? Cancer J. 2011;17(2):89–95. doi:10.1097/PPO.0b013e318212dd3d
4. Zhang D, Fan D. New insights into the mechanisms of gastric cancer multidrug resistance and future perspectives. Future Oncol. 2010;6(4):527–537. doi:10.2217/fon.10.21
5. Di Leva G, Garofalo M, Croce CM. MicroRNAs in cancer. Annu Rev Pathol. 2014;9:287–314. doi:10.1146/annurev-pathol-012513-104715
6. Xia L, Zhang D, Du R, et al. miR-15b and miR-16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. Int J Cancer. 2008;123(2):372–379. doi:10.1002/ijc.23501
7. Wang F, Li T, Zhang B, et al. MicroRNA-19a/b regulates multidrug resistance in human gastric cancer cells by targeting PTEN. Biochem Biophys Res Commun. 2013;434(3):688–694. doi:10.1016/j.bbrc.2013.04.010
8. Zhang Y, Lu Q, Cai X. MicroRNA-106a induces multidrug resistance in gastric cancer by targeting RUNX3. FEBS Lett. 2013;587(18):3069–3075. doi:10.1016/j.febslet.2013.06.058
9. Shang Y, Zhang Z, Liu Z, et al. miR-508-5p regulates multidrug resistance of gastric cancer by targeting ABCB1 and ZNRD1. Oncogene. 2014;33(25):3267–3276. doi:10.1038/onc.2013.297
10. Wang M, Li C, Nie H, et al. Down-regulated miR-625 suppresses invasion and metastasis of gastric cancer by targeting ILK. FEBS Lett. 2012;586(16):2382–2388. doi:10.1016/j.febslet.2012.05.050
11. Zheng G, Liu X, Han F. [Establishment and characterization of doxorubicin-resistant BGC-823/DOX of human gastric cancer cell line]. Zhonghua Wai Ke Za Zhi. 1997;35(5):262–264.
12. Chen C, Ridzon DA, Broomer AJ, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33(20):e179. doi:10.1093/nar/gni178
13. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi:10.1006/meth.2001.1262
14. Crowley LC, Marfell BJ, Scott AP, et al. Quantitation of apoptosis and necrosis by annexin V binding, propidium iodide uptake, and flow cytometry. Cold Spring Harb Protoc. 2016;2016(11):
15. Zhu W, Shan X, Wang T, et al. miR-181b modulates multidrug resistance by targeting BCL2 in human cancer cell lines. Int J Cancer. 2010;127(11):2520–2529. doi:10.1002/ijc.25260
16. Rebucci M, Michiels C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem Pharmacol. 2013;85(9):1219–1226. doi:10.18632/oncotarget.2388
17. Pan ST, Li ZL, He ZX, et al. Molecular mechanisms for tumour resistance to chemotherapy. Clin Exp Pharmacol Physiol. 2016;43(8):723–737. doi:10.1111/1440-1681.12581
18. Dolka I, Krol M, Sapierzynski R. Evaluation of apoptosis-associated protein (Bcl-2, Bax, cleaved caspase-3 and p53) expression in canine mammary tumors: an immunohistochemical and prognostic study. Res Vet Sci. 2016;105:124–133. doi:10.1016/j.rvsc.2016.02.004
19. Czabotar PE, Lessene G, Strasser A, et al. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nature Rev Mol Cell Biol. 2014;15(1):49–63. doi:10.1038/nrm3722
20. Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med. 2014;20(8):460–469. doi:10.1016/j.molmed.2014.06.005
21. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi:10.1016/j.cell.2004.12.035
22. Li X-S, Xu Q, Fu X, et al. ALDH1A1 overexpression is associated with the progression and prognosis in gastric cancer. BMC Cancer. 2014;14(1):705. doi:10.1186/1471-2407-14-705
23. Januchowski R, Wojtowicz K, Zabel M. The role of aldehyde dehydrogenase (ALDH) in cancer drug resistance. Biomed Pharmacother. 2013;67(7):669–680. doi:10.1016/j.biopha.2013.04.005
24. Garrido M, Fonseca PJ, Vieitez JM, Frunza M, Lacave AJ. Challenges in first line chemotherapy and targeted therapy in advanced gastric cancer. Expert Rev Anticancer Ther. 2014;14(8):887–900. doi:10.1586/14737140.2014.915194
25. Fan K, Fan D, Cheng LF, Li C. Expression of multidrug resistance-related markers in gastric cancer. Anticancer Res. 2000;20(6C):4809–4814.
26. Dehghanzadeh R, Jadidi-Niaragh F, Gharibi T, Yousefi M. MicroRNA-induced drug resistance in gastric cancer. Biomed Pharmacother. 2015;74:191–199. doi:10.1016/j.biopha.2015.08.009
27. Riquelme I, Letelier P, Riffo-Campos AL, Brebi P, Roa J. Emerging role of miRNAs in the drug resistance of gastric cancer. Int J Mol Sci. 2016;17(3):424. doi:10.3390/ijms17030424
28. Wu Q, Yang Z, Nie Y, Shi Y, Fan D. Multi-drug resistance in cancer chemotherapeutics: mechanisms and lab approaches. Cancer Lett. 2014;347(2):159–166. doi:10.1016/j.canlet.2014.03.013
29. Shin VY, Chu KM. miRNA as potential biomarkers and therapeutic targets for gastric cancer. World J Gastroenterol. 2014;20(30):10432–10439. doi:10.3748/wjg.v20.i30.10432
30. Brenner B, Hoshen MB, Purim O, et al. MicroRNAs as a potential prognostic factor in gastric cancer. World J Gastroenterol. 2011;17(35):3976–3985. doi:10.3748/wjg.v17.i35.3976
31. Li C, Zou J, Zheng G, et al. MiR-30a Decreases Multidrug Resistance (MDR) of gastric cancer cells. Med Sci Monit. 2016;22:4509.
32. Wang Z, Qiao Q, Chen M, et al. miR-625 down-regulation promotes proliferation and invasion in esophageal cancer by targeting Sox2. FEBS Lett. 2014;588(6):915–921. doi:10.1016/j.febslet.2014.01.035
33. Zhou X, Zhang CZ, Lu SX, et al. miR-625 suppresses tumour migration and invasion by targeting IGF2BP1 in hepatocellular carcinoma. Oncogene. 2015;34(8):965–977. doi:10.1038/onc.2014.35
34. Zhang H, Feng C, Zhang M, et al. miR-625-5p/PKM2 negatively regulates melanoma glycolysis state. J Cell Biochem. 2019;120(3):2964–2972. doi:10.1002/jcb.26917
35. Wu Q, Yang Z, Xia L, et al. Methylation of miR-129-5p CpG island modulates multi-drug resistance in gastric cancer by targeting ABC transporters. Oncotarget. 2014;5(22):11552–11563. doi:10.18632/oncotarget.2594
36. Zhang J, Zhang J, Zhang J, et al. MicroRNA-625 inhibits the proliferation and increases the chemosensitivity of glioma by directly targeting AKT2. Am J Cancer Res. 2017;7(9):1835–1849.
37. Jiang Q, Lu X, Huang P, et al. Expression of miR-652-3p and effect on apoptosis and drug sensitivity in pediatric acute lymphoblastic leukemia. Biomed Res Int. 2018;2018:5724686. doi:10.1155/2018/5724686
38. Duong HQ, Hwang JS, Kim HJ, et al. Aldehyde dehydrogenase 1A1 confers intrinsic and acquired resistance to gemcitabine in human pancreatic adenocarcinoma MIA PaCa-2 cells. Int J Oncol. 2012;41(3):855–861. doi:10.3892/ijo.2012.1516
39. Schafer A, Teufel J, Ringel F, et al. Aldehyde dehydrogenase 1A1–a new mediator of resistance to temozolomide in glioblastoma. Neuro-Oncology. 2012;14(12):1452–1464. doi:10.1093/neuonc/nos270
40. Jiang J, Liu Y, Tang Y, et al. ALDH1A1 induces resistance to CHOP in diffuse large B-cell lymphoma through activation of the JAK2/STAT3 pathway. Onco Targets Ther. 2016;9:5349–5360. doi:10.2147/OTT.S107957
41. Januchowski R, Wojtowicz K, Sterzyska K, et al. Inhibition of ALDH1A1 activity decreases expression of drug transporters and reduces chemotherapy resistance in ovarian cancer cell lines. Int J Biochem Cell Biol. 2016;78:248–259. doi:10.1016/j.biocel.2016.07.017
42. Yang Y, Zhou W, Xia J, et al. NEK2 mediates ALDH1A1-dependent drug resistance in multiple myeloma. Oncotarget. 2014;5(23):11986–11997. doi:10.18632/oncotarget.2388
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.