")
Back to Journals » Cancer Management and Research » Volume 12
miR-613 Suppresses Chemoresistance and Stemness in Triple-Negative Breast Cancer by Targeting FAM83A
Received 16 August 2020
Accepted for publication 10 October 2020
Published 8 December 2020 Volume 2020:12 Pages 12623—12633
DOI https://doi.org/10.2147/CMAR.S276316
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lu-Zhe Sun
Cuiping Liu,1 Yizhou Jiang,2 Baosan Han3
1Department of Breast Surgery, Affiliated Hospital of Chifeng University, Chifeng, Inner Mongolia 024005, People’s Republic of China; 2Department of Breast Surgery, Fudan University Shanghai Cancer Center, Shanghai 200032, People’s Republic of China; 3Department of Surgery, Xin Hua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai 200092, People’s Republic of China
Correspondence: Baosan Han
Xin Hua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, No. 1665 Kongjiang Road, Yangpu District, Shanghai 200092, People’s Republic of China
Tel +86-2165792951
Email [email protected]
Introduction: Triple-negative breast cancer (TNBC) is the most aggressive malignancy of breast cancer, which represents about 20% of all cases. The prognosis of TNBC remains unfavorable due to the lack of targeted therapy and chemoresistance. The aim of this study is to investigate the role of miR-613 in TNBC.
Material and Methods: Quantitative RT-PCT was used to explore the expression of miR-613 in breast cancer clinical samples and cell lines. MTT, colony formation assay, spheroid formation assay and xenograft tumor growth assay were used to investigate the role of miR-613 in vitro and in vivo. Cell apoptosis and surface marker expression were measured by flow cytometry. Dual-luciferase reporter assay was used to explore the function of miR-613 in regulating FAM83A 3ʹUTR. Immunohistochemical staining was used to investigate the expression of FAM83A in TNBC tissues.
Results: We found that miR-613 expression was significantly downregulated in breast cancer tissues and was even lower in TNBC compared with that in other types of breast cancer. A similar result was found in breast cancer cell lines. Further analysis indicated that miR-613 could suppress TNBC cell growth, chemoresistance and stem-cell-like phenotype. Moreover, we also demonstrated that miR-613 suppressed tumorigenesis in vivo. Mechanically, we explored the downstream target of miR-613 and identified that miR-613 could directly bind to the 3ʹUTR of FAM83A, which contributed to the miR-613 mediated tumor suppression. The expression of miR-613 and FAM83A was negatively correlated. Restoring the expression of FAM83A attributed to the chemoresistance and stemness of TNBC cells.
Conclusion: We demonstrated that loss of miR-613 was critical for TNBC malignancy and restoring its expression could be served as a potential approach for TNBC treatment.
Keywords: triple-negative breast cancer, miR-613, FAM83A, chemoresistance, stemness
Introduction
Triple-negative breast cancer (TNBC) is the most malignant tumor of breast cancer, and one of the major causes of cancer-related death accounts for 12–18% of all cases of breast cancer.1,2 Compared with the endocrine and targeted therapy for hormone receptor- and HER2-positive cancer, there are no effective drugs yet and cytotoxic chemotherapy is still the main approach for TNBC.3 The standard procedure for many TNBC patients is neoadjuvant chemotherapy (NAC). However, about 50% of the patients will have resistance and worse outcomes.1,4 Hence, understanding the mechanism of chemoresistance will be critical for clinical treatment.
Stemness plays pivotal roles in tumor invasion, metastasis, and chemo-and immuno-resistance.5,6 It has been demonstrated that the percentage of CD44 cancer stem-like cells is higher in TNBC than that in other types of BC.7 Furthermore, elevated expression of ALDH1, another stem-like marker, was found in TNBC cells and could be an independent prognostic factor in patients.8,9 Hence, targeting the cancer cell stemness might greatly reduce cancer malignancy and improve the overall survival.
miRNAs are short non-coding RNAs that contains about 22 nucleotides. Emerging evidence has been demonstrated that miRNAs are involved in cancer initiation and progression. So, miRNAs could be oncogenes or tumor suppressor genes mainly by regulating targeting genes that contribute to cellular processes such as cell cycle, mobility and survival.10 miR-613 has been demonstrated to be downregulated in several types of cancers including renal cell carcinoma, glioma, lung cancer, and cervical cancer, implying that miR-613 may act as a tumor suppressor gene.11–14 For instance, miR-613 targeted CDK9 to inhibit proliferation and invasion.15 It was also reported that downregulation of miR-613 facilitated liver cancer stem cells, indicating that miR-613 was involved in tumor stemness.16 Moreover, miR-613 suppressed migration and invasion in TNBC via targeting Daam1/RhoA signaling.17 However, its function and mechanism in TNBC remain unclear.
Family with sequence similarity 83 member A (FAM83A) is also known as BJ-TSA-9. Previous studies indicated that FAM83A expression was elevated and served as an oncogene in several types of cancers such as breast cancer, pancreatic cancer, and hepatocellular cancer.18,19 FAM83A is amplified and promotes cancer stem cell-like traits in pancreatic cancer.20 Meanwhile, long non-coding RNA FAM83A-AS1 could help to stabilize FAM83A mRNA, suggesting the post-transcriptional regulation.21 FAM83A was also reported to confer tyrosine kinase inhibitor (TKI) resistance in breast cancer cells.22 Hence, targeting FAM83A might be a therapeutic approach for breast cancer.
In the present study, we identified miR-613 was significantly downregulated in TNBC tissues and cell lines. Gain- and loss-function analysis indicated that miR-613 suppressed cell proliferation, stemness, and tumor formation. Mechanically, FAM83A was found to be a direct target of miR-613 and was responsible for miR-613 mediated tumor suppression.
Materials and Methods
Ethics and Clinical Samples
The study was approved by the Ethics Committees of Xin Hua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine. A total of 132 cases of breast cancer tissues and corresponding adjacent non-cancer tissues were surgically removed and restored in the Department of Surgery, Xin Hua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine. Written informed consent was obtained from all patients involved in this study, and none of the patients had received chemotherapy, radiotherapy or related therapies before surgery. The subtype of each patient was confirmed by two independent pathologists.
Cell Lines and Reagents
The immortal epithelial cell MCF10A and other breast cancer cell lines (MCF7, T47D, BT474, SKBR3, BT20, HCC1937, Hs578T, MDAMB231, MDAMB435) were purchased from American Type Culture Collection (ATCC, USA) and Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Cells were cultured in RPMI1640 medium supplemented with 10% FBS and 100mg/mL streptomycin and 100U/mL penicillin. Cells were confirmed with no mycoplasma contamination and maintained at 37°C with a humid incubator of 5% CO2. Paclitaxel (Taxol) was purchased from MedChemExpress (HY-B0015, NJ, USA).
Cell Transfection
For stable overexpression of miR-613, miR-613 precursor and some of its flanking sequences were cloned into the pEGFP-N1 vector to construct pEGFP-miR-613 vector. The vector was transfected into cells using Lipofectamine 2000 (Invitrogen, CA, USA) according to the manufacture’s protocol. After 48hours of transfection, cells were selected with 500mg/mL G418 for 2 weeks. For knocking down of miR-613, miR-613 antagonism was purchased from Genecopoeia (Guangzhou, China) and transfected into cells using Lipofectamine 2000. For FAM83A overexpression and knockdown, FAM83A cDNA or small interfering RNAs (siRNAs) were purchased from Genecopoeia and transfected into cells using Lipofectamine 2000. The primer used to amplify miR-613 precursor was 5ʹ-GGGCTTTATTGGTTGATTTC-3ʹ and 5ʹ-GGAAGAACACCATGTGAGA-3ʹ. The siRNAs targeting FAM83A were 5ʹ-GGAGAGAUAUACUGUGCCA-3ʹ and 5ʹ-GGAAAUUCGCUGGCCAAAU-3ʹ.
Quantitative Real-Time PCR
Total RNA was extracted from tissue samples and cell lines with Trizol reagent following the protocol as described previously.23 The primers used for detecting gene expression were used as follows: FAM83A, 5ʹ-GGAGATGTGTGACAAAGTCCAG-3ʹ, 5ʹ-CCAGCGAATTTCCTGCCTG-3ʹ; ALDH1A1, 5ʹ-GCACGCCAGACTTACCTGTC-3ʹ, 5ʹ-CCTCCTCAGTTGCAGGATTAAAG-3ʹ; CD44, 5ʹ-CTGCCGCTTTGCAGGTGTA-3ʹ, 5ʹ-CATTGTGGGCAAGGTGCTATT-3ʹ; MUC1, 5ʹ-TGCCGCCGAAAGAACTACG-3ʹ, 5ʹ-TGGGGTACTCGCTCATAGGAT-3ʹ; SOX2, 5ʹ-GCCGAGTGGAAACTTTTGTCG-3ʹ, 5ʹ-GGCAGCGTGTACTTATCCTTCT-3ʹ; BMI1, 5ʹ-CCACCTGATGTGTGTGCTTTG-3ʹ, 5ʹ-TTCAGTAGTGGTCTGGTCTTGT-3ʹ; ABCG2, 5ʹ-ACGAACGGATTAACAGGGTCA-3ʹ, 5ʹ-CTCCAGACACACCACGGAT-3ʹ, β-actin, 5ʹ-CATGTACGTTGCTATCCAGGC-3ʹ, 5ʹ-CTCCTTAATGTCACGCACGAT-3ʹ. The β-actin or U6 was used as an internal control for normalization.
Western Blot
Western blot was performed according to the protocol. Briefly, cells were lysed by RIPA buffer containing proteinase and phosphatase inhibitor cocktail and PSMF. Total protein was quantified by the BCA method. An equal amount of protein (30µg) was loaded into SDS-PAGE and transferred into PVDF membranes. The membrane was incubated with primary antibody overnight at 4°C. After washing with PBST, the membrane was incubated with the secondary antibody for 1hour at room temperature, and visualized by a chemiluminescent detection system (Bio-Rad). The primary antibodies used were as follows: anti-FAM83A (PA5-46441, ThermoFisher Scientific), anti-cleaved PARP (ab32064, Abcam Inc.), anti-β-actin (ab179467, Abcam Inc.).
Cell Proliferation, Colony Formation and Viability Assay
For cell proliferation, 1500 cells were seeded into 96-well plates. Twenty microliters of MTT (5mg/mL) was added into the well at each time point and incubated for 3hours at 37°C. Then, the medium was removed and MTT formazan was resolved in 150µL DMSO. Absorbance at 570nm was detected by a microplate reader. For colony formation, 2000 cells were seeded into 6-well plates and cultured for about 10 days. The colonies were stained with 0.1% crystal violet for 20 min, and washed with PBS, and counted and calculated under a microscope. For IC50 detection, 2000 cells were seeded into 96-well plate and treated with indicated Taxol in the gradient concentration (1, 4, 16, 64, 256nM) for 48 hours. The cell viability was detected using MTT methods.
Spheroid Formation Assay
Tumor spheroid was performed as described previously.24 2000 Cells were cultured in the ultra-low adhesive plate and suspended in DMEM/F12 medium supplemented with B27 (Gibco, USA), 20ng/mL EGF, and 10ng/mL FGF. After 21 days, the spheroid was analyzed and photographed.
Flow Cytometry
For cell apoptosis, indicated cells were seeded into 6-well plates and treated with chemodrugs and cultured in 1% FBS medium for 48 hours. Cells were harvested and stained with Annexin-V/7-AAD according to the manufacture’s instruction. The cells were detected using BD Calibur. For stemness marker EpCAM detection, indicated cells were stained with FITC-EpCAM, and positively stained cells were analyzed by flow cytometry.
Xenograft Tumor Model
Mice study was approved by the Ethics Committee of Xin Hua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine (Approval No. XHEC‐C‐2018‐037) and performed according to the Guideline of the Care and Use of Laboratory Animals in Shanghai Jiao Tong University School of Medicine.106 miR-613 overexpressing BT20 cells and control cells were suspended in HBSS buffer and mixed with Matrigel (BD Bioscience, 1:1), then, subcutaneously injected into the flank of 6-week old nude mice. Tumor volume was measured at the indicated day. After 4 weeks, the mice were sacrificed and xenograft tumors were dissected for further analysis.
Immunohistochemical Staining (IHC)
IHC was performed on the mouse xenograft tumor tissue slides and patients’ samples. Briefly, tumor tissue was formalin-fixed, paraffin-embedded, and then cut into 4µm slides. The slides were deparaffinized and rehydrated with xylene and gradient ethanol. Endogenous peroxidase was inactivated with 3% H2O2. Then, the slides were washed with PBS and incubated with primary antibody overnight at 4°C. Slides were washed with PBS and incubated with HRP-conjugated secondary antibody for 45min and developed by DAB. At last, the slides were counterstained with hematoxylin.
Luciferase Reporter Assay
Wild-type 3ʹ UTR of FAM83A was amplified by PCR from genomic DNA. The mutant form was synthesized by the point mutation kit (Takara, Dalian, China). Both sequences were cloned into pGL-3 basic vector. Then, the vectors and miR-613 mimics or antagonist were co-transfected into cells with the Renilla vector, respectively. After 48hours of culturing, cells were trypsinized and the luciferase activity was examined by the dual-luciferase assay system (Promega, USA) on a Luminometer. The luciferase activity of each sample was normalized to Renilla activity according to the previous study.25
Statistical Analysis
The statistical analysis was performed using SPSS21.0 (IBM, Chicago, USA). In vitro experiments were performed at least three times. Data were presented as mean ± S.D. The expression of miR-613 in tissue samples was measure by the Mann–Whitney test. Student’s t-test and two-way ANOVA were used to test the significance of each group. A p value less than 0.05 was considered statistically significant.
Results
miR-613 is Significantly Downregulated in TNBC Tissues and Cell Lines
To investigate the expression of miR-613 in breast cancer clinical samples, we performed qRT-PCR in 132 cases of breast cancer tissues and paired adjacent non-cancer tissues. We noticed that miR-613 was significantly downregulated in cancer tissues compared with that in normal tissues (Figure 1A). We further examined the expression of miR-613 in different subtypes of breast cancer, including Luminal A/B (99 cases), HER2 (9 cases), and TNBC (24 cases). Intriguingly, we found that miR-613 was notably decreased in TNBC tissues rather than the other three subtypes (Figure 1B). Meanwhile, we detected miR-613 expression in breast cancer cell lines and found similar results that miR-613 was more decreased in TNBC cell lines compared to that in other types (Figure 1C). Collectively, these data suggested miR-613 played pivotal roles in breast cancer, especially TNBC.
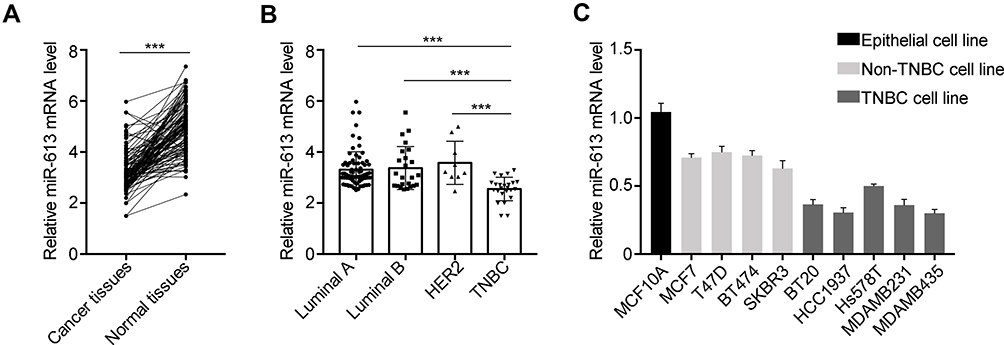 |
Figure 1 Expression of miR-613 in breast cancer tissues and cell lines. (A) qRT-PCR analysis of miR-613 in 132 cases of breast cancer samples and adjacent non-cancer tissues. (B) The expression of miR-613 in each subtype of breast cancer, including Luminal A (75 cases), Luminal B (24 cases), HER2 (9 cases) and TNBC (24 cases). (C) The expression of miR-613 in cell lines. ***p<0.001. |
miR-613 Suppresses Cell Proliferation and Induces Sensitivity to Chemotherapy
Given the results that miR-613 was more likely to be a tumor suppressor gene in TNBC, we stably overexpressed miR-613 in BT20 cells and inhibited miR-613 in Hs578T by transfecting miRNA antagonist. The cell proliferation assay indicated that overexpression of miR-613 significantly inhibited cell growth while knocking down of miR-613 promoted cell growth (Figure 2A). Also, the colony formation assay showed similar results (Figure 2B).
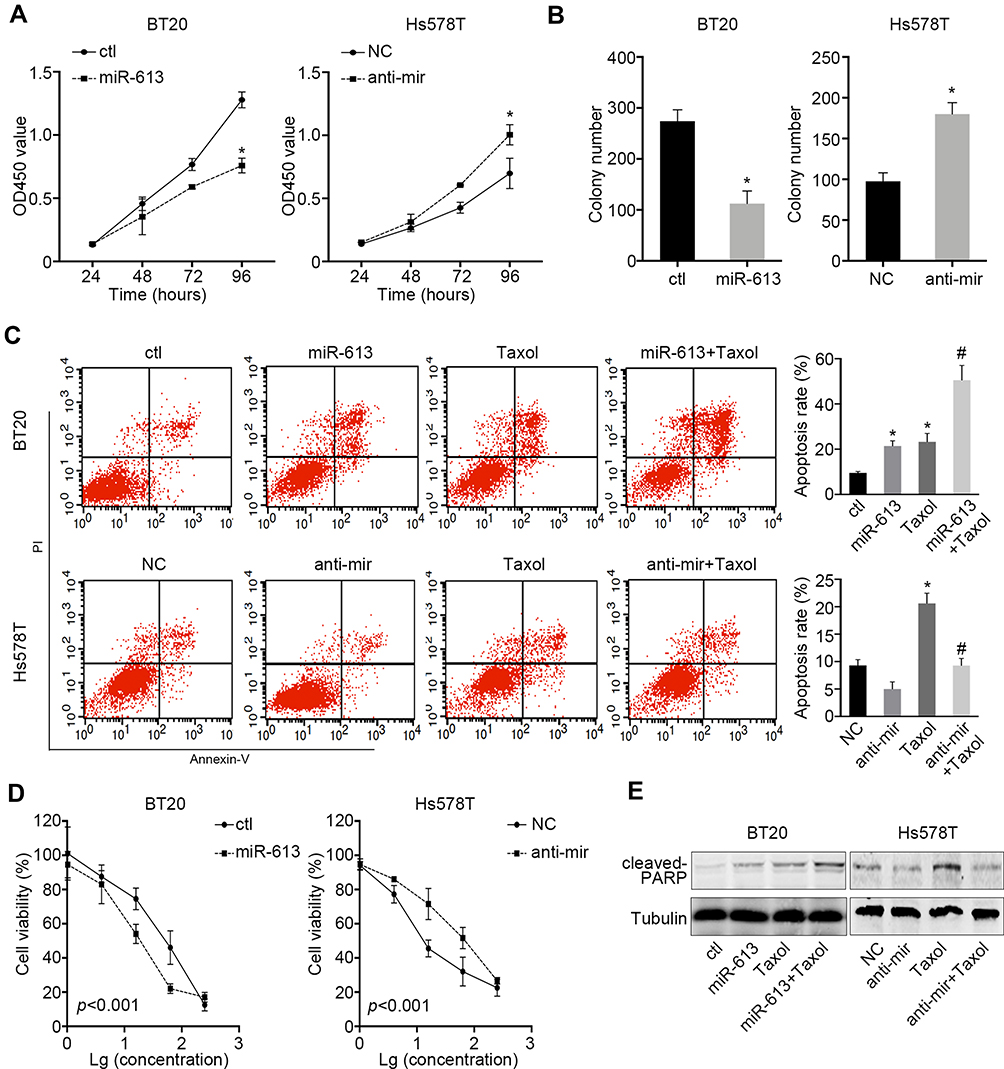 |
Figure 2 miR-613 suppresses proliferation and enhances cellular sensitivity to chemotherapy. (A) Proliferation assay of miR-613 overexpression or miR-613 inhibition cells with control, respectively. (B) Colony formation assay of miR-613 overexpression or miR-613 inhibition cells with control, respectively. (C) Taxol (20nM) were used to treat miR-613 overexpression BT20 cells and inhibition Hs578T cells with control cells, respectively, for 2 days and Annexin V/PI were double stained and measured by flow cytometry. (D) Gradient concentrations (1, 4, 16, 64, 256nM) of Taxol were used to treat cells with miR-613 overexpression or inhibition with control cells, respectively for 2 days and cell viability was measured. Significance between control and miR-613 overexpression or inhibition groups was measured by two-way ANOVA method. (E) Cells with miR-613 overexpression or inhibition with control cells, respectively, were treated with Taxol and indicated protein was measured by western bolt. *p<0.05; #p<0.05, compared to Taxol group. |
Meanwhile, we treated cells in low nutrient supply (1% FBS) and Taxol and then, measured the inducted apoptosis by flow cytometry. We demonstrated that cells treated with Taxol in the presence of miR-613 overexpression had a significantly high apoptosis rate. However, knocking down of miR-613 could impair Taxol-induced cell apoptosis (Figure 2C). We also treated miR-613 overexpression and knocking down cells, and control cells respectively with gradient Taxol and found that miR-613 sensitized cells to Taxol, evidenced by decreased IC50 values (Figure 2D). Furthermore, we performed Western blot and found that cleaved PARP, an apoptosis marker, was also notably upregulated in cells treated with Taxol and miR-613 overexpression, while decreased after miR-613 knockdown (Figure 2E).
miR-613 Suppresses TNBC Cells Stemness
TNBC was characterized by the stemness phenotype. So, we further examined the self-renewal ability of cells overexpressing miR-613 or knocking down of miR-613. Sphere formation assay indicated that miR-613 suppressed tumor spheroid formation (Figure 3A and B). Moreover, we determined cell surface stem marker EpCAM by FACS in miR-613 overexpression cells or knocking down cells, with control cells, respectively. The results indicated that overexpression of miR-613 significantly decreased EpCAM expression (Figure 3C). However, inhibition of miR-613 increased EpCAM expression about 10 folds compared to NC cells (Figure 3D). We also assessed the expression of stemness-associated genes and found that miR-613 suppressed these genes’ expression while knocking down of miR-613 increased gene expression (Figure 3E and F).
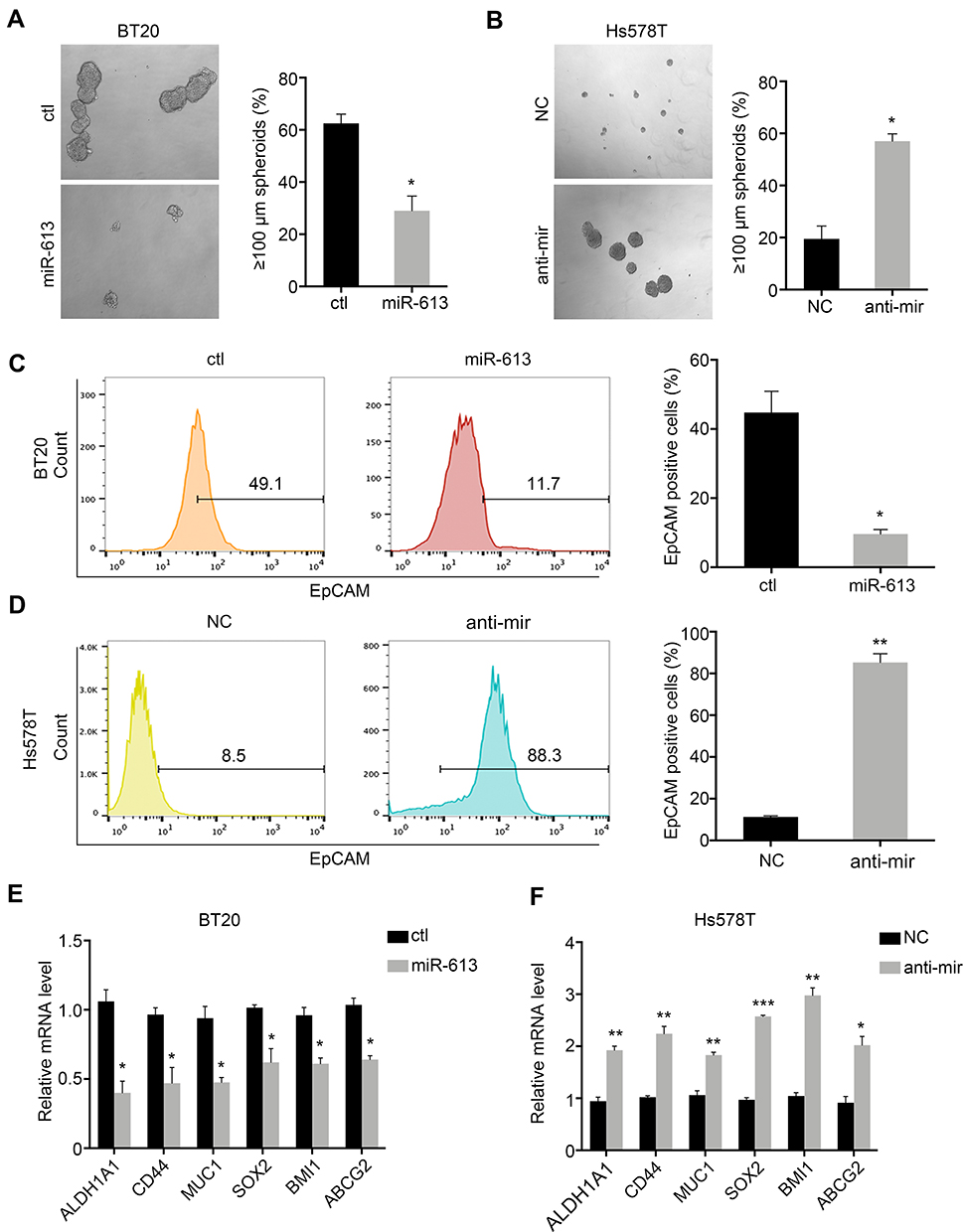 |
Figure 3 miR-613 suppresses tumor stemness. Spheroid formation assay of miR-613 overexpression (A) and inhibition cells (B) with control cells, respective. Flow cytometry detection of EpCAM expression on miR-613 overexpression (C) and inhibition cells (D) with control cells, respective. The expression of stem-associated genes in cells with miR-613 overexpression (E) and inhibition (F) compared with control cells, respectively. *p<0.05, **p<0.01, ***p<0.001. |
miR-613 Suppresses Tumor Growth in vivo
To further explore the role of miR-613 in vivo, we performed the xenograft mouse model using miR-613 stably expressing cells and control cells, respectively. And 1×106 cells were subcutaneously injected into the female nude mice, and after 7 days, we started to monitor the tumor growth every 4 days. We noticed that the ectopic expression of miR-613 significantly suppressed tumor growth in vivo, as showed in tumor volume and tumor weight (Figure 4A–C). Moreover, we performed IHC analysis of Ki67 to confirm the inhibition of tumor growth effect of miR-613 overexpression (Figure 4D).
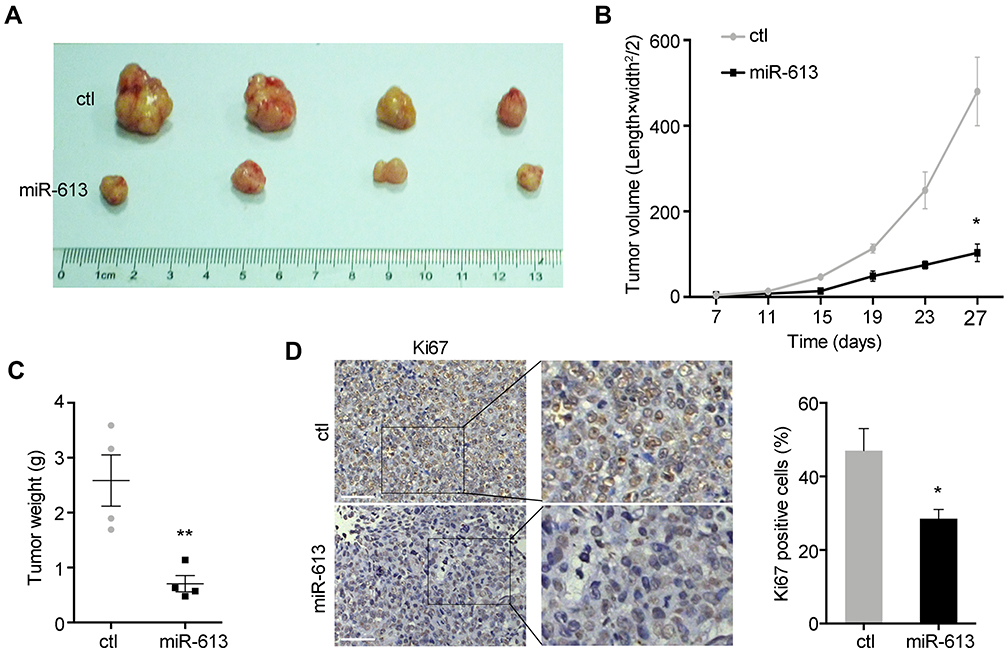 |
Figure 4 miR-613 suppresses tumor growth in vivo. (A) Xenograft tumor derived from miR-613 overexpressing cells and control cells. The growth rate (B) and weight (C) of xenograft tumors were measured. (D) The IHC staining of Ki67 was performed in tumor slides (bar: 100µm). Statistical analysis of Ki67 staining. *p<0.05, **p<0.01. |
FAM83A is a Direct Target of miR-613
We further explored the potential underlying mechanism of miR-613 in tumorigenesis suppression. FAM83A was reported to be an oncogene involved in tumor stemness and chemoresistance. We explored the miRBase and Targetscan databases and found that miR-613 could bind to the 3ʹ UTR of FAM83A (Figure 5A). Then, we constructed the wild type (wt) and mutant type (mut) of the 3ʹ UTR of FAM83 and performed luciferase reporter assay with miR-613 transfection. We found that overexpression of miR-613 significantly suppressed the luciferase activity of the vector containing wild-type sequence while had no effect on the vector containing the mutant sequence (Figure 5B). In parallel, we also performed luciferase assay after miR-613 knocking down and found similar results, that knocking down of miR-613 elevated luciferase activity, but no effect was found in mutant type (Figure 5C). Then, we examined the mRNA and protein levels of FAM83A and demonstrated that FAM83A was downregulated after miR-613 overexpression, while decreased after miR-613 knocking down (Figure 5D and E). More importantly, we performed IHC in 24 cases of TNBC tissues with miR-613 high or low expression and found the expression of FAM83A was negatively correlated with miR-613 expression in clinical samples (Figure 5F and G).
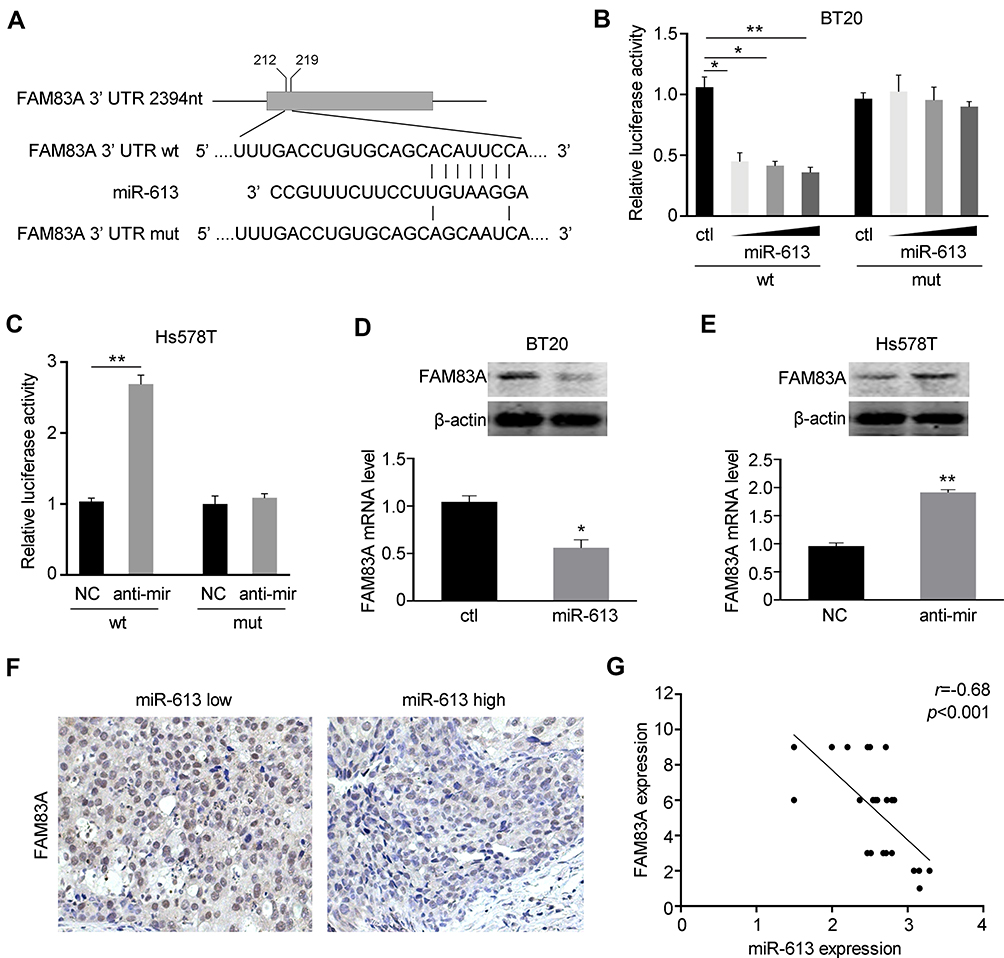 |
Figure 5 miR-613 targets FAM83A in breast cancer. (A) The sequences of wild type (wt) or mutant (mut) 3ʹUTR of FAM83A were indicated. (B) BT20 cells were co-transfected with miR-613 and pGL3-basic containing wild type or mutant type of 3ʹUTR, respectively. Dual-luciferase reporter assay was performed to measure the luciferase activity. (C) Hs578T cells were co-transfected with miR-613 inhibitor and pGL3-basic containing wild type or mutant type of 3ʹUTR, respectively. Dual-luciferase reporter assay was performed to measure the luciferase activity. (D) The mRNA and protein levels of FAM83A were examined after miR-613 overexpression. (E) The mRNA and protein levels of FAM83A were examined after miR-613 inhibition. (F) Representative images of IHC staining of FAM83A in breast cancer samples with miR-613 high or low expression. (G) The correlation between miR-613 and FAM83A expression in 24 cases of TNBC samples. *p<0.05, **p<0.01. |
FAM83A is Involved in miR-613-Mediated Tumor Suppression
To further determine whether suppression of FAM83A was responsible for miR-613 mediated tumor inhibition, we overexpressed FAM83A in miR-613 expressing cells and found that restored FAM83A expression could significantly increase the cell proliferation rate, and on the contrary, knocking down of FAM83A decreased cell proliferation which was increased by the miR-613 antagonist (Figure 6A). The colony formation assay indicated similar results (Figure 6B). Meanwhile, we detected the apoptosis rate of cells after treating with Taxol and found that FAM83A could reduce Taxol induced apoptosis (Figure 6C and D).
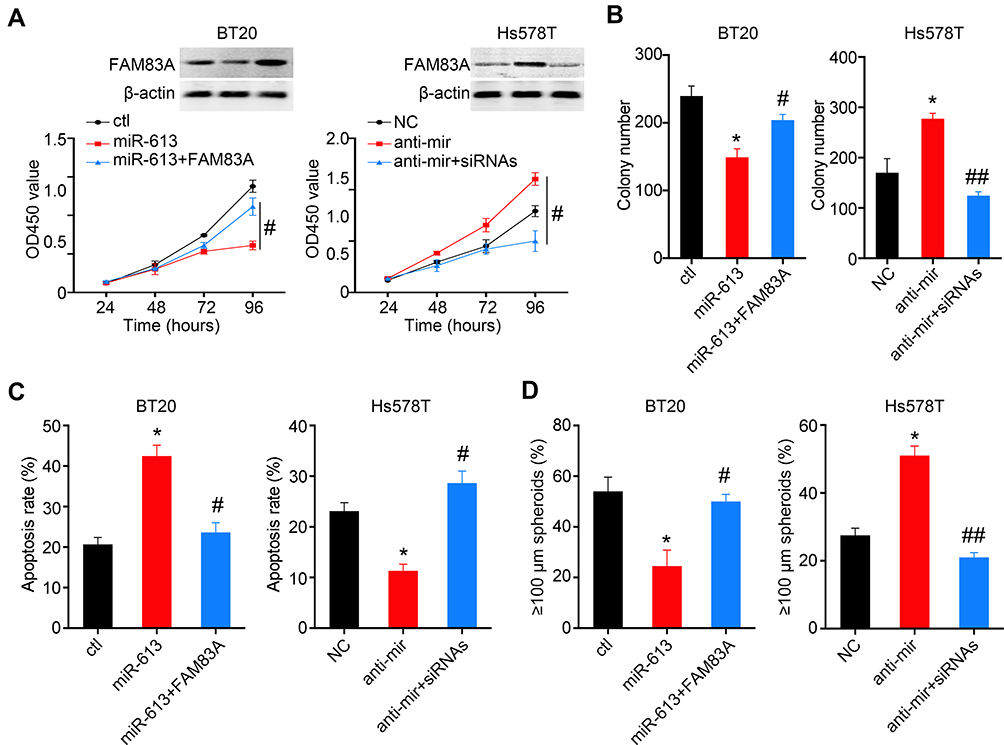 |
Figure 6 FAM83A partially reverse miR-613 mediated tumor suppression function. (A) BT20 cells with miR-613 overexpression were transfected with FAM83A plasmid and Hs578T cells with miR-613 inhibition were transfected with FAM83A siRNAs. The cell proliferation assay was performed in the aforementioned cells. Western blot is used to validate the expression of FAM83A. The colony formation assay (B), Apoptosis assay (C) and spheroid formation assay (D) were performed in the aforementioned cells. *p<0.05 compared to control group; #p<0.05, ##p<0.01 compared to miR-613 or anti-miR group. |
Discussion
The finding of the present study provides new insight into a potential tumor suppressor role for miR-613 in TNBC, and highlight that deregulated miR-613/FAM83A axis contributes to the tumor stemness-like traits, and chemoresistance. We demonstrated miR-613 was significantly downregulated in TNBC and overexpression of miR-613 inhibited tumor growth, stemness and chemoresistance by targeting FAM83A. Hence, our results uncover a mechanism leading to TNBC malignancy and imply a therapeutic target for TNBC.
The stemness-like traits of tumor cells contribute to poor prognosis, metastasis and chemoresistance, suggesting that targeting this subtype of tumor cells could have great benefit for TNBC treatment.26,27 Recently Single-cell RNA sequencing data revealed that gene signatures including AKT signaling, CDH1 targets, hypoxia, EMT, and angiogenesis were upregulated in the chemoresistant tumor cells in which miR-613 might be most involved.2,28 For instance, miR-613 could target VEGFA to inhibit angiogenesis.12 More importantly, we employed EpCAM as the stemness marker and found miR-613 significantly decreased the proportion of EpCAM-positive cells, and a group of stem-associated genes was downregulated, indicating that miR-613 had a pivotal role in regulating tumor stemness. Meanwhile, it is reported that EpCAM inhibition could sensitize chemoresistant leukemia to immune therapy.29–31 So, the further direction would be to investigate the potential of miR-613 with immunotherapy.
FAM83A is well studied in a variety of types of cancers.32 Emerging evidence has shown that FAM83A played critical roles in tyrosine kinase inhibitors (TKIs) resistance in breast cancer.33 Mechanically, FAM83A could interact and cause phosphorylation of c-RAF and PI3K p85 to bypass the TKI caused suppression of EGFR.22 Also, Chen et al demonstrated that FAM83A promoted pancreatic progression via the activation of TGF-β and Wnt/β-catenin pathways, which conferred self-renewal and metastatic abilities of primary epithelial cells.20 Therefore, in our study, miR-613 significantly suppressed tumor growth both in vitro and in vivo, and rescuing the expression of FAM83A could partially enhance the cell proliferation rate and stemness traits.
In summary, this study demonstrated that in TNBC, miR-613 negatively regulated FAM83A expression by directly targeting its 3ʹ UTR. MiR-613-mediated disruption of FAM83A impaired cancer stemness and tumorigenesis in vitro and in vivo. Since the NAC might induce chemoresistance, potential targeting FAM83A by miR-613 might increase the sensitivity of TNBC cells and improve the outcomes of TNBC.
Abbreviations
BC, breast cancer; TNBC, triple-negative breast cancer; NAC, neoadjuvant chemotherapy; CD, cell cluster; TKI, tyrosine kinase inhibitor; CDK, cyclin-dependent kinase; EGF, epidermal growth factor; FGF, fibroblast growth factors; DMSO, dimethyl sulfoxide; PVDF, polyvinylidene fluoride; IC50, half maximal inhibitory concentration; NC, negative control; UTR, untranslated region; TGF, transforming growth factor.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363(20):1938–1948. doi:10.1056/NEJMra1001389
2. Kim C, Gao R, Sei E, et al. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell. 2018;173(4):879–893 e813. doi:10.1016/j.cell.2018.03.041
3. Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13(11):674–690. doi:10.1038/nrclinonc.2016.66
4. Liedtke C, Mazouni C, Hess KR, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26(8):1275–1281. doi:10.1200/JCO.2007.14.4147
5. Kong T, Ahn R, Yang K, et al. CD44 promotes PD-L1 expression and its tumor-intrinsic function in breast and lung cancers. Cancer Res. 2020;80(3):444–457. doi:10.1158/0008-5472.CAN-19-1108
6. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–111. doi:10.1038/35102167
7. Idowu MO, Kmieciak M, Dumur C, et al. CD44(+)/CD24(-/low) cancer stem/progenitor cells are more abundant in triple-negative invasive breast carcinoma phenotype and are associated with poor outcome. Hum Pathol. 2012;43(3):364–373. doi:10.1016/j.humpath.2011.05.005
8. Li H, Ma F, Wang H, et al. Stem cell marker aldehyde dehydrogenase 1 (ALDH1)-expressing cells are enriched in triple-negative breast cancer. Int J Biol Markers. 2013;28(4):e357–364. doi:10.5301/JBM.5000048
9. Ohi Y, Umekita Y, Yoshioka T, et al. Aldehyde dehydrogenase 1 expression predicts poor prognosis in triple-negative breast cancer. Histopathology. 2011;59(4):776–780. doi:10.1111/j.1365-2559.2011.03884.x
10. Treiber T, Treiber N, Meister G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat Rev Mol Cell Biol. 2019;20(1):5–20.
11. Song H, Nan Y, Wang X, Zhang G, Zong S, Kong X. MicroRNA613 inhibits proliferation and invasion of renal cell carcinoma cells through targeting FZD7. Mol Med Rep. 2017;16(4):4279–4286. doi:10.3892/mmr.2017.7076
12. Yu X, Wang W. Tumor suppressor microRNA613 inhibits glioma cell proliferation, invasion and angiogenesis by targeting vascular endothelial growth factor A. Mol Med Rep. 2017;16(5):6729–6735. doi:10.3892/mmr.2017.7422
13. Tian Y, Yu M, Sun L, et al. Long noncoding RNA00887 reduces the invasion and metastasis of nonsmall cell lung cancer by causing the degradation of miRNAs. Oncol Rep. 2019;42(3):1173–1182.
14. Li WT, Wang BL, Yang CS, Lang BC, Lin YZ. MiR-613 promotes cell proliferation and invasion in cervical cancer via targeting PTPN9. Eur Rev Med Pharmacol Sci. 2018;22(13):4107–4114.
15. Lu Y, Tang L, Zhang Q, Zhang Z, Wei W. MicroRNA-613 inhibits the progression of gastric cancer by targeting CDK9. Artif Cells Nanomed Biotechnol. 2018;46(5):980–984. doi:10.1080/21691401.2017.1351983
16. Li B, Liu D, Yang P, Li HY, Wang D. miR-613 inhibits liver cancer stem cell expansion by regulating SOX9 pathway. Gene. 2019;707:78–85. doi:10.1016/j.gene.2019.05.015
17. Xiong H, Yan T, Zhang W, et al. miR-613 inhibits cell migration and invasion by downregulating Daam1 in triple-negative breast cancer. Cell Signal. 2018;44:33–42. doi:10.1016/j.cellsig.2018.01.013
18. Liu C, Peng X, Li Y, et al. Positive feedback loop of FAM83A/PI3K/AKT/c-Jun induces migration, invasion and metastasis in hepatocellular carcinoma. Biomed Pharmacother. 2019;123:109780. doi:10.1016/j.biopha.2019.109780
19. Parameswaran N, Bartel CA, Hernandez-Sanchez W, et al. A FAM83A positive feed-back loop drives survival and tumorigenicity of pancreatic ductal adenocarcinomas. Sci Rep. 2019;9(1):13396. doi:10.1038/s41598-019-49475-5
20. Chen S, Huang J, Liu Z, Liang Q, Zhang N, Jin Y. FAM83A is amplified and promotes cancer stem cell-like traits and chemoresistance in pancreatic cancer. Oncogenesis. 2017;6(3):e300. doi:10.1038/oncsis.2017.3
21. He J, Yu J. Long noncoding RNA FAM83A-AS1 facilitates hepatocellular carcinoma progression by binding with NOP58 to enhance the mRNA stability of FAM83A. Biosci Rep. 2019;39(11). doi:10.1042/BSR20192550
22. Lee SY, Meier R, Furuta S, et al. FAM83A confers EGFR-TKI resistance in breast cancer cells and in mice. J Clin Invest. 2012;122(9):3211–3220. doi:10.1172/JCI60498
23. Ma Z, Cai H, Zhang Y, Chang L, Cui Y. MiR-129-5p inhibits non-small cell lung cancer cell stemness and chemoresistance through targeting DLK1. Biochem Biophys Res Commun. 2017;490(2):309–316. doi:10.1016/j.bbrc.2017.06.041
24. Qian Y, Wang B, Ma A, et al. USP16 downregulation by carboxyl-terminal truncated HBx promotes the growth of hepatocellular carcinoma cells. Sci Rep. 2016;6:33039. doi:10.1038/srep33039
25. Zhu Y, Tang H, Zhang L, et al. Suppression of miR-21-3p enhances TRAIL-mediated apoptosis in liver cancer stem cells by suppressing the PI3K/Akt/Bad cascade via regulating PTEN. Cancer Manag Res. 2019;11:955–968. doi:10.2147/CMAR.S183328
26. Park SY, Choi JH, Nam JS. Targeting cancer stem cells in triple-negative breast cancer. Cancers. 2019;11(7):965. doi:10.3390/cancers11070965
27. Xu X, Zhang L, He X, et al. TGF-beta plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT and apoptosis. Biochem Biophys Res Commun. 2018;502(1):160–165. doi:10.1016/j.bbrc.2018.05.139
28. Karaayvaz M, Cristea S, Gillespie SM, et al. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat Commun. 2018;9(1):3588. doi:10.1038/s41467-018-06052-0
29. Zheng X, Fan X, Fu B, et al. EpCAM inhibition sensitizes chemoresistant leukemia to immune surveillance. Cancer Res. 2017;77(2):482–493. doi:10.1158/0008-5472.CAN-16-0842
30. Jenkins SV, Nima ZA, Vang KB, et al. Triple-negative breast cancer targeting and killing by EpCAM-directed, plasmonically active nanodrug systems. NPJ Precis Oncol. 2017;1(1):27. doi:10.1038/s41698-017-0030-1
31. Kubo M, Umebayashi M, Kurata K, et al. Catumaxomab with activated T-cells efficiently lyses chemoresistant EpCAM-positive triple-negative breast cancer cell lines. Anticancer Res. 2018;38(7):4273–4279. doi:10.21873/anticanres.12724
32. Bartel CA, Jackson MW. HER2-positive breast cancer cells expressing elevated FAM83A are sensitive to FAM83A loss. PLoS One. 2017;12(5):e0176778. doi:10.1371/journal.pone.0176778
33. Cipriano R, Graham J, Miskimen KL, et al. FAM83B mediates EGFR- and RAS-driven oncogenic transformation. J Clin Invest. 2012;122(9):3197–3210. doi:10.1172/JCI60517
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.