")
Back to Journals » OncoTargets and Therapy » Volume 9
miR-590-5p regulates gastric cancer cell growth and chemosensitivity through RECK and the AKT/ERK pathway
Authors Shen B, Yu S, Zhang Y, Yuan Y, Li X, Zhong J, Feng J
Received 20 April 2016
Accepted for publication 3 August 2016
Published 3 October 2016 Volume 2016:9 Pages 6009—6019
DOI https://doi.org/10.2147/OTT.S110923
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr William C. Cho
Bo Shen,* Shaorong Yu,* Yan Zhang, Yuan Yuan, Xiaoyou Li, Jian Zhong, Jifeng Feng
Department of Oncology, Jiangsu Cancer Hospital Affiliated to Nanjing Medical University, Nanjing, Jiangsu, People’s Republic of China
*These authors contributed equally to this work
Background: The aim of this study was to determine the role of miRNA-590-5p in gastric cancer (GC) progression.
Methods: Quantitative real-time polymerase chain reaction was performed to measure endogenous miR-590-5p levels in GC cells and tissues. Overexpression or knockdown of miR-590-5p in GC cells was performed by transfection with mimics or an inhibitor, respectively. MTT, matrigel transwell, and Western blot assays were used to assess the effects of miR-590-5p on cell proliferation, invasion, chemosensitivity of GC cells, and the AKT pathway, respectively. In silico prediction and luciferase reporter activity were used to identify potential targets of miR-590-5p. A xenograft model was also established to evaluate the function of miR-590-5p in vivo.
Results: The expression of miR-590-5p was significantly increased in GC cells and tissues, and upregulated miR-590-5p was associated with increased tumor size, lymph node metastasis, and poor survival. Overexpression of miR-590-5p promoted cell proliferation and invasion and reduced the sensitivity of GC cells to cisplatin and paclitaxel. In contrast, inhibition of miR-590-5p had the opposite effects on GC cells. RECK was identified as a direct target of miR-590-5p. Knockdown of RECK accelerated cell proliferation and motility and decreased the drug sensitivity. Furthermore, reintroduction of RECK inhibited the oncogenic effects of miR-590-5p by suppressing cell proliferation and invasion and increasing drug sensitivity. We found that the AKT/ERK and STAT3 signaling pathways were activated by miR-590-5p overexpression. The chemoresistance of miR-590-5p was also verified by in vivo analysis.
Conclusion: In summary, we suggest that the miR-590-5p/RECK/AKT axis contributes to GC and may serve as a promising therapeutic target for treatment.
Keywords: miR-590, RECK, invasion, prognosis, AKT pathway, gastric cancer
Introduction
Gastric cancer (GC), the second most common cause of cancer-related death, is also the fourth most common type of human malignancy worldwide.1 Although treatments have improved in past years, the 5-year survival rate among GC patients is still unsatisfactory.2,3 Metastasis and chemoresistance are the major challenges blocking effective GC treatment. Thus, understanding the molecular mechanisms of invasion and identifying potential novel chemotherapy targets are indispensable for improving the prognosis of GC.
miRNAs are a class of endogenous, small, non-coding regulatory RNAs that negatively regulate gene expression at the post-transcriptional level. miRNAs exert their oncogenic or tumor-suppressive effects through base-pairing with the 3′ untranslated region (3′-UTR) of target mRNAs4 and have been widely demonstrated to be critical regulators of cell proliferation, migration, invasion, and apoptosis.5–7 Previous studies have reported that some miRNAs are closely correlated with the progression of GC.8–10 For example, Nishida et al found that miR-125a-5p is a relevant prognostic marker and could inhibit proliferation of GC cells.10 Hu et al showed that miR-449a inhibited GC cell proliferation and enhanced chemosensitivity by downregulating BCL2 and CCND1.11 Taken together, the results indicate that miRNAs are potential biomarkers for diagnosis or therapeutic targets of GC.
RECK, which is suppressed strongly in human malignancy, is frequently reported as a tumor suppressor.12 Loss of RECK is correlated with metastasis in lung cancer, hepatoblastoma, and osteosarcoma.13–15 RECK inhibited tumorigenicity of GC by suppressing NOTCH1 signaling.16 Hypoxia- and RAS-signaling cooperatively downregulated the expression of RECK, thereby promoting malignant cell behavior.17 Collectively, RECK functions as a multi-role regulator involvement in tumorigenesis.
miR-590-5p has been reported to be involved in cell growth of liver cancer and cervical cancer.18,19 However, the function and underlying mechanism of miR-590-5p in gastric carcinogenesis remain unknown. In this study, we found that miR-590-5p was upregulated in GC tissues, and ectopic expression of miR-590-5p promoted the colony formation, proliferation, and invasion of GC cells. Most importantly, we found that miR-590-5p could reduce the sensitivity of GC cells to cisplatin (cDDP) and paclitaxel (PTX). RECK was identified as a direct target of miR-590-5p and is involved in miR-590-5p-regulated invasion and chemosensitivity of GC cells.
Materials and methods
Tissue specimens and cell lines
Tissue specimens of GC and adjacent nontumor tissues were obtained from 50 patients who were diagnosed and underwent surgery at Jiangsu Cancer Hospital Affiliated with Nanjing Medical University. This study was approved by the ethics committee of Nanjing Medical University, and written informed consent was obtained from all patients before surgery. After resection, all specimens were snap-frozen in liquid nitrogen immediately and then stored at −80°C. All clinicopathological data, such as sex, age, TNM stage, differentiation and lymph node metastasis (LNM), were collected from the medical records and are summarized in Table S1. By regular visits or telephone interviews, the patients’ overall survival was also determined.
The human GC cell lines SGC-7901, MKN-28, MKN-45, BGC-823, AGS, and MGC-803 were all obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The normal human gastric epithelial cell line GES-1 (Chinese Academy of Sciences, Shanghai, People’s Republic of China) was also used in this study. Cells were cultured in Roswell Park Memorial Institute-1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum and antibiotics (1% penicillin/streptomycin; Thermo Fisher Scientific) in a humidified chamber with an atmosphere of 5% CO2 at 37°C.
Quantitative real-time polymerase chain reaction (qRT-PCR) of miRNAs
The miRNAs were isolated and purified with a miRNA isolation system (Exiqon, Vedbaek, Denmark). For qRT-PCR, the miScript II RT Kit (Qiagen, Germany) was used to generate cDNA, and qRT-PCR was performed using a miScript SYBR Green PCR Kit (Qiagen NV) according to the manufacturer’s instructions. The qRT-PCR analysis was conducted on 7,500 Real-Time PCR System (ABI, Foster City, CA, USA). The miRNA expression was calculated by normalization to the internal control U6 using the 2−ΔΔCt method.
Transfection and plasmid construction
The miR-590-5p mimics, mimic control, miR-590-5p inhibitor, and inhibitor control were synthesized by GenePharma (Shanghai, People’s Republic of China). For cell transfection, cells were seeded in six-well plates (2×105/well), and transfection was performed using Lipofectamine 2000 (Thermo Fisher Scientific). For luciferase activity analysis, the 3′-UTR of RECK containing the putative binding sites for miR-590-5p was amplified and cloned into the pGL3-luciferase reporter plasmid (Promega Corporation, Madison, WI, USA). Mutations in the miR-590-5p binding sites of RECK 3′-UTR were generated using a QuikChange Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA, USA). For stably overexpressing miR-590-5p cells, the lentiviruses expressing miR-590-5p and control viruses were used to infect SGC-7901 cells.
The siRNA 5′-AAGACCCAGCCCUUGCCUCAA-3′ (sense) was used to knock down the expression of RECK, and the RNA 5′-AACGUUGCGAUAGCGUAGUAC-3′ was used as a control. The RECK coding region with and without the 3′-UTR was obtained from Hanbio (Shanghai, People’s Republic of China).
Cell proliferation assays
For proliferation assay, the transfected GC cells were seeded in 96-well plates (2,000 cells per well) and maintained at 37°C and 5% CO2 for 4 days. The reaction was stopped by addition of 150 μL dimethyl sulfoxide, and cell proliferation was determined by measuring the optical density at 490 nm using a spectrophotometric plate reader (Thermo Fisher Scientific).
Cell invasion assay
Matrigel transwell assays were used to assess the invasive ability of GC cells. Briefly, at 36 hours post-transfection, 3×104 cells in 300 μL serum-free medium were placed into the upper chambers precoated with 150 mg matrigel (BD Biosciences, San Jose, CA, USA). Then, 0.5 mL medium containing 10% fetal bovine serum was added to the lower chambers as a chemo-attractant. After incubation at 37°C for 24 hours, cells that migrated to the bottom of the membrane were fixed, stained, and visualized under a microscope.
MTS assay
The sensitivity of cells to cDDP was determined using a Cell Titer 96® AQueous One Solution Cell Proliferation Assay kit (Promega Corporation). Cells (3×103 cells/well) were seeded onto 96-well plates for 24 hours before use. The medium was exchanged with fresh medium containing cDDP at different concentrations, and cells were then cultured for an additional 72 hours. Then, 20 μL MTS was added to each well. Two hours later, the absorbance at 490 nm was recorded for each well on a BioTek Synergy 2 (BioTek Instruments Inc, Winooski, VT, USA). cDDP was obtained from Calbiochem (Darmstadt, Germany) and was stored at a concentration of 5 mg/mL at −20°C. Freshly prepared PTX (Sigma-Aldrich Co., St Louis, MO, USA) was added at a final concentration ranging from 5 to 100 nM.
Luciferase reporter assay
In a 96-well plate, HEK-293 cells were co-transfected with 0.2 mg/mL of wild-type or mutant-RECK-3′-UTR plasmid and 50 ng of Renilla luciferase plasmid with miR-590-5p or an empty vector control using Lipofectamine 2000 (Thermo Fisher Scientific). After 24 hours, the cells were lysed, and luciferase activity was determined using a Dual-Luciferase reporter assay system (Promega Corporation). Firefly luciferase activity was then normalized to the corresponding Renilla luciferase activity.
Western blot analysis
Protein concentrations were measured using a BCA Protein Assay Kit (Pierce, Rockford, IL, USA). Equal amounts of proteins (20 μg) were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories Inc., Hercules, CA, USA). The membranes were blocked by incubation in Tris-buffered saline with Tween 20 (25 mM Tris, pH 7.4, 150 mM NaCl, 0.1% Tween 20) containing 5% fat free milk for 1 hour at room temperature and were then immunoblotted with primary antibodies against RECK (R&D Systems, Inc., Minneapolis, MN, USA), AKT, p-AKT (Ser473), ERK, p-ERK (Cell Signaling Technology, Danvers, MA, USA), pSTAT3, STAT3, and β-actin (Affinity, Ossipee, NH, USA), followed by incubation with HRP-conjugated secondary antibodies (Santa Cruz Biotechnology Inc., Dallas, TX, USA). Blots were detected using an enhanced chemiluminescence detection system (Pierce).
In vivo xenograft models
All animal experiments in this study were approved by ethics committee of Nanjing Medical University, and the national Animal Care and Use guidelines were followed. Thirty-six mice (4 weeks old) were used in this study and equally divided into three groups. One group was injected with the miR-590-expressing vector (3×106), one group with cDDP, and one group with both miR-590 and cDDP. When the tumors were palpable, cDDP (5 mg/kg) was peritoneally injected into the mice every 4 days. Tumor volume was measured and calculated as tumor volume = length × width2/2. Mice were sacrificed on the 14th, 21st, and 28th day, and the harvested tumors were subjected to Western blot assays.
Statistical analysis
Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA) and SPSS 14.0 (SPSS Inc., Chicago, IL, USA) were used to analyze the experimental data. The data are presented as the mean ± standard error of mean, and the differences between groups were analyzed using Student’s t-test with only two groups or one-way analysis of variance when more than two groups were compared. A Mann–Whitney test was used to assess the expression of miR-590-5p between two groups, and a Kruskal–Wallis test was used for more than two groups. Kaplan–Meier and log-rank tests were used in the overall survival analysis. A P-value of <0.05 was considered statistically significant.
Results
Upregulated miR-590-5p is correlated with poor prognosis
First, we determined the endogenous miR-590-5p levels in GC cells and compared them with the miR-590-5p expression in the normal control cell line GES-1. The expression of miR-590-5p in GC cells was significantly higher than that in GES-1 cells (Figure 1A). Subsequently, we measured the expression of miR-590-5p in GC patients. Similar results showed that the miR-590-5p level in primary tumors was significantly increased compared to that in adjacent non-tumor tissues (0.0174±0.0010 vs 0.0157±0.0009, P=0.0003, Figure 1B).
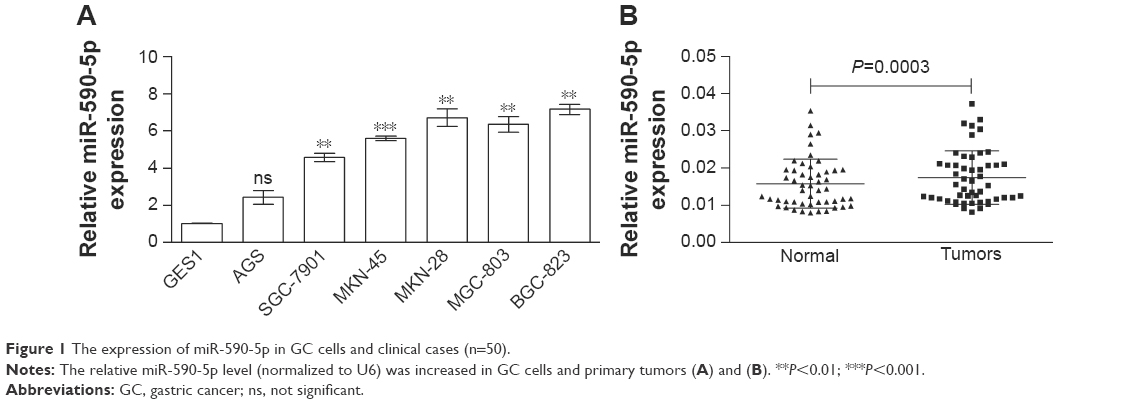 | Figure 1 The expression of miR-590-5p in GC cells and clinical cases (n=50). |
We separated GC cases according to the different clinicopathological characteristics of patients, such as female vs male, tumor stage I/II vs stage III/IV, with LNM vs without LNM. We found that the expression of miR-590-5p was gradually increased along with increasing tumor size (0.0139±0.0010 vs 0.0162±0.0016 vs 0.0215±0.0018, P=0.012, Figure 2A). The expression of miR-590-5p was markedly higher in patients with LNM than that in patients without LNM (0.0195±0.0014 vs 0.0152±0.0013, P=0.028, Figure 2B). For survival analysis, the mean value of miR-590-5p expression in all GC tumors was used as a threshold value to classify the 50 patients into the high or low miR-590-5p expression group. Kaplan–Meier survival analysis showed that the low miR-590-5p group had a significantly higher overall survival compared to that of the high miR-590-5p group (P=0.025, Figure 2C). These results suggested that miR-590-5p is involved in gastric tumorigenesis, and its upregulation is inversely associated with prognosis.
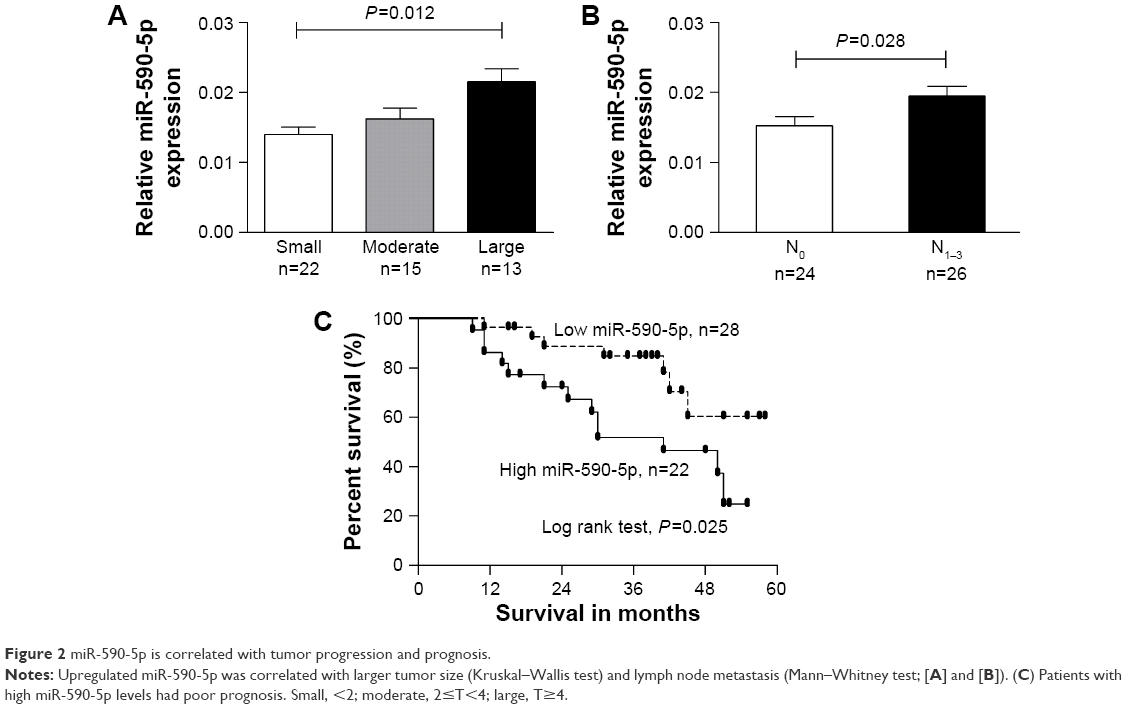 | Figure 2 miR-590-5p is correlated with tumor progression and prognosis. |
miR-590-5p promotes cell proliferation and invasion and reduces sensitivity of GC cells to cDDP and PTX in vitro
Because the expression of miR-590-5p was correlated with tumor size and LNM, we explored the effect of miR-590-5p on cell proliferation and invasion. SGC-7901 cells expressing relatively low levels of miR-590-5p and BGC-823 cells expressing relatively high levels of miR-590-5p were transfected with miR-590-5p mimics and inhibitor, respectively, which increased the level of miR-590-5p in SGC-7901 cells by 10.28-fold and decreased that in BGC-823 cells by 3.03-fold compared to that of matched control cells (Figure 3A).
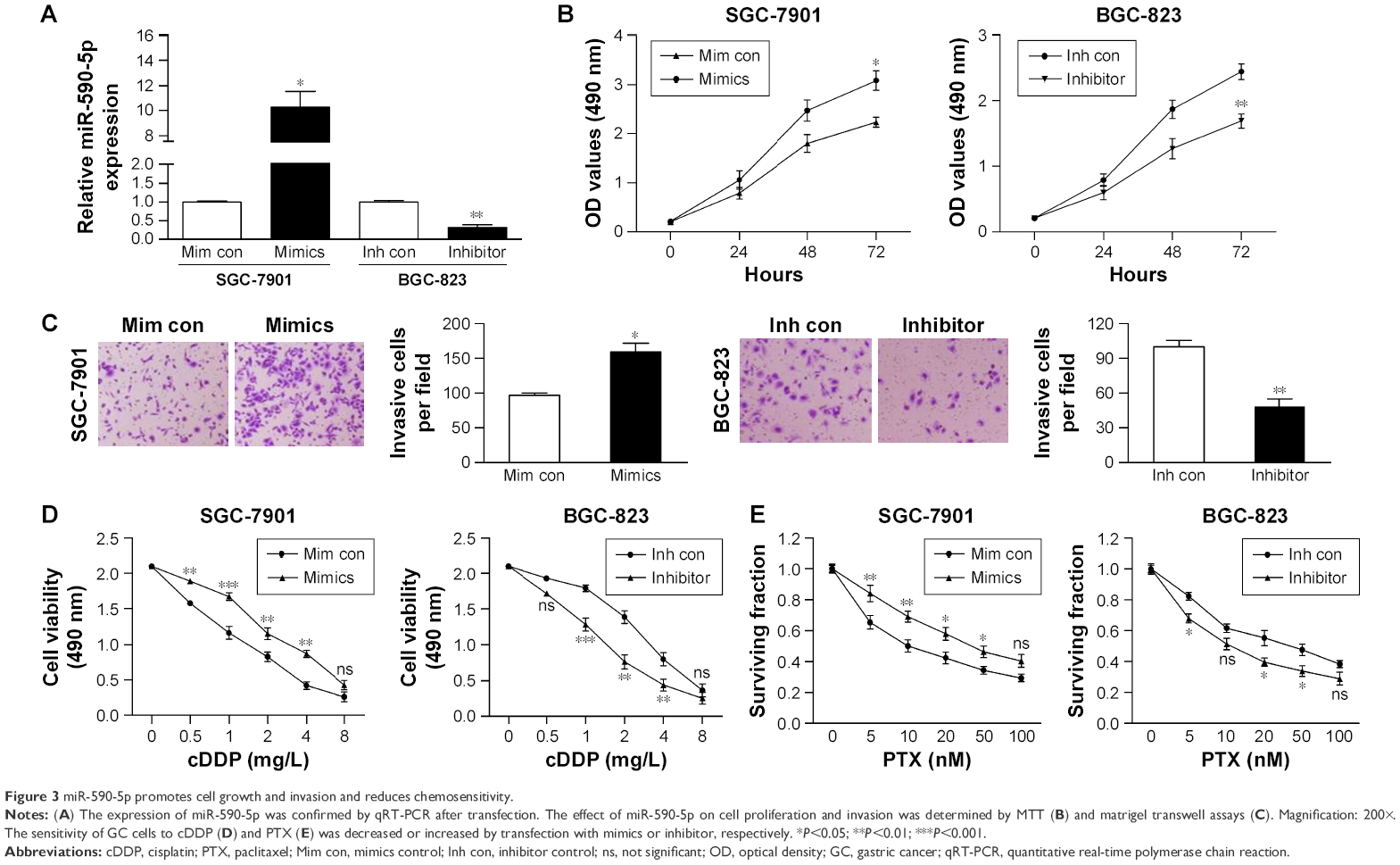 | Figure 3 miR-590-5p promotes cell growth and invasion and reduces chemosensitivity. |
MTT assays indicated that overexpression of miR-590-5p significantly promoted the proliferation of SGC-7901 cells. In contrast, knockdown of miR-590-5p significantly suppressed the proliferation of BGC-823 cells compared to control cells (Figure 3B). Matrigel transwell assays showed that miR-590-5p markedly increased the invasion of SGC-7901 cells, whereas inhibition of miR-590-5p impaired this activity in BGC-823 cells (Figure 3C).
We also explored the effects of ectopic miR-590-5p expression on cell chemosensitivity. Here, we found that overexpression of miR-590-5p via transfection of mimics significantly reduced the sensitivity of SGC-7901 cells to cDDP and PTX (Figure 3D and E). Consistent with these results, downregulation of miR-590-5p noticeably increased the chemosensitivity of BGC-823 cells. These data collectively indicated that miR-590-5p functions as an oncogene and desensitizes GC cells to chemotherapeutics.
RECK is a direct target of miR-590-5p
We used the bioinformatics algorithms TargetScan, miRDB, and microRNA to search for potential targets of miR-590-5p. All of these databases predicted RECK as a target of miR-590-5p, and the 3′-UTR of RECK mRNA contains a highly conserved binding site from positions 1,138 to 1,145 with a miR-590-5p seed sequence (Figure 4A). Luciferase activity analysis showed that miR-590-5p significantly inhibited the luciferase activity of the RECK-3′-UTR-WT reporter, but did not affect the RECK-3′-UTR-Mut reporter (Figure 4B). Furthermore, Western blot analysis showed that the RECK protein expression was downregulated in GC cells compared to GES-1 cells and was substantially reduced by miR-590-5p overexpression in both SGC-7901 and AGS cells (Figure 4C).
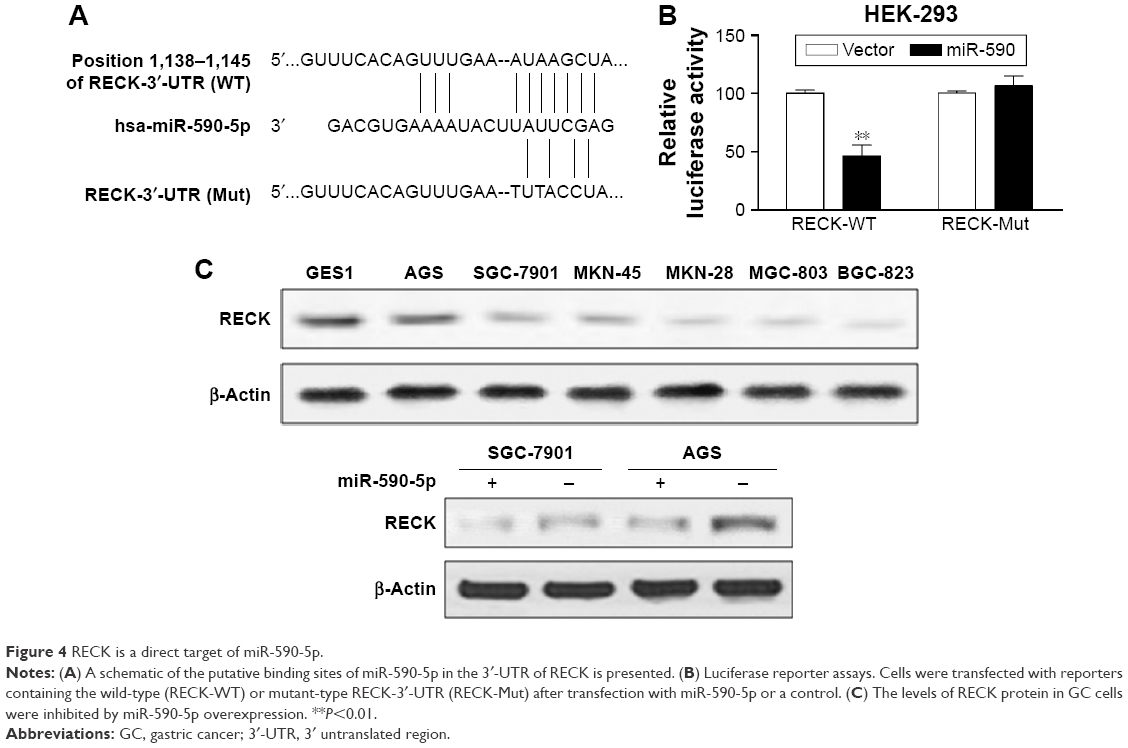 | Figure 4 RECK is a direct target of miR-590-5p. |
miR-590-5p modulates cell growth, invasion, and chemosensitivity by targeting RECK
To verify whether the oncogenic effects of miR-590-5p are mediated through RECK, we knocked down RECK by RNA interference, and the protein level of RECK was significantly suppressed after RECK knockdown (Figure 5A). MTT assays indicated that knockdown of RECK in SGC-7901 cells significantly enhanced cell growth and invasion compared with control cells (Figure 5B and C). Additionally, knockdown of RECK markedly decreased the sensitivity of SGC-7901 cells to cDDP (Figure 5D).
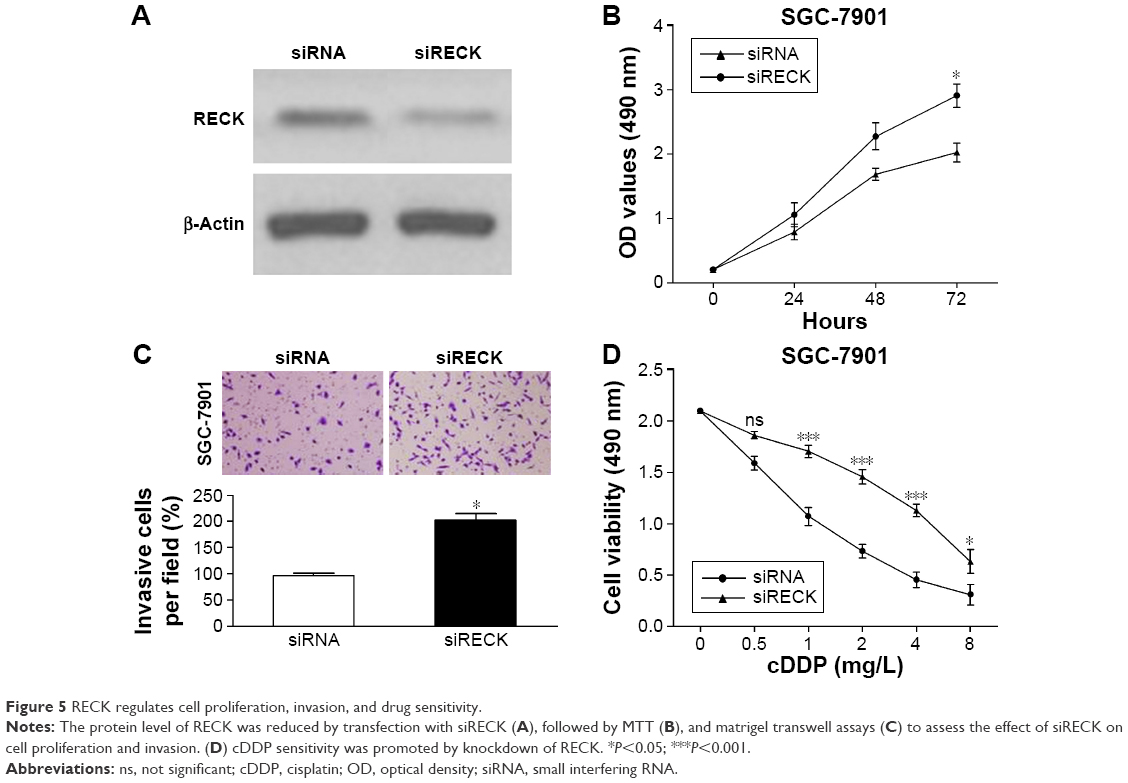 | Figure 5 RECK regulates cell proliferation, invasion, and drug sensitivity. |
We rescued the expression of RECK in miR-590-5p mimic-transfected cells to demonstrate the functional relationship between miR-590-5p and RECK. We transfected the RECK-expressing vector (lacking 3′-UTR) into SGC-7901 cells, and the protein expression level of RECK was determined by Western blot assays (Figure 6A). As shown in Figure 6B and C, increased RECK in SGC-7901 cells inhibited the effect of miR-590-5p on cell proliferation and invasion. The chemosensitivity of SGC-7901 cells was also enhanced by RECK protein overexpression (Figure 6D). We evaluated whether miR-590-5p could regulate the AKT pathway, which is generally activated and associated with the progression and multi-drug resistance of cancer.20,21 As shown in Figure 6E, the phosphorylation of AKT (Ser473) and ERK (Thr202/Thr204) was promoted by miR-590-5p mimics. The total protein levels of AKT and ERK were not changed. Furthermore, STAT3 signaling was also activated by overexpression of RECK (Figure 6E).
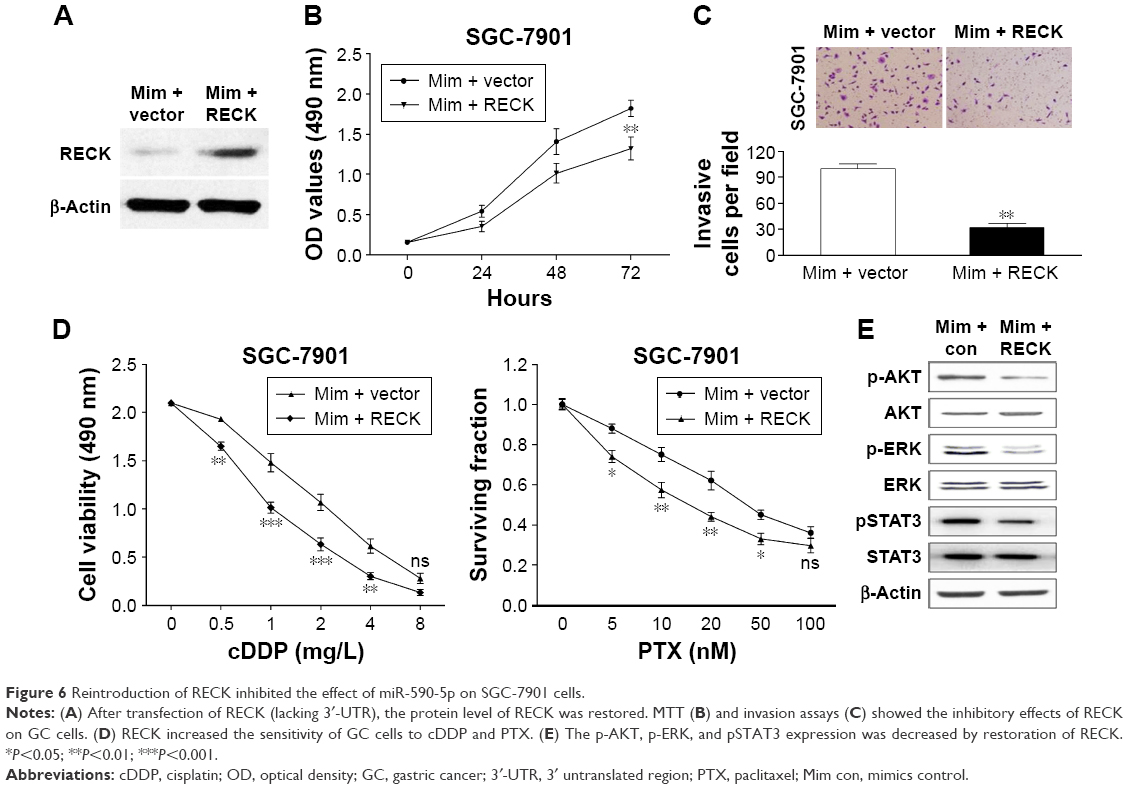 | Figure 6 Reintroduction of RECK inhibited the effect of miR-590-5p on SGC-7901 cells. |
miR-590-5p promotes tumor growth in vivo
To further confirm the role of miR-590-5p in reducing the sensitivity of GC cells to chemotherapeutics in vivo, the stable miR-590-5p-expressing SGC-7901 cells were inoculated into the flanks of four nude mice (Figure 7A). The results showed that the tumors from the miR-590-5p-expressing cells were significantly larger than those derived from the vector control (Figure 7B). The tumor weight was also increased in the miR-590-5p-overexpressing cells (Figure 7C). We analyzed the expression of miR-590, RECK, pAKT, pERK, and pSTAT3 in xenograft tumors. In miR-590-overexpressing tumors, the levels of miR-590-5p were enhanced along with the activated pAKT, pERK, and pSTAT3, while the RECK protein was downregulated (Figure 7D and E).
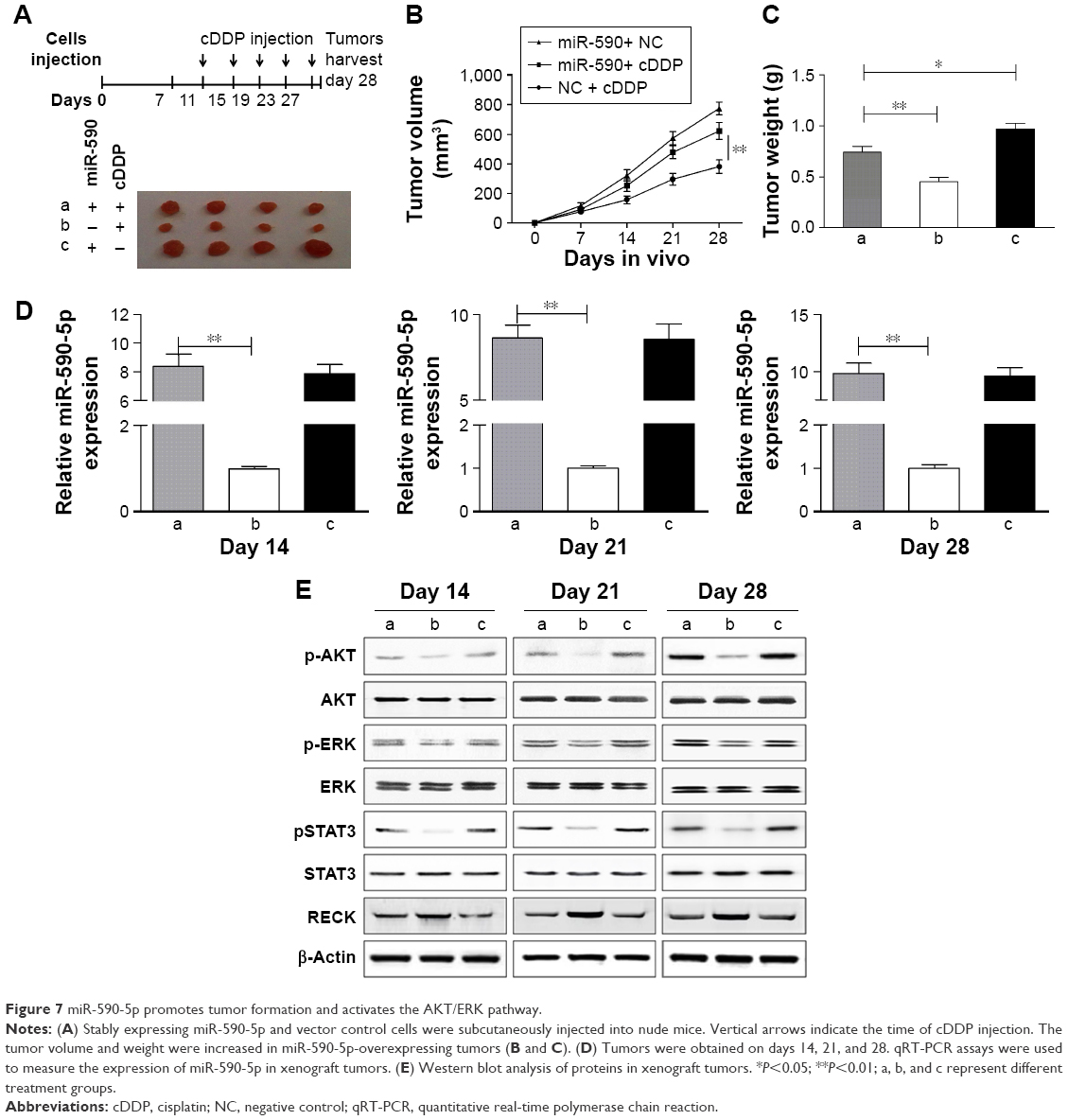 | Figure 7 miR-590-5p promotes tumor formation and activates the AKT/ERK pathway. |
Discussion
GC progression is a multistep process involving genetic and epigenetic alteration of proto-oncogenes and tumor-suppressor genes. Hence, identification of the exact role of miRNAs in GC progression and chemoresistance would provide novel insights for diagnosis and therapy of GC patients.
In this study, upregulated miR-590-5p was observed in GC cells and primary tumors, suggesting the oncogenic role of miR-590-5p in GC pathogenesis. Previous studies also reported upregulated miR-590-5p in cells from hepatocellular carcinoma, cervical cancer, and renal carcinoma.18,19,22 Therefore, we speculated that miR-590 has a broad role in various types of tumors. We showed that upregulated miR-590-5p was correlated with tumor progression and poor survival, similar to a previous study showing that miR-590-5p was positively correlated with LNM.23 Further studies based on a large number of clinical cases may provide more information on miR-590-5p as a prognostic factor.
Subsequently, we found that miR-590-5p contributed to cell growth and invasion, which was consistent with the observations in previous studies.18,19 Our data expanded the knowledge of miR-590-5p in carcinogenesis. More importantly, we found that miR-590-5p attenuated the chemosensitivity of GC cells, suggesting the potential function of miR-590-5p as a drug-related gene in cancer treatment. In former studies, miR-590-5p was one of three miRNA transcripts determined to predict complete or incomplete response to neoadjuvant chemoradiotherapy in rectal cancer24 or serve as predictive markers of chemo-responsiveness in Wilms’ tumors.25 Alterations in the circulating miR-590-5p levels during chemotherapy indicated the response of head and neck cancer cells.26 These data and ours implied that miR-590-5p may be a promising therapeutic target. Further studies examining the correlations between miR-590-5p and chemotherapy would be beneficial for the improvement of treatment and prognosis of patients.
RECK is usually reported as a tumor suppressor, particularly an inhibitor of metastasis, in many types of cancer.14,15,27 In this study, the suppressive effect of RECK on GC cells was confirmed, and RECK was inhibited by miR-590-5p. Additionally, other miRNAs, including miR-221, miR-222, miR-374-5p, and miR-25, also regulated RECK by binding to its 3′-UTR and contributing to the growth and invasion of GC.28–30 RECK may be a critical regulator of miRNA-related tumor cell behavior. Furthermore, RECK was involved in miR-590-related AKT/ERK and STAT3 signaling activation in our present study. The anti-tumor activity of RECK was documented in breast cancer by inhibiting erbB signaling, which is associated with chemoresistance.31 RECK was downregulated by miR-96 and ultimately led to proliferation and chemoresistance in esophageal cancer.32 The activated AKT/ERK signaling pathway may explain the alterations in cell growth and drug sensitivity in our study. Overall, we elucidated a novel mechanism responsible for gastric tumorigenesis and found that the miR-590/RECK axis may be a useful therapeutic target.
Conclusion
Taken together, our findings highlight the important function of miR-590-5p in GC progression and chemotherapy. Dysregulated miR-590-5p targets RECK and AKT/ERK signaling to promote GC progression and acquired chemoresistance. Therefore, the miR-590-5p/RECK/AKT axis was identified as a new molecular mechanism and potential therapeutic tool for GC treatment.
Acknowledgments
This study was funded by the Subject of Jiangsu Provincial Commission of Health (No H201411) and Wu Jieping Medical Fund (No 320.6750.13231).
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
Coburn NG. Lymph nodes and gastric cancer. J Surg Oncol. 2009;99(4):199–206. | ||
De Vita F, Vecchione L, Galizia G, et al. Perspectives in adjuvant therapy of gastric cancer. Oncology. 2010;77 Suppl 1:38–42. | ||
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. | ||
Wang R, Wang ZX, Yang JS, Pan X, De W, Chen LB. MicroRNA-451 functions as a tumor suppressor in human non-small cell lung cancer by targeting ras-related protein 14 (RAB14). Oncogene. 2011;30(23):2644–2658. | ||
Deng M, Tang H, Zhou Y, et al. miR-216b suppresses tumor growth and invasion by targeting KRAS in nasopharyngeal carcinoma. J Cell Sci. 2011;124(Pt 17):2997–3005. | ||
Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med. 2012;4(3):143–159. | ||
Tang L, Chen F, Pang EJ, Zhang ZQ, Jin BW, Dong WF. MicroRNA-182 inhibits proliferation through targeting oncogenic ANUBL1 in gastric cancer. Oncol Rep. 2015;33(4):1707–1716. | ||
Shen B, Zhang Y, Yu S, et al. MicroRNA-339, an epigenetic modulating target is involved in human gastric carcinogenesis through targeting NOVA1. FEBS Lett. 2015;589(20 Pt 8):3205–3211. | ||
Nishida N, Mimori K, Fabbri M, et al. MicroRNA-125a-5p is an independent prognostic factor in gastric cancer and inhibits the proliferation of human gastric cancer cells in combination with trastuzumab. Clin Cancer Res. 2011;17(9):2725–2733. | ||
Hu J, Fang Y, Cao Y, Qin R, Chen Q. miR-449a Regulates proliferation and chemosensitivity to cisplatin by targeting cyclin D1 and BCL2 in SGC7901 cells. Dig Dis Sci. 2014;59(2):336–345. | ||
Weaver VM. Membrane-associated MMP regulators: novel cell adhesion tumor suppressor proteins? Dev Cell. 2002;2(1):6–7. | ||
Yeh HH, Tseng YF, Hsu YC, et al. Ras induces experimental lung metastasis through up-regulation of RbAp46 to suppress RECK promoter activity. BMC Cancer. 2015;15:172. | ||
Xu M, Wang HF, Zhang HZ. Expression of RECK and MMPs in hepatoblastoma and neuroblastoma and comparative analysis on the tumor metastasis. Asian Pac J Cancer Prev. 2015;16(9):4007–4011. | ||
Wang L, Ge J, Ma T, et al. Promoter hypermethylation of the cysteine protease RECK may cause metastasis of osteosarcoma. Tumour Biol. 2015;36(12):9511–9516. | ||
Hong KJ, Wu DC, Cheng KH, Chen LT, Hung WC. RECK inhibits stemness gene expression and tumorigenicity of gastric cancer cells by suppressing ADAM-mediated Notch1 activation. J Cell Physiol. 2014;229(2):191–201. | ||
Loayza-Puch F, Yoshida Y, Matsuzaki T, Takahashi C, Kitayama H, Noda M. Hypoxia and RAS-signaling pathways converge on, and cooperatively downregulate, the RECK tumor-suppressor protein through microRNAs. Oncogene. 2010;29(18):2638–2648. | ||
Jiang X, Xiang G, Wang Y, et al. MicroRNA-590-5p regulates proliferation and invasion in human hepatocellular carcinoma cells by targeting TGF-β RII. Mol Cells. 2012;33(6):545–551. | ||
Chu Y, Ouyang Y, Wang F, et al. MicroRNA-590 promotes cervical cancer cell growth and invasion by targeting CHL1. J Cell Biochem. 2014;115(5):847–853. | ||
Zhao T, Hu F, Liu X, Tao Q. Blockade of telomerase reverse transcriptase enhances chemosensitivity in head and neck cancers through inhibition of AKT/ERK signaling pathways. Oncotarget. 2015;6(34):35908–35921. | ||
Li Y, Jia L, Ren D, et al. Axl mediates tumor invasion and chemosensitivity through PI3K/Akt signaling pathway and is transcriptionally regulated by slug in breast carcinoma. IUBMB Life. 2014;66(7):507–518. | ||
Xiao X, Tang C, Xiao S, Fu C, Yu P. Enhancement of proliferation and invasion by MicroRNA-590-5p via targeting PBRM1 in clear cell renal carcinoma cells. Oncol Res. 2013;20(11):537–544. | ||
Yang X, Wu X. miRNA expression profile of vulvar squamous cell carcinoma and identification of the oncogenic role of miR-590-5p. Oncol Rep. 2016;35(1):398–408. | ||
Kheirelseid EA, Miller N, Chang KH, et al. miRNA expressions in rectal cancer as predictors of response to neoadjuvant chemoradiation therapy. Int J Colorectal Dis. 2013;28(2):247–260. | ||
Watson JA, Bryan K, Williams R, et al. miRNA profiles as a predictor of chemoresponsiveness in Wilms’ tumor blastema. PLoS One. 2013;8(1):e53417. | ||
Summerer I, Niyazi M, Unger K, et al. Changes in circulating microRNAs after radiochemotherapy in head and neck cancer patients. Radiat Oncol. 2013;8:296. | ||
Clark JC, Thomas DM, Choong PF, Dass CR. RECK – a newly discovered inhibitor of metastasis with prognostic significance in multiple forms of cancer. Cancer Metastasis Rev. 2007;26(3–4):675–683. | ||
Liu W, Song N, Yao H, Zhao L, Liu H, Li G. miR-221 and miR-222 simultaneously target RECK and regulate growth and invasion of gastric cancer cells. Med Sci Monit. 2015;21:2718–2725. | ||
Xie J, Tan ZH, Tang X, et al. MiR-374b-5p suppresses RECK expression and promotes gastric cancer cell invasion and metastasis. World J Gastroenterol. 2014;20(46):17439–17447. | ||
Zhao H, Wang Y, Yang L, Jiang R, Li W. MiR-25 promotes gastric cancer cells growth and motility by targeting RECK. Mol Cell Biochem. 2014;385(1–2):207–213. | ||
Hong KJ, Hsu MC, Hung WC. RECK impedes DNA repair by inhibiting the erbB/JAB1/Rad51 signaling axis and enhances chemosensitivity of breast cancer cells. Am J Cancer Res. 2015;5(8):2422–2430. | ||
Xia H, Chen S, Chen K, Huang H, Ma H. MiR-96 promotes proliferation and chemo- or radioresistance by down-regulating RECK in esophageal cancer. Biomed Pharmacother. 2014;68(8):951–958. |
Supplementary material
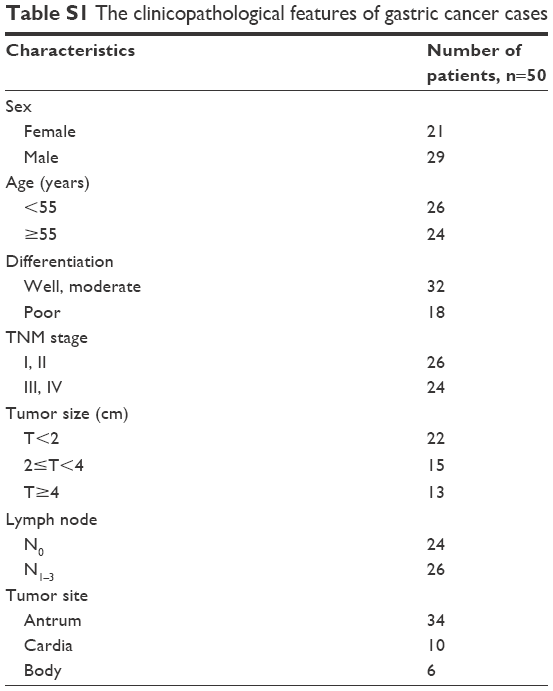 | Table S1 The clinicopathological features of gastric cancer cases |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.