")
Back to Journals » OncoTargets and Therapy » Volume 11
miR-488 inhibits cell growth and metastasis in renal cell carcinoma by targeting HMGN5
Received 7 November 2017
Accepted for publication 27 February 2018
Published 18 April 2018 Volume 2018:11 Pages 2205—2216
DOI https://doi.org/10.2147/OTT.S156361
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianmin Xu
This paper has been retracted.
Xin Wei, Lili Yu, Xiangbo Kong
Department of Urology, China-Japan Union Hospital of Jilin University, Changchun, China
Purpose: microRNAs are thought to play crucial roles in tumorigenesis. Dysregulation of miR-488 has been implicated to be involved in several cancer progressions. However, the biological functions of miR-488 in renal cell carcinoma (RCC) remain unclear. This study aimed to explore the molecular mechanism underlying the role of miR-488 in RCC development.
Materials and methods: The expression levels of miR-488 were detected in 38 paired RCC tumor samples and cell lines by quantitative real-time polymerase chain reaction method. miR-488 was upregulated by mimics transfection in RCC cell lines. MTT, colony formation, transwell assay, flow cytometry assay, and a xenograft model were performed to determine cell proliferation, invasion, migration, epithelial-to-mesenchymal transition, and apoptosis in vitro and in vivo. Moreover, the potential target of miR-488 was verified by dual-luciferase reporter assay, quantitative real-time polymerase chain reaction, and Western blot. The correlation between miR-488 expression and its target gene expression was confirmed by Spearman’s correlation analysis in 38 selected RCC tissue samples.
Results: We found that miR-488 was remarkably downregulated in human RCC samples and cell lines compared with paired normal tissues and cell lines. Functional investigations revealed that overexpression of miR-488 significantly suppressed cell proliferation, invasion, and migration, and promoted cell apoptosis in RCC cells. Nucleosome binding protein 1 (high-mobility group nucleosome binding domain 5 [HMGN5]) was identified as a direct target of miR-488, and an inverse relationship was found between miR-488 expression and HMGN5 mRNA levels in RCC specimens. Rescue experiments suggested that restoration of HMGN5 partially abolished miR-488-mediated cell proliferation and invasion inhibition in RCC cells through regulating phosphatidylinositol 3-kinase/protein kinase B/the mammalian target of rapamycin and epithelial-to-mesenchymal transition signaling pathways.
Conclusion: These data indicated that miR-488 acted as a tumor suppressor in RCC proliferation and invasion by targeting HMGN5, which might provide potential therapeutic biomarker for RCC patients.
Keywords: renal cell carcinoma, miRNA-488, HMGN5, metastasis, proliferation
Introduction
Renal cell carcinoma (RCC), originating in the renal cortex, is the most common neoplasm of the adult kidney and the third leading cause of cancer-related mortality worldwide.1 The incidence rate of newly diagnosed RCC patients is increasing every year, which accounts for ~3% of new adult malignancy cases globally.2 Because RCC is resistant to adjuvant therapies like chemotherapy and radiotherapy, surgical resection still remains the only curative treatment for RCC in clinics.3 However, after the curative nephrectomy, about 40% of patients diagnosed with localized RCC will develop recurrence and metastasis, leading to poor prognosis in these patients.4 RCC is characterized by multiple histologic and diverse molecular alterations even in a single patient, which highlights the heterogeneity and complexity of this disease.5 Recently, the novel therapies for distant metastatic RCC have been obviously improved,6 yet the 5-year survival rate of these advanced RCC patients is still <10%.7 Therefore, there is urgent need to develop effective prognostic and therapeutic agents for the treatment of RCC patients in clinic.
microRNAs (miRNAs) belong to a class of endogenous, small, and noncoding RNAs, with ~19–22 nucleotides in length, that posttranscriptionally regulate gene expressions by binding to the 3′-untranslated region (3′-UTR) of their target mRNAs, which results in the translational repression or mRNA clearance of these target genes.8 It has been postulated that the expressions of >10,000 mRNAs are thought to be mediated by miRNAs in the human genome.9 Increasing evidence has demonstrated that miRNAs play critical roles in a variety of crucial biological processes including cell growth, apoptosis, metastasis, angiogenesis, and differentiation.10 Recently, aberrant expressions of miRNAs are identified to be involved in the pathogenesis of multiple human cancers, including RCC.10 It has been demonstrated that miRNAs expression profiles contribute to RCC progression at different stages through inducing oncogenes and suppressing tumor suppressor genes.11 Thus, miRNAs are referred to as a group of potential cancer biomarkers, which may provide benefits to RCC patients at the aspects of diagnosis, prognosis, and even therapeutics.
The aberrant expression of miR-488 has been reported to be involved in the pathological processes of various diseases. Song et al have showed that miR-488 plays a positive role in chondrocyte differentiation and inhibits the pathogenesis of osteoarthritis.12 Muinos-Gimeno et al have found that altered expression of miR-488 is associated with panic disorder and regulates the physiological pathways related to anxiety.13 Recently, miR-488 has been found to function as a tumor suppressor during different cancer developments. Fang et al have indicated that exogenous overexpression of miR-488 could inhibit proliferation and cisplatin sensitivity in none of the small-cell lung cancer cells.14 Lv et al have illustrated that downregulation of miR-488 increases cell proliferation and migration, and accelerates the progression of colorectal cancer.15 Zhao et al have suggested that miR-488 acts as a tumor suppressor by inhibiting cell proliferation and migration in gastric cancer.16 However, the association of miR-488 with the progression and development of RCC is still unknown.
High-mobility group nucleosome binding domain 5 (HMGN5/NSBOP1) is referred as a ubiquitous nucleosome binding protein and located at the Xq13.3 region of the human genome.17 It has the characteristic structure of a nucleosome location signal (NLS) in the N-terminal, a highly conserved nucleosomal binding domain, and a negatively charged C-terminal domain.18 HMGN5 specifically interacts with nucleosome core particles and participates in the cellular processes of replication, transcription, DNA repair, and recombination.19 Accumulating studies have demonstrated that HMGN5 is identified as a candidate oncogene in diverse cancers, including gliomas, prostate cancer, breast cancer, and osteosarcoma.20 Notably, a recent study has illustrated that silencing of HMGN5 inhibits the growth and invasion of clear cell RCC cells in vitro and in vivo.21 Nevertheless, whether specific miRNAs are involved in the molecular regulation of HMGN5 during RCC progression remains largely unknown.
The present study was aimed to investigate the function of miR-488 in the pathogenesis of RCC and illuminate the underlying molecular mechanisms by which miR-488 regulated RCC progression. In this study, we found that miR-488 expression was significantly downregulated in RCC tissue samples and cell lines. Functional experiments showed that miR-488 acted as a tumor suppressor by inhibiting cell proliferation, migration, and invasion, and by promoting apoptosis in RCC cells in vitro and in vivo. Moreover, HMGN5 was identified as a direct target of miR-488, and restoration of HMGN5 could partially attenuate the inhibitory effect of miR-488 on RCC cell proliferation and invasion. Therefore, we supposed that these findings might give insight into developing a new therapeutic biomarker for RCC treatment.
Materials and methods
Collection of human samples
RCC tissue samples and paired adjacent normal tissues were obtained from 38 patients who underwent radical nephrectomy at the Department of Urology, China-Japan Union Hospital of Jilin University, between May 2014 and March 2016. None of the patients had received radiotherapy or chemotherapy prior to surgical resection. The collected samples were immediately stored in liquid nitrogen. Written informed consent was obtained from all the patients, and this study was approved by the Institution Research Ethics Committee of Jilin University.
Cell culture
Five human RCC cell lines including ACHN, Caki-1, 786-O, 769-P, and A498, and the normal human proximal tubule epithelial cell line HK-2 were obtained from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). All cells were cultured in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific) and 1% penicillin/streptomycin at 37°C in a 5% CO2 humidified incubator.
Cell transfection
The miR-488 mimics (miR-488) and the corresponding negative control (miR-NC) were synthesized and obtained from GenePharma (Shanghai, China). pcDNA3.1-HMGN5 recombinant plasmid was also constructed by GenePharma to restore HMGN5 expression. miR-488, miR-NC, pcDNA3.1-HMGN5, or pcDNA3.1 empty vector was transfected into RCC cell lines with Lipofectamine 2000 (Thermo Fisher Scientific) for 24 h according to the manufacturer’s protocols.
Quantitative real-time polymerase chain reaction assay
Total RNA from tissues or cell lines was isolated by using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. cDNA was synthesized using PrimeScript RT reagent kit (Takara, Tokyo, Japan). Detection of miR-488 was performed by using a TaqMan miRNA assay in accordance with the manufacturer’s protocols (Qiagen, Shanghai, China) under ABI 7500 Real-time PCR system (Bio-Rad Laboratories Inc., Hercules, CA, USA). U6 was used as the internal control for miRNA. The mRNA expression level of HMGN5 expression was measured by SYBR Green Master Mix (Thermo Fisher Scientific) using ABI 7500 Real-time PCR system (Bio-Rad Laboratories Inc.). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the internal control for mRNA. The relative expression level of miR-488 or HMGN5 was calculated with 2−ΔΔCt method.
Western blot
Cells were collected and lysed with radio immunoprecipitation assay buffer according to the manufacturer’s instruction. The protein concentration was measured by BCA protein assay kit (Boster, Wuhan, China). Twenty micrograms of proteins was separated on 12% SDS-PAGE and electrophoretically transferred onto polyvinylidene fluoride membrane (EMD Millipore, Billerica, MA, USA). The membranes were blocked with 5% nonfat skim milk for 1 h at the room temperature, and then incubated with primary antibodies against HMGN5, E-cadherin, N-cadherin, vimentin, phosphorylated (p)-AKT, p-mTOR, P13K, AKT, and mTOR (Abcam, Cambridge, UK) overnight at 4°C. After rinsing, the membranes were incubated with the corresponding secondary horseradish peroxidase-conjugated secondary antibodies (Abcam) for 2 h at 37°C. The protein bands were visualized with ECL chemiluminescent kit (Thermo Fisher Scientific) and quantified by ImageJ software. GAPDH was used for normalization.
MTT assay
MTT assay was performed to examine cell viability. After transfection with miR-488 mimics or miR-NC for 24 h, 768-O and A498 cells were seeded into 96-well plates at the density of 5×103 cells per well and cultured for 24, 48, 72, or 96 h. Subsequently, 20 μL of MTT solution (0.5 mg/mL; Sigma-Aldrich Co., St Louis, MO, USA) was added into each well and incubated for 4 h, followed by additional incubation with 200 μL of dimethyl sulfoxide for 15 min. Optical density was determined by a microplate reader at a wavelength of 490 nm.
Colony formation assay
For colony formation assay, after transfection with miR-488 mimics or miR-NC for 24 h, 768-O and A498 cells were plated in 6-well plates (5×108 cells/well) and incubated for 14 days. Subsequently, cells were fixed with ice-cold methanol for 30 min at room temperature and then stained with 1% crystal violet. The number of colonies was detected and counted under a light microscope (Olympus, Tokyo, Japan). The percentage of the colony formation was measured by adjusting miR-NC to 100%.
Cell invasion and migration assays
Cell invasion and migration assays were performed using Transwell chambers (pore size, 8.0 μm; BD Biosciences, San Jose, CA, USA). For invasion assay, 4×104 transfected cells were suspended in 200 mL of serum-free medium and placed in the upper chamber, which was precoated with Matrigel (BD Biosciences). For migration assay, 2×104 transfected cells were suspended in 200 mL of serum-free medium and placed in the upper chamber. Six hundred microliters of medium with 10% FBS was added to the bottom chamber as a chemoattractant. After incubation at 37°C with 5% CO2 for 24 h, the cells remaining in the upper chamber were removed with a cotton swab. The cells on the bottom chambers were fixed in methanol and stained with 0.2% crystal violet (Sigma-Aldrich Co.). The number of invading or migrating cells was counted randomly in five fields of each membrane by a light microscope (Olympus).
Cell apoptosis
Cell apoptosis was determined with Annexin V-FITC Apoptosis Detection kit (BD Biosciences). Briefly, after transfecting with miR-488 mimics or NC for 24 h, 2×105 RCC cells were harvested and resuspended with 200 μL of binding buffer. Five microliters of Annexin V-FITC and 10 μL of propidium iodide were added and incubated for 15 min in the dark at room temperature. Then, the cells were resuspended with 500 μL binding buffer, and cell apoptosis was detected by using a flow cytometer (BD Biosciences).
Dual-luciferase reporter assay
Bioinformatics databases (TargetScan, PicTar, and miRanda) were used to predict the potential target gene of miR-488, and HMGN5 was selected for further analysis. Wild-type (WT) 3′-UTR region of HMGN5 containing the predicted target site of miR-488 and the mutant (MU) sites were amplified and cloned into the pmirGLO dual-luciferase miRNA expression vector (Promega Corporation, Fitchburg, WI, USA). Next, 5×104 RCC cells were plated in a 24-well plate and cotransfected with WT or MU 3′-UTR of HMGN reporter plasmid, and miR-488 mimics or miR-NC using Lipofectamine 2000 according to the manufacturer’s instruction. After transfection for 24 h, the luciferase activity was measured by a dual-luciferase reporter assay system (Promega Corporation). Renilla luciferase was used for normalization.
Xenograft mouse model
Male BALB/c nude mice (5 weeks old) were obtained from the Laboratory Animal Center of Jilin University (Changchun, China) and raised under special pathogen-free conditions with 12 h light/dark cycle. All animal experiments were approved by the Animal Care and Use Ethics Committee of Jilin University and were performed in accordance with the guidelines of the Animal Welfare Act of the National Institutes of Health (NIH Publications no 80-23), revised 2010. Animals were randomly divided into two groups (n=8 for each group), 786-O cells with miR-488 mimics or miR-NC transfection were suspended in serum-free DMEM medium (3×106 cells) and subcutaneously injected into the left flank of each mice. Xenograft growth was monitored every 7 days for 35 consecutive days after injection. Tumor volume was calculated as V=0.5×length×width2 by calipers. Thirty-five days after injection, mice were sacrificed, and tumor tissues were isolated and weighted. Then, part of the tumor tissues were immediately stored at −80°C for quantitative real-time polymerase chain reaction (qRT-PCR) and Western blot analysis.
Statistical analysis
Statistical analysis was performed using the Graphpad Prism 6.0 (GraphPad Software, La Jolla, CA, USA) and expressed as mean±SD. Student’s t-test was used to analyze the difference between two groups, and one-way analysis of variance followed by Newman–Keuls test was used to analyze difference among three or more groups. Spearman’s correlation test was used for analyzing the correlations between the miR-488 and HMNG5 expression levels in 35 RCC specimens. P<0.05 was considered to be statistically significant. All experiments were done at least three independent times.
Results
miR-488 was downregulated in RCC tissue samples and cell lines
qRT-PCR was performed to detect the miR-488 expression levels in RCC tissues and their paired normal tissues from 35 patients. The results showed that the expression levels of miR-488 were significantly reduced in RCC tissues compared with those in normal tissue samples (P<0.01, Figure 1A). In addition, the expression levels of miR-488 were also evaluated in RCC cell lines (ACHN, Caki-1, 786-O, 769-P, and A498) and the normal human proximal tubule epithelial cell line HK-2. We found that miR-488 expression in RCC cell lines was lower than that in HK-2 cells (P<0.05, Figure 1B). The difference varied among the five RCC cell lines, of which 786-O and A498 cell lines showed lower miR-488 expressions.
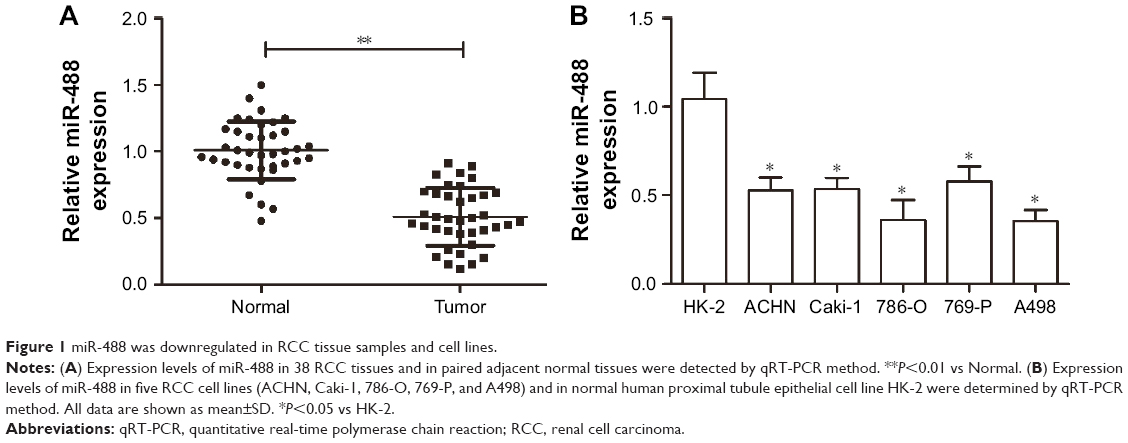 | Figure 1 miR-488 was downregulated in RCC tissue samples and cell lines. |
miR-488 inhibited cell proliferation and promoted apoptosis in RCC cells
To determine the influence of miR-488 on cell proliferation and apoptosis, 786-O and A498 cell lines were selected to undergo the overexpression experiments. The efficiencies of miR-488 mimics transfection were confirmed in 786-O and A498 cell lines by using qRT-PCR assay (P<0.01, respectively, Figure 2A). MTT and colony formation assays showed that overexpression of miR-488 remarkably suppressed cell viability and colony formation abilities in 786-O and A498 cell lines with miR-488 transfection when compared with those in NC groups (all P<0.01, Figure 2B and C). Furthermore, compared with NC groups, the percentage of apoptotic cells was significantly increased by miR-488 overexpression in miR-488 groups (P<0.01, Figure 2D). Thus, these data indicated that overexpression of miR-488 could inhibit cell proliferation and promote apoptosis in RCC cells.
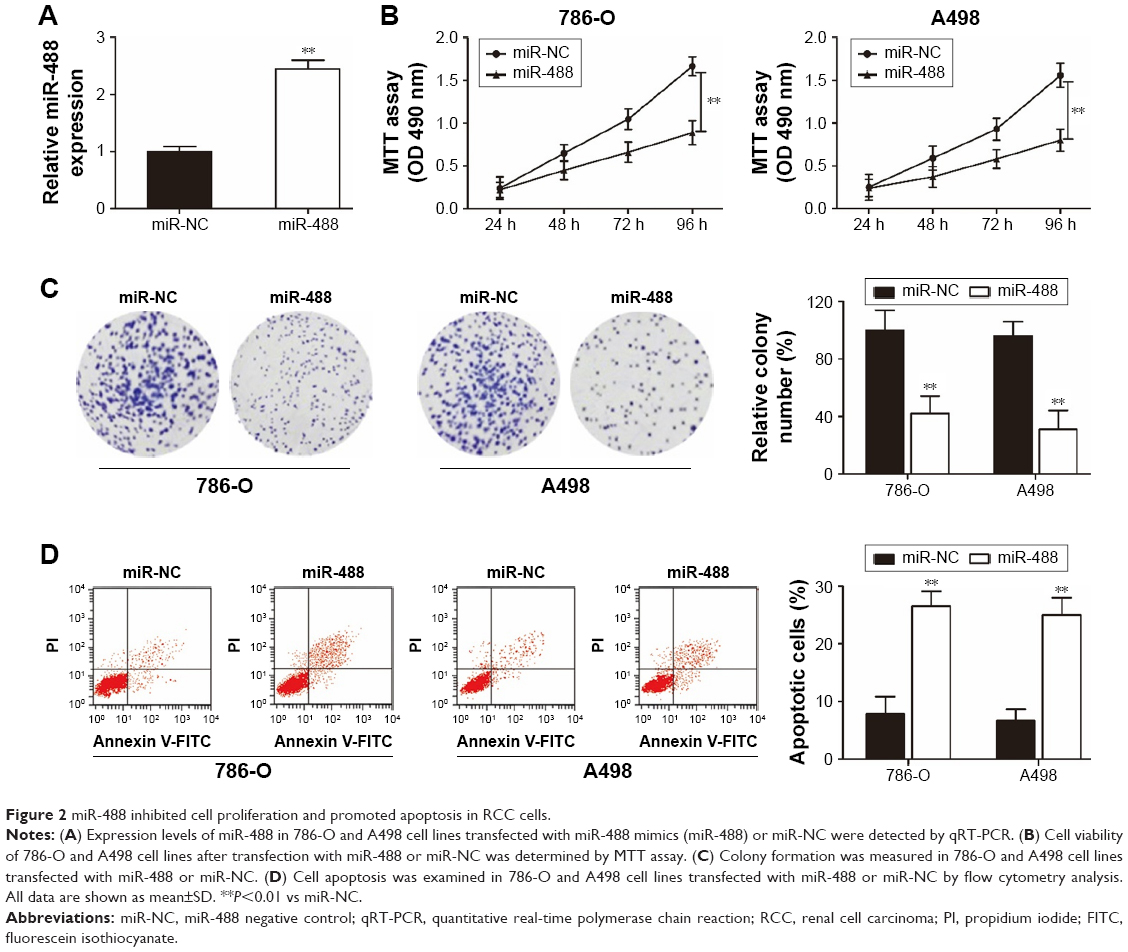 | Figure 2 miR-488 inhibited cell proliferation and promoted apoptosis in RCC cells. |
miR-488 suppressed cell invasion, migration, and EMT in RCC cells
Transwell assays were performed to investigate the effect of miR-488 on RCC cell invasion and migration. We found that overexpression of miR-488 apparently inhibited cell migratory and invasive abilities in 786-O and A498 cell lines with miR-488 transfection when compared with those in NC groups (all P<0.01, Figure 3A and B). Since epithelial–mesenchymal transition (EMT) plays a crucial role in the metastasis of various cancer cells, the expressions of EMT-related protein were examined by Western blot in this study. The results showed that, compared with NC groups, overexpression of miR-488 in 786-O and A498 cell lines markedly downregulated the expression levels of N-cadherin and vimentin, while upregulated the expression levels of E-cadherin (all P<0.05, Figure 3C), which lead to the inhibition of EMT.
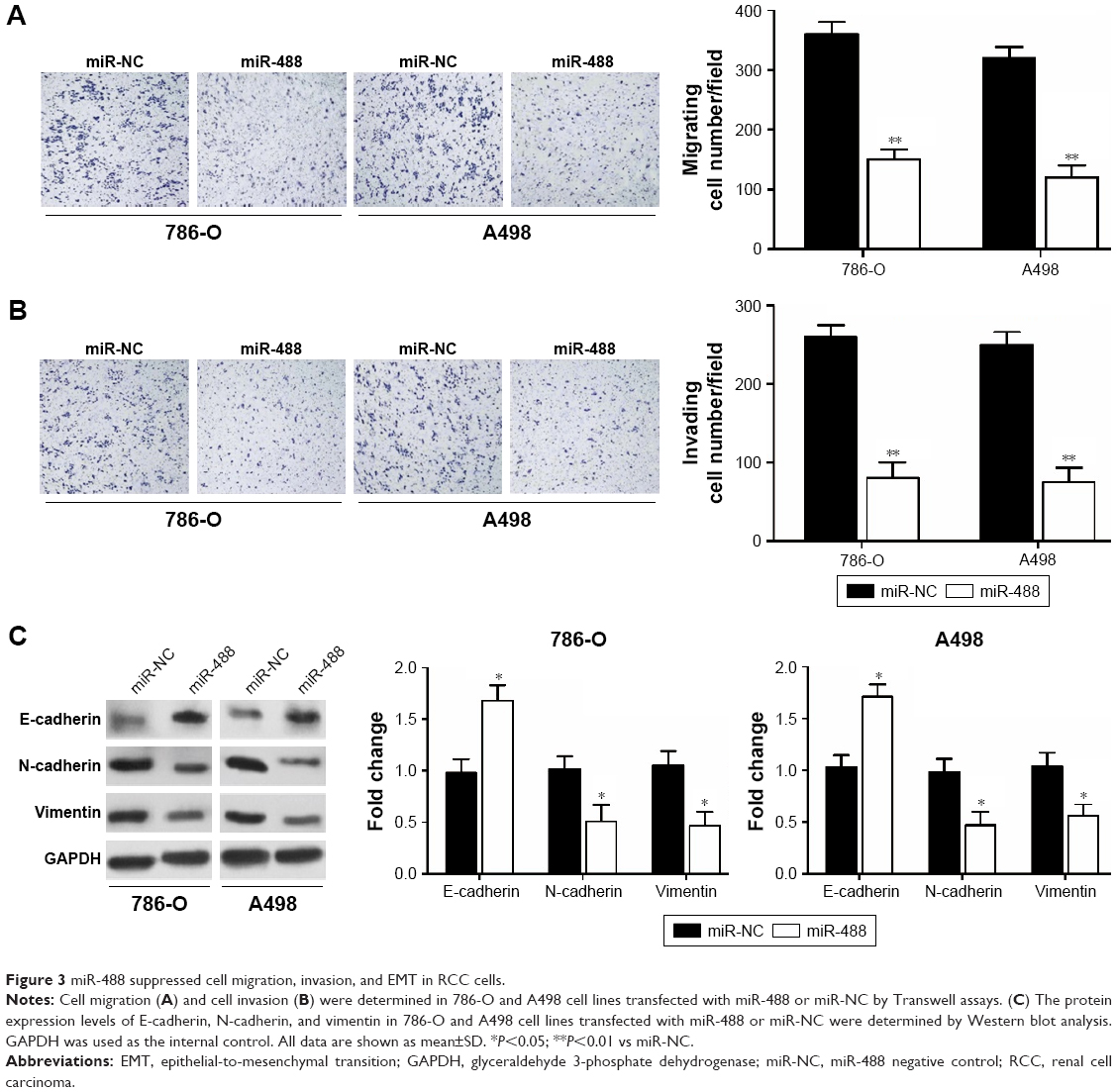 | Figure 3 miR-488 suppressed cell migration, invasion, and EMT in RCC cells. |
miR-488 directly targeted HMGN5 in RCC cells
To unravel the molecular mechanism by which miR-488 regulates RCC cells, bioinformatic databases (TargetScan, miRanda, and PicTar) were used to predict the potential miR-488 targets. The results showed that HMGN5 was a potential target of miR-488 (Figure 4A). To verify the potential interaction between HMGN5 and miR-488, WT 3′-UTR region containing the potential binding sites or MU of HMGN5 was cloned into the pmirGLO vectors. A dual-luciferase reporter assay revealed that the luciferase activity was significantly reduced in 786-O and A498 cells cotransfected with WT 3′-UTR HMGN5 and miR-488 mimics (P<0.01, respectively, Figure 4B), whereas no significant effect was observed in MU 3′-UTR HMGN5 transfection groups. qRT-PCR and Western blot further confirmed that overexpression of miR-488 dramatically inhibited HMGN5 expression in 786-O and A498 cells both at mRNA and protein levels (all P<0.05, Figure 4C). In addition, we also found that mRNA expression levels of HMGN5 in 35 RCC tissues were much higher than those in paired adjacent normal tissues (P<0.01, Figure 4D). Spearman’s rank correlation analysis showed that there was an inverse correlation between the HMGN5 mRNA expression and miR-488 expression in RCC tissues (P<0.001, r2=0.228; Figure 4E). Therefore, these findings demonstrated that HMGN5 was a direct target of miR-488 in RCC.
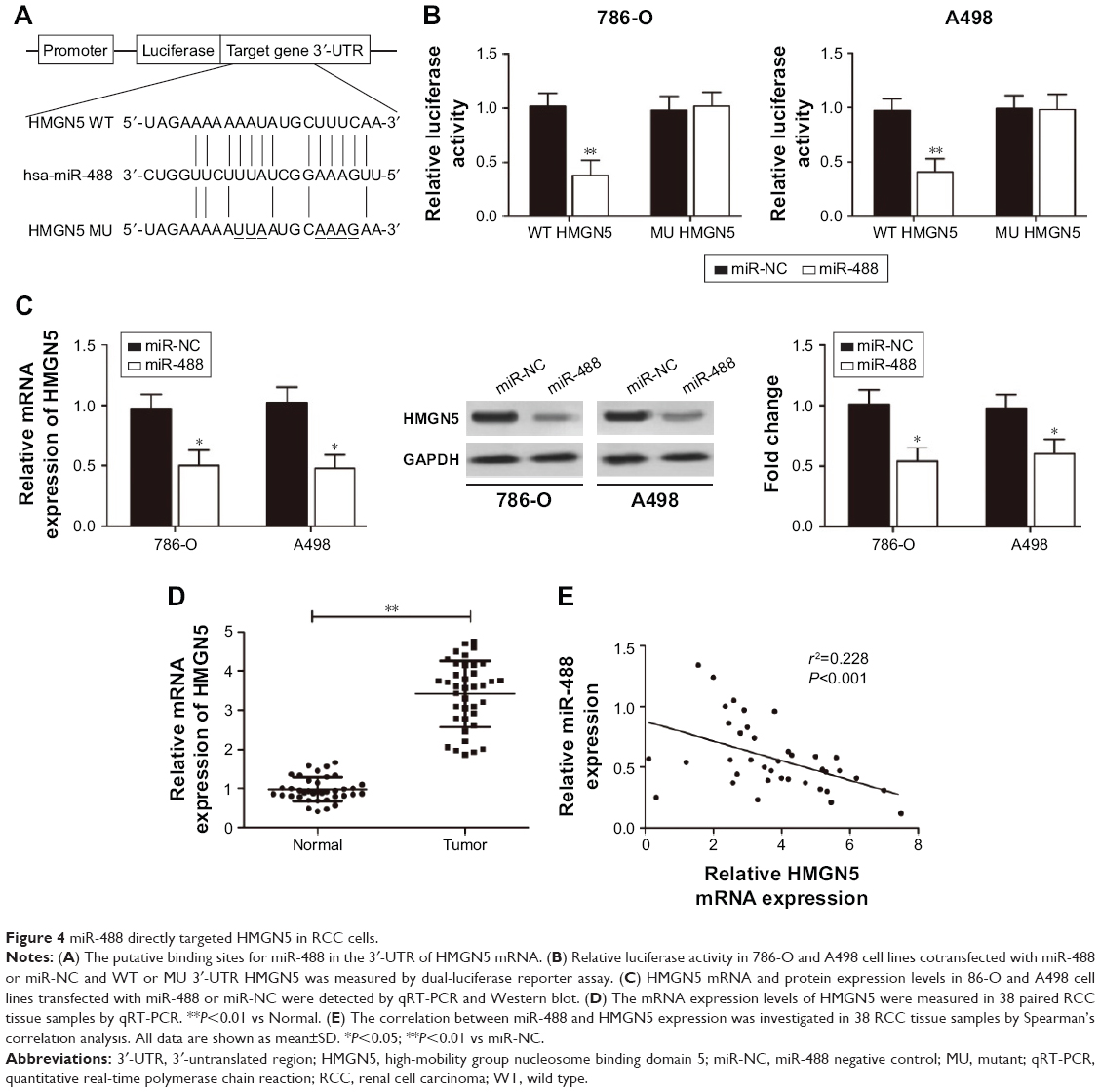 | Figure 4 miR-488 directly targeted HMGN5 in RCC cells. |
Restoration of HMGN5 expression rescued the miR-488-mediated inhibitory effect on RCC cells
To determine whether HMGN5 was a candidate target to regulate the biological functions of miR-488 in RCC cells, HMGN5 overexpressing vector was constructed and transfected into 786-O and A498 cells. qRT-PCR and Western blot assays showed that forced expression levels of HMGN5 significantly rescued the decreased mRNA and protein levels of HMGN5 induced by miR-488 in 786-O and A498 cells (all P<0.05, Figure 5A). Furthermore, restoration of HMGN5 expression also partially reversed the suppressive effect of miR-488 on cell viability, colony formation, invasion, migration, and EMT in 786-O and A498 cells (all P<0.05, Figure 5B–F). Taken together, these data indicated that miR-488 exerted its inhibitory effects in RCC cells through repressing HMGN5.
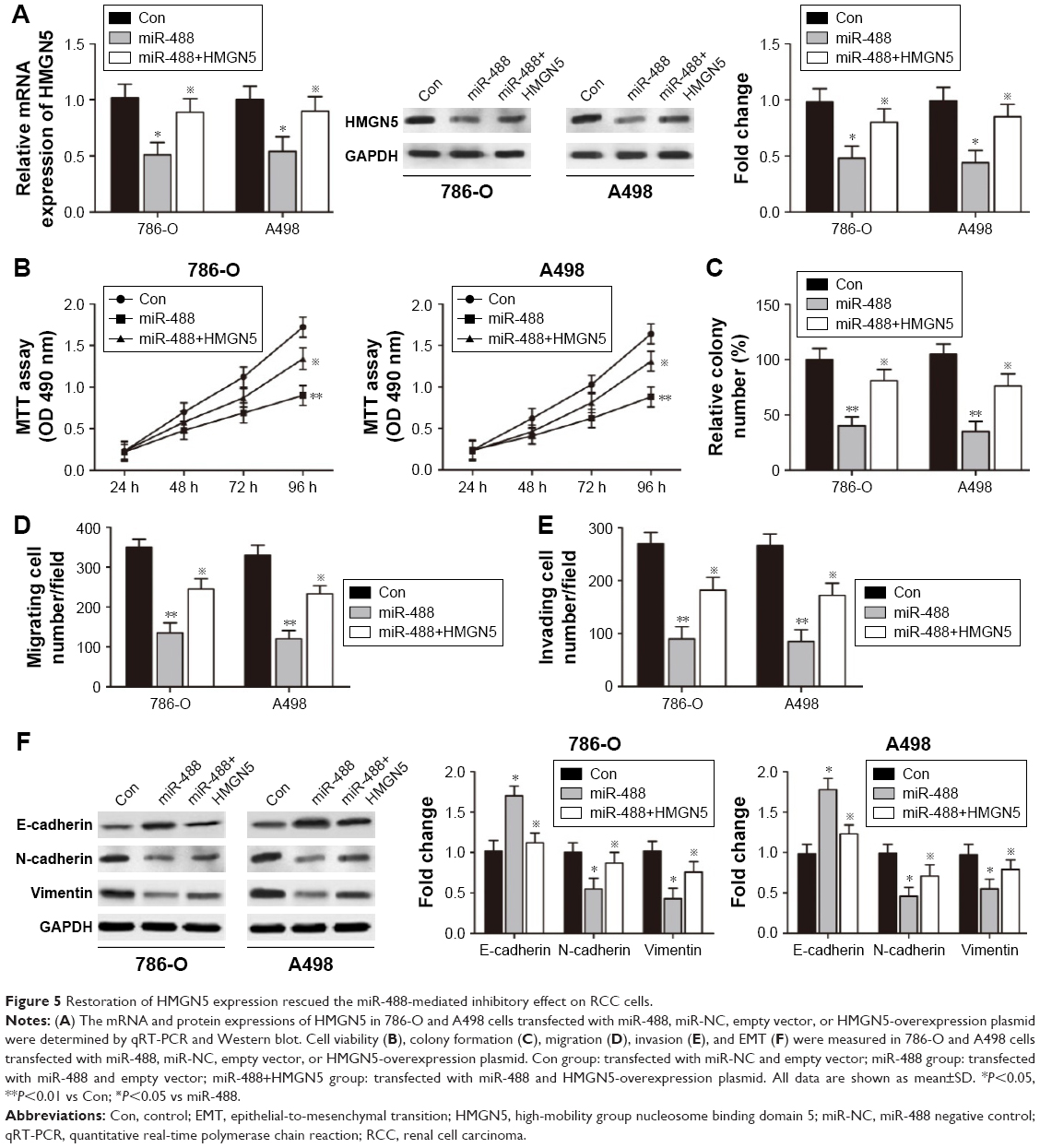 | Figure 5 Restoration of HMGN5 expression rescued the miR-488-mediated inhibitory effect on RCC cells. |
 P<0.05 vs miR-488.
P<0.05 vs miR-488.miR-488 suppressed RCC growth in vivo by inhibiting HMGN5
To investigate the role of miR-488 in RCC tumorigenicity in vivo, xenograft mouse model was established by injecting 786-O cells with miR-488 mimics or miR-NC transfection. The results showed that miR-488 overexpression remarkably inhibited tumor growth in vivo (P<0.01, Figure 6A). The size and weight of tumors isolated from miR-488 mice were much lower than those of miR-NC group (P<0.01, Figure 6B and C). In addition, the expression levels of miR-488 and HMGN5 were also determined in the isolated tumors. As shown in Figure 6D, when compared with miR-NC group, miR-488 expression level was significantly enhanced in miR-488 group (P<0.01, Figure 6D), while HMGN5 mRNA and protein levels were significantly inhibited in miR-488 group (P<0.05, Figure 6E and F). Furthermore, Western blot assay also revealed that the protein levels of N-cadherin, vimentin, p-AKT, p-mTOR, and P13K were obviously reduced, while that of E-cadherin was significantly increased compared with those in miR-NC group (all P<0.05, Figure 6F), but not in the AKT and mTOR expressions. These findings suggested that miR-488/HMGN5 axis inhibited RCC tumorigenicity in vivo by regulating P13K/AKT/mTOR signaling pathway.
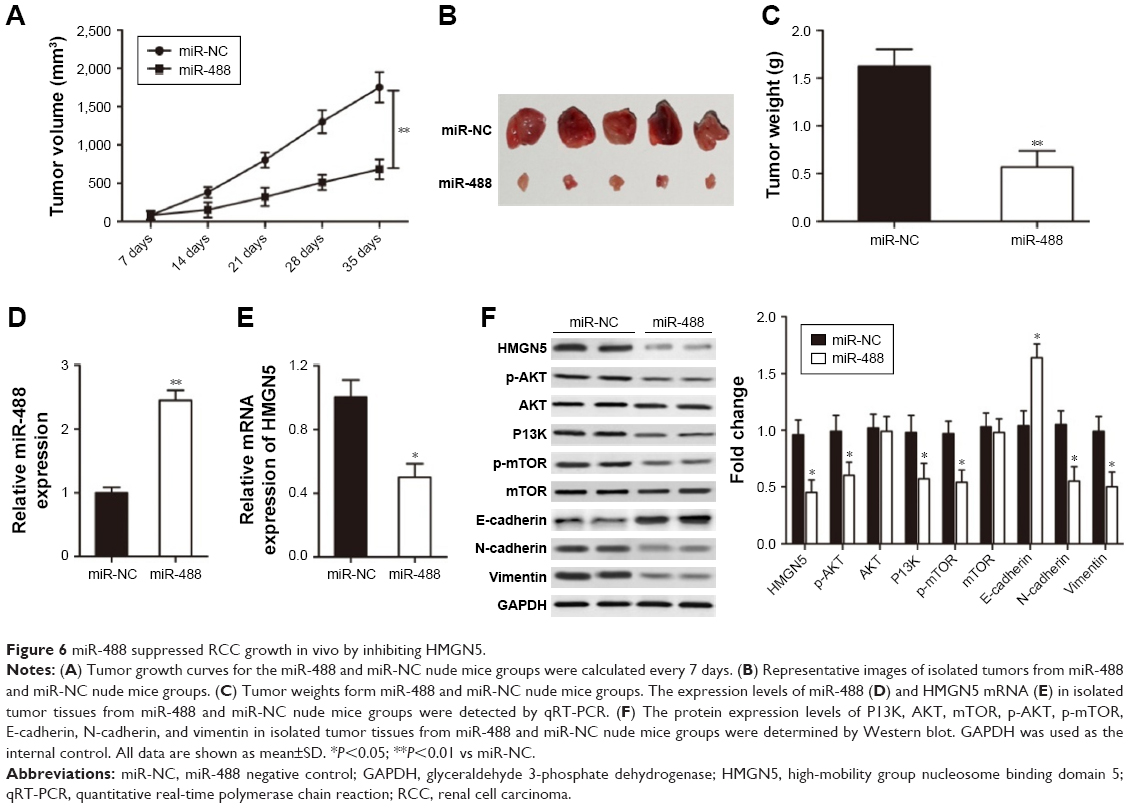 | Figure 6 miR-488 suppressed RCC growth in vivo by inhibiting HMGN5. |
Discussion
RCC is the most lethal type of urologic cancer and accounts for ~90% of all renal malignancies worldwide.22 Metastatic RCC resulted in the death of nearly 40% patients every year, which makes it the tough therapeutic challenge to overcome.23 Recently, improved understanding of molecular mechanism of RCC has received new prospects and more specific focus on the treatment of this disease.24 Targeted therapies that aim to block molecular pathways have showed significant antitumor growth and survival outcomes, which obviously ameliorate the poor prognosis of advanced RCC patients.25 So far, three kinds of targeted therapies including multitargeted tyrosine kinase inhibitor, the mTOR complex 1 kinase inhibitor, and the humanized anti-vascular endothelial growth factor monoclonal antibody have been developed and widely used in clinics.26 However, increasing evidence demonstrates that resistance to targeted therapeutic strategy has emerged in multiple tumors, which results from the secondary mutation of the target protein or pathway that avoids the target site suppression.27
Recently, it has been well identified that a variety of miRNAs could act either as an oncogenic or tumor suppressor in the development and progression of RCC, indicating that investigation and measurement of these kinds of small nucleotides might provide available insight into the diagnostic and therapeutic improvement of RCC.28,29 For example, Cui et al have suggested that miR-99a functions as a tumor suppressor by inducing G1 phase cell cycle arrest and inhibiting tumorigenicity in RCC.30 Li et al have showed that miR-21 acts as an oncogene in RCC by promoting cancer cell hyperplasia and contributes to tumor cell transformation and metastasis.31 Machackova et al have proved the oncogene role of miR-429 on the metastasis and poor overall survival of RCC patients.32 To our knowledge, this is the first study that demonstrated that miR-488 played a tumor suppressor role in RCC by inhibiting RCC growth both in vitro and in vivo by targeting HMGN5.
The dysregulation of miR-488 has been reported to be involved in the tumorigenesis and progression in several human tumors.14 In the current study, we observed that the expression levels of miR-488 were significantly downregulated in clinic RCC tissue samples and cell lines when compared with adjacent normal tissues and cell line. Overexpression of miR-488 in RCC cells suppressed cell proliferation due to the reduced cell viability and colony formation capacity, as well as promoted apoptosis in vitro. Furthermore, xenografted tumor experiment also confirmed the inhibitory role of miR-488 on RCC tumorigenesis in vivo. In addition, forced expression of miR-488 was identified to contribute to the inhibition of invasive and migratory abilities of RCC cells by abrogating EMT (as indicated by upregulation of E-cadherin and downregulation of N-cadherin and vimentin). Accordingly, these data suggested that miR-488 might function as a tumor suppressor in the development and progression of RCC.
Since miRNAs exert their biological functions through binding to the 3′-UTR of their target genes, the underlying mechanism by which miR-488 regulated RCC progression was further explored in this study. The bioinformatics databases (TargetScan, PicTar, and miRanda) were used to help find the putative targets for miR-488. Among these theoretical targets, HMGN5 was selected as one of the possible target gene due to its oncogenic characteristics. HMGN5 has been demonstrated to be overexpressed in a variety of human cancers and acted as an oncogene in carcinogenesis.33 For instance, Gan et al showed that knockdown of HMGN5 inhibited the growth and invasion of 5,637 human urothelial bladder cancer cells both in vitro and in vivo.34 Weng et al found that HMGN5 was upregulated in breast cancer and played an oncogenic role by promoting cell proliferation and invasion, and activating apoptosis.35 Ji et al also revealed that knockdown of HMNG1 suppressed the proliferation and invasion of clear cell RCC cells in vitro and in vivo.21 Furthermore, recent researchers have reported that HMGN5 is a direct target of several miRNAs that contribute to cell proliferation, invasion, and apoptosis during various cancer progressions. Yao et al showed that miR-186 suppressed the growth and metastasis of bladder cancer by targeting HMGN5.36 Wei et al illustrated that miR-340 inhibited tumorigenic potential of prostate cancer cells by targeting HMGN5.37 Li et al suggested that suppression of HMGN5 by miR-326 impeded cell proliferation and invasion in non-small-cell lung cancer cells.38 Meng et al elucidated that miR-140-5p promoted autophagy and regulated chemoresistance mediated by HMGN5 in osteosarcoma.20
Accordingly, it prompted us to demonstrate that miR-488 regulates RCC progression may be partially via targeting HMGN5. In the current study, our results confirmed that miR-488 directly binds to the 3′-UTR of HMGN5, as its overexpression was correlated with the inhibition of luciferase activity in dual-luciferase reporter system driven by the 3′-UTR of HMGN5. qRT-PCR and Western blot further verified that overexpression of miR-488 resulted in reduced expressions of HMGN5 at both mRNA and protein levels in RCC cells. Furthermore, Spearman’s correlation analysis identified the negative association between miR-488 and HMGN5 in RCC tissue samples. Importantly, rescue experiments and functional assays demonstrated that restoration of HMGN5 partially abolished the inhibitory effect of miR-488 on proliferation and metastasis of RCC cells. Taken together, these findings implicated that posttranscriptional modulation of HMGN5 by miR-488 might be a pivotal molecular mechanism underlying RCC cancer progression.
The downstream molecular signalings involved in the regulation of miR-488/HMGN5 axis on RCC development still remains largely unknown. Accumulating evidence has demonstrated that P13K/AKT/mTOR signaling pathway is closely associated with the tumorigenesis in multiple malignancies.39 The activation of P13K/AKT/mTOR pathway could stimulate various oncogenic systems including EMT and metastasis by transmitting signals from the cell membrane into the nucleus.40 During EMT program, epithelial cells obtain migratory and invasive features through transferring epithelial characteristics into mesenchymal phenotype.41 A recent study has reported that HMGN5 promotes tumor proliferation and invasion in osteosarcoma through the P13K/AKT signaling pathway.42 Consistently, our study also demonstrated that overexpression of miR-488 inhibited the proliferative and metastatic abilities of RCC cells in vivo through suppressing the activation of P13K/AKT/mTOR pathway and EMT, indicating that P13K/AKT/mTOR pathway might be involved in the regulation of miR-488/HMGN5 axis during tumor proliferation and metastasis in RCC progression.
In conclusion, this study implicated the downregulation of miR-488 expression in human RCC tissues and cell lines, and that forced expression of miR-488 could remarkably inhibit cell proliferation, invasion, migration, EMT, and promote apoptosis in vitro and in vivo by targeting HMGN5. Furthermore, miR-488 suppressed the activation of P13K/AKT/mTOR signaling pathway in RCC cells. Therefore, these data suggested that miR-488 functioned as a novel tumor suppressor that regulated RCC progression through P13K/AKT/mTOR signaling pathway by targeting HMGN5, which might provide valuable evidence for therapeutic target development for RCC.
Disclosure
The authors report no conflicts of interest in this work.
References
Garcia JA, Cowey CL, Godley PA. Renal cell carcinoma. Curr Opin Oncol. 2009;21(3):266–271. | ||
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. | ||
Cohen HT, McGovern FJ. Renal-cell carcinoma. N Engl J Med. 2005;353(23):2477–2490. | ||
Rini BI. Metastatic renal cell carcinoma: many treatment options, one patient. J Clin Oncol. 2009;27(19):3225–3234. | ||
Lam JS, Shvarts O, Leppert JT, Figlin RA, Belldegrun AS. Renal cell carcinoma 2005: new frontiers in staging, prognostication and targeted molecular therapy. J Urol. 2005;173(6):1853–1862. | ||
Rodriguez-Vida A, Hutson TE, Bellmunt J, Strijbos MH. New treatment options for metastatic renal cell carcinoma. ESMO Open. 2017;2(2):e000185. | ||
Patil S, Manola J, Elson P, et al. Improvement in overall survival of patients with advanced renal cell carcinoma: prognostic factor trend analysis from an international data set of clinical trials. J Urol. 2012;188(6):2095–2100. | ||
Zhang Y, Wang Z, Gemeinhart RA. Progress in microRNA delivery. J Control Release. 2013;172(3):962–974. | ||
Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34(Database issue):D140–D144. | ||
Huang Y, Shen XJ, Zou Q, Wang SP, Tang SM, Zhang GZ. Biological functions of microRNAs: a review. J Physiol Biochem. 2011;67(1):129–139. | ||
Li M, Wang Y, Song Y, et al. MicroRNAs in renal cell carcinoma: a systematic review of clinical implications (Review). Oncol Rep. 2015;33(4):1571–1578. | ||
Song J, Kim D, Jin EJ. MicroRNA-488 suppresses cell migration through modulation of the focal adhesion activity during chondrogenic differentiation of chick limb mesenchymal cells. Cell Biol Int. 2011;35(2):179–185. | ||
Muinos-Gimeno M, Espinosa-Parrilla Y, Guidi M, et al. Human microRNAs miR-22, miR-138-2, miR-148a, and miR-488 are associated with panic disorder and regulate several anxiety candidate genes and related pathways. Biol Psychiatry. 2011;69(6):526–533. | ||
Fang C, Chen YX, Wu NY, et al. MiR-488 inhibits proliferation and cisplatin sensibility in non-small-cell lung cancer (NSCLC) cells by activating the eIF3a-mediated NER signaling pathway. Sci Rep. 2017;7(1):40384. | ||
Lv Y, Shi Y, Han Q, Dai G. Histone demethylase PHF8 accelerates the progression of colorectal cancer and can be regulated by miR-488 in vitro. Mol Med Rep. 2017;16(4):4437–4444. | ||
Zhao Y, Lu G, Ke X, et al. miR-488 acts as a tumor suppressor gene in gastric cancer. Tumour Biol. 2017;37(7):8691–8698. | ||
King LM, Francomano CA. Characterization of a human gene encoding nucleosomal binding protein NSBP1. Genomics. 2001;71(2):163–173. | ||
Rochman M, Malicet C, Bustin M. HMGN5/NSBP1: a new member of the HMGN protein family that affects chromatin structure and function. Biochim Biophys Acta. 2010;1799(1–2):86–92. | ||
Postnikov Y, Bustin M. Regulation of chromatin structure and function by HMGN proteins. Biochim Biophys Acta. 2010;1799(1–2):62–68. | ||
Meng Y, Gao R, Ma J, et al. MicroRNA-140-5p regulates osteosarcoma chemoresistance by targeting HMGN5 and autophagy. Sci Rep. 2017;7(1):416. | ||
Ji SQ, Yao L, Zhang XY, Li XS, Zhou LQ. Knockdown of the nucleosome binding protein 1 inhibits the growth and invasion of clear cell renal cell carcinoma cells in vitro and in vivo. J Exp Clin Cancer Res. 2012;31(1):22. | ||
Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socioeconomic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev. 2008;34(3):193–205. | ||
Flanigan RC, Campbell SC, Clark JI, Picken MM. Metastatic renal cell carcinoma. Curr Treat Options Oncol. 2003;4(5):385–390. | ||
Pal SK, Nelson RA, Vogelzang N. Disease-specific survival in de novo metastatic renal cell carcinoma in the cytokine and targeted therapy era. PLoS One. 2013;8(5):e63341. | ||
Su D, Stamatakis L, Singer EA, Srinivasan R. Renal cell carcinoma: molecular biology and targeted therapy. Curr Opin Oncol. 2014;26(3):321–327. | ||
Pracht M, Berthold D. Successes and limitations of targeted therapies in renal cell carcinoma. Prog Tumor Res. 2014;41(1):98–112. | ||
Abe H, Kamai T. Recent advances in the treatment of metastatic renal cell carcinoma. Int J Urol. 2013;20(10):944–955. | ||
Gu L, Li H, Chen L, et al. MicroRNAs as prognostic molecular signatures in renal cell carcinoma: a systematic review and meta-analysis. Oncotarget. 2015;6(32):32545–32560. | ||
Kurozumi A, Goto Y, Okato A, Ichikawa T, Seki N. Aberrantly expressed microRNAs in bladder cancer and renal cell carcinoma. J Hum Genet. 2017;62(1):49–56. | ||
Cui L, Zhou H, Zhao H, et al. MicroRNA-99a induces G1-phase cell cycle arrest and suppresses tumorigenicity in renal cell carcinoma. BMC Cancer. 2012;12(1):546. | ||
Li X, Xin S, He Z, et al. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor PDCD4 and promotes cell transformation, proliferation, and metastasis in renal cell carcinoma. Cell Physiol Biochem. 2014;33(6):1631–1642. | ||
Machackova T, Mlcochova H, Stanik M, et al. MiR-429 is linked to metastasis and poor prognosis in renal cell carcinoma by affecting epithelial-mesenchymal transition. Tumour Biol. 2016;37(11):14653–14658. | ||
Shi Z, Tang R, Wu D, Sun X. Research advances in HMGN5 and cancer. Tumour Biol. 2016;37(2):1531–1539. | ||
Gan Y, Tan J, Yang J, et al. Knockdown of HMGN5 suppresses the viability and invasion of human urothelial bladder cancer 5637 cells in vitro and in vivo. Med Oncol. 2015;32(4):136. | ||
Weng M, Song F, Chen J, et al. The high-mobility group nucleosome-binding domain 5 is highly expressed in breast cancer and promotes the proliferation and invasion of breast cancer cells. Tumour Biol. 2015;36(2):959–966. | ||
Yao K, He L, Gan Y, Zeng Q, Dai Y, Tan J. MiR-186 suppresses the growth and metastasis of bladder cancer by targeting NSBP1. Diagn Pathol. 2015;10(1):146. | ||
Wei P, Qiao B, Li Q, et al. microRNA-340 suppresses tumorigenic potential of prostate cancer cells by targeting high-mobility group nucleosome-binding domain 5. DNA Cell Biol. 2016;35(1):33–43. | ||
Li D, Du X, Liu A, Li P. Suppression of nucleosome-binding protein 1 by miR-326 impedes cell proliferation and invasion in non-small cell lung cancer cells. Oncol Rep. 2016;35(2):1117–1124. | ||
Bjelogrlic SK, Srdic T, Radulovic S. Mammalian target of rapamycin is a promising target for novel therapeutic strategy against cancer. J BUON. 2006;11(3):267–276. | ||
Gu J, Liang Y, Qiao L, et al. URI expression in cervical cancer cells is associated with higher invasion capacity and resistance to cisplatin. Am J Cancer Res. 2015;5(4):1353–1367. | ||
Ding XM. MicroRNAs: regulators of cancer metastasis and epithelial-mesenchymal transition (EMT). Chin J Cancer. 2014;33(3):140–147. | ||
Zhou X, Yuan B, Yuan W, Wang C, Gao R, Wang J. The expression and clinical significance of high mobility group nucleosome binding domain 5 in human osteosarcoma. Tumour Biol. 2014;35(7):6539–6547. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.