")
Back to Journals » OncoTargets and Therapy » Volume 13
miR-34c Targets MET to Improve the Anti-Tumor Effect of Cisplatin on Ovarian Cancer
Received 20 November 2019
Accepted for publication 3 March 2020
Published 5 April 2020 Volume 2020:13 Pages 2887—2897
DOI https://doi.org/10.2147/OTT.S239425
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjay Singh
Shiying Yang,1 Zhen Li,2 Rui Luo3
1Department of Gynecology and Obstetrics, Rizhao People’s Hospital, Rizhao City 276800, Shandong Province, People’s Republic of China; 2Reproductive Medicine Center, Qingdao Women and Children Hospital, Qingdao City 266011, Shandong Province, People’s Republic of China; 3Department of Gynecology, Linyi People’s Hospital, Linyi City 276000, Shandong Province, People’s Republic of China
Correspondence: Rui Luo Email [email protected]
Background: Cisplatin is a commonly used drug for the treatment of various types of malignant cancers, including ovarian cancer. However, resistance to cisplatin is still a considerable obstacle to achieve a satisfactory therapeutic effect. The purpose of this study is to develop a strategy to sensitize ovarian cancer cells to cisplatin-induced cytotoxicity.
Methods: miR-34c levels in ovarian cancer tissues and cell lines were tested by qRT-PCR analysis. In vitro assays, the effect of miR-34c on cisplatin was evaluated by using MTT. Expression of MET and phosphorylation of PI3K and AKT were tested by Western blot assays. Conjugation with Bad and Bcl-xl was evaluated through immunoprecipitation. Flow cytometry analysis was performed to measure the apoptotic rate of ovarian cancer cells.
Results: Downregulation of miR-34c was observed in ovarian cancer tissues and cell lines. However, miR-34c overexpression was found to sensitize ovarian cancer cells to cisplatin treatment in vitro and in vivo. Mechanically, we found that miR-34c targeted the MET gene, thereby inhibiting the phosphorylation of PI3K and AKT to activate Bad. As a result, miR-34c reduced resistance of ovarian cancer cells to cisplatin-induced apoptosis.
Conclusion: miR-34c/MET axis promotes cisplatin-induced cytotoxicity against ovarian cancer by targeting the MET/PI3K/AKT/Bad pathway.
Keywords: ovarian cancer, miR-34c, cisplatin, MET, Bad
Introduction
Ovarian cancer is a common malignant disease in women. Due to the rapid development of ovarian cancer, it is the leading cause of death in gynecologic cancers. Moreover, the 5-year survival rate of ovarian cancer patients is very low.1,2 Despite the development of medical technology, surgery and chemotherapy are still the main approaches for prolonging the life of patients with ovarian cancer.3,4 However, ovarian cancer cells usually exhibit resistance to chemotherapy and it is necessary to develop strategies to increase their sensitivity to chemotherapeutic drugs.5,6
Cisplatin is a platinum-based cytotoxic agent that is commonly used for the treatment of various types of malignant cancers including ovarian cancer. As a cytotoxic agent, cisplatin cross-links with DNA and subsequently inhibits DNA replication and transcription and thus induces apoptosis in cancer cells.7–10 However, large doses and the long-term administration of cisplatin usually induce serious side effects in cancer patients, including nephrotoxicity, gastrointestinal reactions, and bone marrow suppression.11,12 Furthermore, resistance to cisplatin is still a considerable obstacle to achieve a satisfactory therapeutic effect in patients with ovarian cancer.13,14
MicroRNAs (miRNAs) are a group of endogenous and non-coding RNAs with 18–25 nucleotides in length. They act as negative regulators of gene expression via RNA binding at the 3′ untranslated region (UTR) of their target mRNAs.15,16 The expression profile of miRNAs has been found to regulate cell growth, differentiation, apoptosis and tumorigenesis. Thus, cancer cells usually exhibit aberrant miRNA expression patterns.17–19 Recently, studies have demonstrated that miRNA dysregulation is responsible for the development of chemoresistance in multiple cancers including ovarian cancer. Correcting aberrant miRNA expression may represent a potential strategy to increase the chemosensitivity of cancer cells.13,20,21 In the present study, we found that the expression of miR-34c was decreased in ovarian cancer cells. Furthermore, miR-34c overexpression promoted cisplatin-induced cytotoxicity in ovarian cancer cells by targeting the MET/PI3K/AKT/Bad pathway.
Materials and Methods
Cell Lines and Patient Specimens
The human normal ovarian surface epithelial cell line HOSEpiC and human ovarian cancer cell lines SKOV3 and A2780 were purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). Cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco, Carlsbad, CA) containing 10% fetal bovine serum (Gibco) at 37°C in an incubator with an atmosphere of 5% CO2. Human ovarian cancer tissues and their corresponding paracancerous tissues (tissues 2 cm away from the focus) were obtained from 25 patients with primary ovarian cancer and collected at the Linyi people’s Hospital between June 2017 and December 2018. We have obtained the written informed consent from all of the patients. The experimental protocols were approved by the ethics committee of the Linyi people’s Hospital.
Detection of miR-34c Expression
miR-34c expression in ovarian cancer cell lines and tissues was detected by quantitative real-time reverse transcription PCR (qRT-PCR) analysis. Briefly, total RNA was extracted from ovarian cancer cell lines and tissues by using the TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA). Subsequently, the extracted RNA was reversely transcribed by a One Step PrimeScript miRNA cDNA Synthesis Kit (TaKaRa Bio, Inc., Otsu, Japan) according to the manufacturer’s instructions. miR-34c expression was quantified using SYBR Premix Ex Taq (TaKaRa) on an ABI PRISM 7900 Sequence Detection System (Applied Biosystems, Foster City, CA) and normalized to U6 small nuclear RNA.
Transient Transfection
Negative control oligonucleotide (NCO, 5′-AGAUGCUUAUAGCGUGUGCGUAG-3′) and miR-34c mimics (5′-AGGCAGUGUAGUUAGCUGAUUGC-3′) were purchased from GenePharma Co., Ltd. (Shanghai, China). MET small interfering RNA (siRNA) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA; sc-29397). A MET eukaryotic expression plasmid was generated by cloning the open reading frame of the MET gene into the pcDNA3.1 plasmid (Life Technologies, Carlsbad, CA). RNA oligonucleotides (50 pmol/mL) or plasmids (2 μg/mL) were transient transfected into ovarian cancer cells by using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions, followed by incubation for 24 h.
Cell Viability Assay
Transfected ovarian cancer cells were seeded into 96-well plates overnight followed by incubation with different concentrations of cisplatin for 48 h. Subsequently, 20 μL MTT reagent (Sigma-Aldrich, St. Louis, MO) (5 mg/mL) was added to the culture medium and the cells were incubated for 4 h at 37°C in a humidified incubator with 5% CO2. The absorbance value of each well was determined at 570 nm by using a microplate reader (Sunrise Microplate Reader, Tecan, Mannedorf, Switzerland). Relative cell viability in the experimental groups was normalized to the NCO group.
Luciferase Reporter Assay
The 3′ UTR of MET containing a putative miR-34c binding site was cloned downstream of the firefly luciferase gene in the pGL3 Luciferase Reporter Vector (Promega, Madison, WI). A mutant plasmid was created by mutating the seed region of the miR-34c binding site (CACUGCCU) by using a site-directed mutagenesis kit (TaKaRa). For the luciferase reporter assay, the cells were seeded in a 48-well plate followed by co-transfection with miR-34c (or NCO, 50 pmol/mL), recombinant pGL3 plasmid (2 μg/mL), and Renilla luciferase pRL-TK vector (100 ng/mL; Promega) for 24 h. Luciferase activity was measured by the Dual-Luciferase Reporter System (Promega) according to the manufacturer’s instruction. Relative luciferase activity in the miR-34c groups was expressed as the firefly/Renilla luciferase ratio and normalized to the NCO groups.
Western Blot Analysis
Ovarian cells were lysed using RIPA buffer on ice for 30 min followed by collection of the supernatants. For the detection of cytochrome c, we collected the cytoplasmic fraction of cancer cells in the absence of mitochondria by using a Mitochondria/Cytosol Fraction Kit (BioVision, Milpitas, CA). The protein samples were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel and transferred to a polyvinylidene fluoride membrane (Millipore, Bedford, MA). Subsequently, the membrane was blocked in 5% skim milk for 1 h at room temperature and incubated with the following primary antibodies (Cell Signaling Technology, Danvers, MA): anti-MET, anti-GAPDH, anti-PI3K, anti-AKT, anti-Bad, anti-phosphorylated (p)-PI3K, AKT, and Bad, anti-Bcl-xl, anti-cytochrome c, and anti-caspase-9 and −3. The membrane was washed and incubated with a horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology) for 2 h at room temperature. Proteins on the membrane were visualized by using an enhanced chemiluminescence detection kit (Pierce, Rockford, IL).
Co-Immunoprecipitation
Ovarian cells were lysed by using NP-40 buffer (Beyotime Biotechnology, Shanghai, China) on ice for 30 min, followed by collection of the supernatants. Subsequently, a primary antibody against Bad was added to the supernatants and incubated overnight at 4°C. Protein A agarose beads (Cell Signaling Technology) were added to the supernatants and incubated for 2 h. The proteins binding to the agarose beads were separated by boiling in SDS sample buffer and detected by Western blot analysis.
Detection of Mitochondrial Membrane Potential and Cell Apoptosis
For the detection of mitochondrial membrane potential (MMP), the cells were harvested through centrifugation at 2000 r.p.m for 5min. Subsequently, cells were washed with PBS for twice and then stained with 5 μM 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl imidacarbo cyanine iodide (JC-1; Molecular Probes; Thermo Fisher Scientific, Inc.) for 20 min at 37°C. Cells emitting red fluorescence were considered as cells with high MMP. For the detection of apoptosis, the cells were harvested through centrifugation at 2000 r.p.m for 5min. Subsequently, cells were washed with PBS for twice and then stained with Annexin V/propidium iodide by using an Annexin V-FITC Apoptosis Detection Kit (Sigma-Aldrich) according to the manufacturer’s instruction. Annexin V-positive cells were considered as apoptotic cells and necrotic cells. MMP and cell apoptosis were analyzed by using flow cytometry (Becton Dickinson, Franklin Lakes, NJ).
In vivo Tumorigenicity
The precursor sequence of miR-34c (5′-AGUCUAGUUACUAGGCAGUGUAGUUAGCUGAUUGCUAAUAGUACCAAUCACUAACCACACGGCCAGGUAAAAAGAUU-3′) was inserted into a lentivirus by GeneChem Co., Ltd. (Shanghai, China). Subsequently, 5.0 × 105 transducing units of recombinant (miR-34c-LV) or empty lentivirus (empty LV) were then transfected into SKOV3 cells. To establish the miR-34c-overexpressing ovarian cancer model in mice, 5.0 × 106 transfected SKOV3 cells were inoculated subcutaneously into nude mice (BALB/c, nu/nu; 17–22 g in weight; Shanghai Super-B&K Laboratory Animal Corp., Ltd., Shanghai, China). For in vivo treatment, the mice were treated with cisplatin i.p. twice a week (2 mg/kg) after the xenografts reached 0.5 cm in diameter. At 28 days post-inoculation, the mice were sacrificed by neck dislocation. The animal experiments were conducted in strict accordance with the guidelines of the Experimental Animal Care and approved by the Ethics Committee of Linyi people’s Hospital.
Statistical Analysis
Experimental data were obtained from at least three independent experiments and are represented as the mean ± standard deviation. Differences between two groups were analyzed by two-tailed Student’s t-tests. In addition, one-way analysis of variance and Bonferroni’s post hoc test were used to determine differences among multiple groups. Statistical analysis was performed by using SPSS 15.0 software (SPSS Inc., Chicago, IL). P < 0.05 was considered to be statistically significant.
Results
miR-34c Is Downregulated in Ovarian Cancer Cells
To investigate the potential role of miR-34c in ovarian cancer, 25 primary ovarian cancer tumor tissues and their paracancerous tissues were collected from patients and analyzed. We found that miR-34c expression was significantly lower in ovarian cancer tissues than that in paracancerous tissues (Figure 1A). We then analyzed miR-34c expression in ovarian cancer cell lines. We found that the ovarian cancer cell lines SKOV3 and A2780 expressed obviously lower levels of miR-34c compared to the HOSEpiC cell line, which is a human normal ovarian surface epithelial cell line (Figure 1B). These results suggested that miR-34c may negatively regulate the development of ovarian cancer.
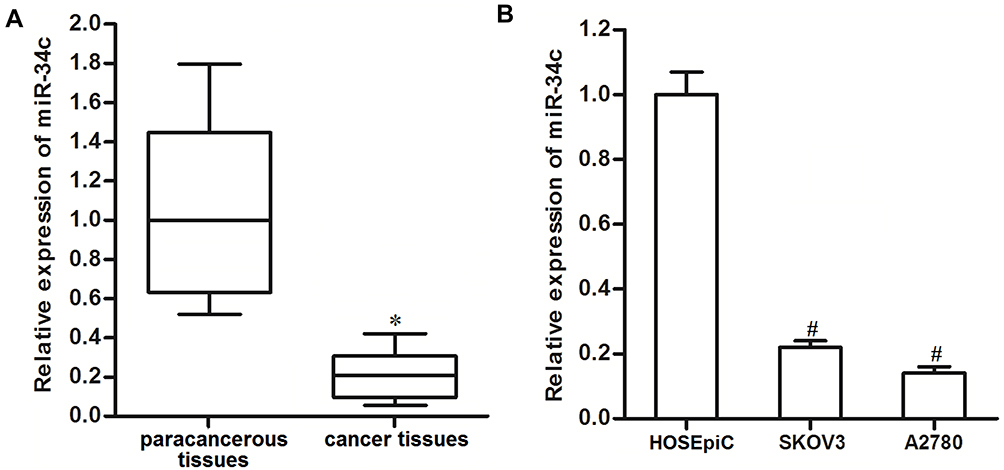 |
Figure 1 Downregulation of miR-34c in ovarian cancer. (A) Expression of miR-34c in 25 primary ovarian cancer tumor tissues and paracancerous tissues was detected by qRT-PCR. (B) Expression of miR-34c in the SKOV3 and A2780 cell lines was measured by qRT-PCR. Notes: Data was expressed as mean±SD. *P<0.05 vs paracancerous tissues, #P<0.05 vs HOSEpiC.Abbreviation: qRT-PCR, quantitative Real-Time Polymerase Chain Reaction. |
miR-34c Overexpression Sensitizes Ovarian Cancer Cells to Cisplatin-Induced Cytotoxicity
To correct the aberrant expression of miR-34c in ovarian cancer cells, we transfected SKOV3 and A2780 cells with miR-34c mimic. QRT-PCR analysis showed that transfection with miR-34c mimic increased the cellular level of miR-34c in both cell lines (Figure 2A). Next, we treated the transfected cells with different concentrations of cisplatin. Interestingly, we discovered that miR-34c overexpression sensitized ovarian cancer cells to cisplatin-induced cytotoxicity (Figure 2B). Intuitively, we demonstrated that the half maximal inhibitory concentration (IC50) of cisplatin to ovarian cancer was clearly decreased by transfection with miR-34c (Figure 2C). Therefore, miR-34c may be used as a sensitizer to enhance the anti-tumor effect of cisplatin on ovarian cancer in vitro.
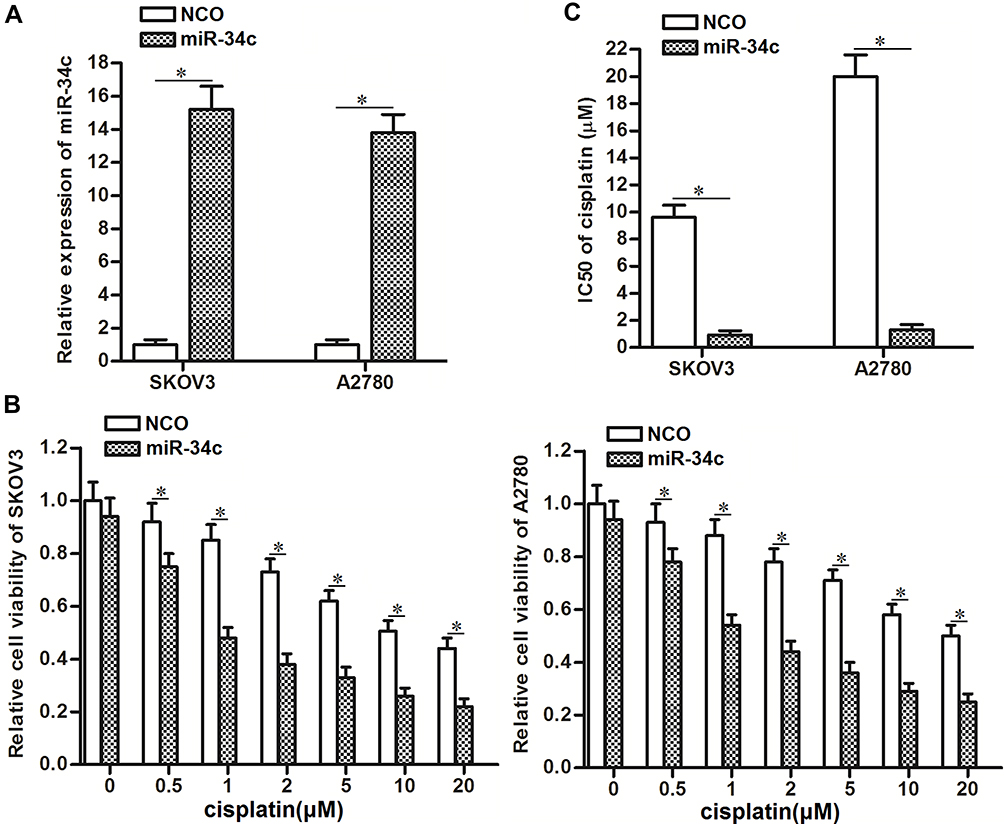 |
Figure 2 Overexpression of miR-34c sensitizes ovarian cancer cells to cisplatin-induced cytotoxicity in vitro. (A) Transfection with miR-34c mimic (50 pmol/mL) increased the cellular level of miR-34c in the SKOV3 and A2780 cell lines. (B) Transfection with miR-34c (50 pmol/mL) increased the cytotoxicity of various concentrations of cisplatin in the SKOV3 and A2780 cell lines. (C) Effect of miR-34c on the IC50 of cisplatin in the SKOV3 and A2780 cell lines. Notes: Data was expressed as mean±SD. *P<0.05. Abbreviation: IC50, 50% inhibiting concentration. |
miR-34c Targets MET in Ovarian Cancer Cells
To investigate the mechanism by which miR-34c sensitized ovarian cancer cells to cisplatin-induced cytotoxicity, we predicted potential targets of miR-34c using public databases (TargetScan, miRanda, and PicTar). Among the candidates, the gene for MET was commonly indicated to contain complementary sequences paired with miR-34c (Figure 3A). Furthermore, contrary to the decreased levels of miR-34c, we found that MET expression was upregulated in ovarian cancer tissues and cell lines (Figure 3B). Thus, we investigated the potential miR-34c/MET axis in ovarian cancer. Western blot analysis showed that transfection with miR-34c clearly decreased the expression of MET in SKOV3 and A2780 ovarian cancer cells (Figure 3C). Furthermore, luciferase reporter assays showed that co-transfection with miR-34c mimic decreased the luciferase activity of pGL3 reporters containing the wild-type 3′ UTR of MET. However, miR-34c had no effect on pGL3 reporters with the mutant 3′ UTR of MET (Figure 3D). These results indicated that miR-34c targeted MET in ovarian cancer cells.
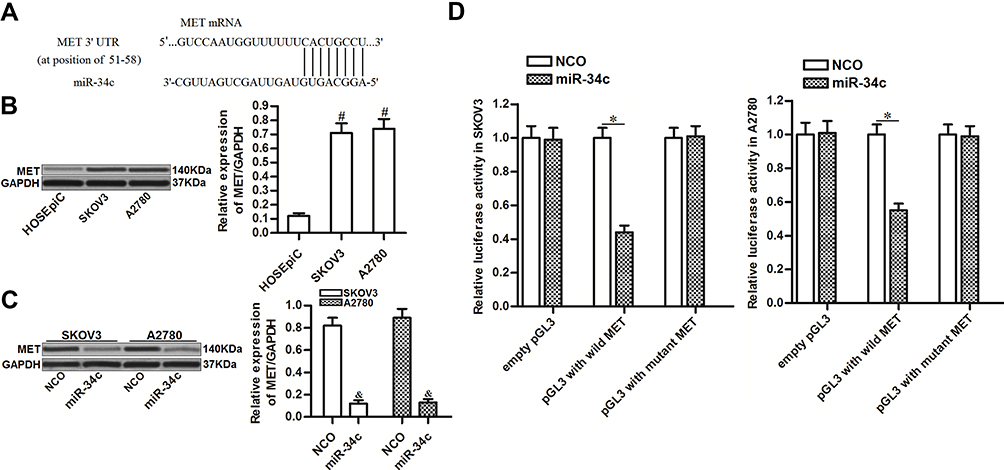 |
Figure 3 miR-34c targets MET in ovarian cancer cells. (A) Complementary sequence in the 3′ UTR of MET paired with miR-34c. (B) MET expression in HOSEpiC and ovarian cancer cell lines (SKOV3 and A2780). (C) Effect of miR-34c transfection (50 pmol/mL) on the level of MET protein in SKOV3 and A2780 cells. (D) A Dual-Luciferase Reporter Assay System was used to measure luciferase activity in SKOV3 and A2780 cells after co-transfection with the pGL3 reporter (2 μg/mL) and miR-34c mimic (50 pmol/mL). Notes: Data was expressed as mean±SD. #P<0.05 vs HOSEpiC, &P<0.05 vs NCO group, *P<0.05. Abbreviations: MET, hepatocyte growth factor receptor; 3’ UTR, 3’untranslated region; NCO, negative control oligonucleotide. |
miR-34c Enhances the Cytotoxicity of Cisplatin in Ovarian Cancer by Suppressing MET
To investigate the potential role of miR-34c/MET axis in regulating cisplatin sensitivity in ovarian cancer, we changed the expression of MET in SKOV3 and A2780 cells through transfection with MET siRNA or plasmid, respectively (Figure 4A). We found that MET overexpression clearly reduced the effect of miR-34c on sensitizing the ovarian cancer cell lines to cisplatin-induced cytotoxicity (Figure 4B). On the contrary, knockdown of MET enhanced the cytotoxicity of cisplatin to SKOV3 and A2780 cells, which was similar to the effect of miR-34c (Figure 4C). These results indicated that the expression profile of MET was associated with cisplatin sensitivity in ovarian cancer. Overexpression of miR-34c decreased the expression of MET to sensitize ovarian cancer cells to cisplatin-induced cytotoxicity.
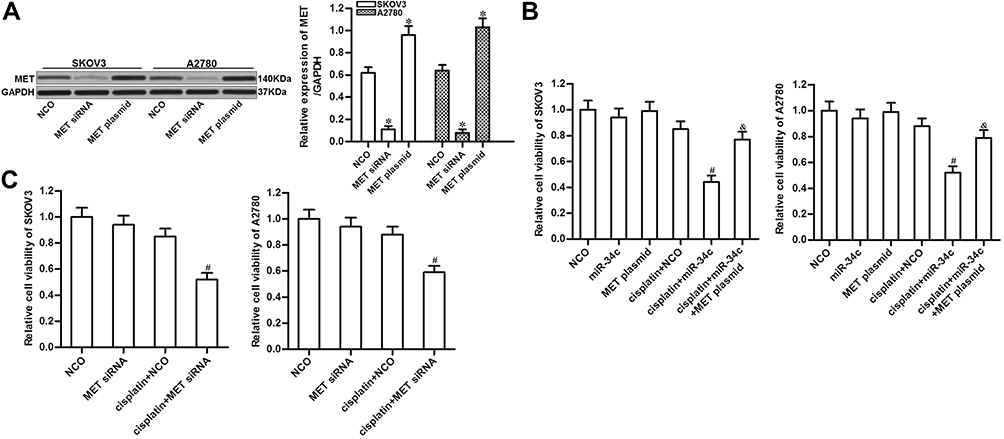 |
Figure 4 Role of the miR-34c/MET axis in regulating cisplatin sensitivity in SKOV3 and A2780 cells. (A) Transfection efficiency of MET siRNA (50 pmol/mL) and plasmid (2 μg/mL) in SKOV3 and A2780 cells. (B) Effect of the MET plasmid on miR-34c-promoted cell death in cisplatin-treated (1 μM) SKOV3 and A2780 cells. (C) Effect of MET siRNA on the sensitivity of SKOV3 and A2780 cells to cisplatin (1 μM). Notes: Data was expressed as mean±SD. *P<0.05 vs. NCO group, #P<0.05 vs. cisplatin + NCO group, &P<0.05 vs. cisplatin + miR-34c group. Abbreviations: MET, hepatocyte growth factor receptor; siRNA, small interfering RNA. |
The miR-34c/MET Axis Regulates the PI3K/AKT/Bad Pathway in Ovarian Cancer Cells
MET is an upstream regulator of the PI3K/AKT pathway.22 Thus, we investigated the role of the miR-34c/MET axis in regulating this highly oncogenic pathway. Interestingly, miR-34c overexpression inhibited the phosphorylation of PI3K and AKT, no matter whether or not SKOV3 and A2780 cells were treated with cisplatin. Furthermore, miR-34c treatment also reduced the phosphorylation of Bad, which is a substrate of AKT.23 On the other hand, the enforced expression of MET inhibited the entire PI3K/AKT/Bad pathway in miR-34c and cisplatin co-treated SKOV3 and A2780 cells (Figure 5A). These findings indicated the effect of the miR-34c/MET axis on the PI3K/AKT/Bad pathway in ovarian cancer cells. Next, we tested the function of Bad in ovarian cancer cells after treatment with miR-34c and cisplatin. We found that miR-34c, but not cisplatin, promoted the interaction between Bad and Bcl-xl. However, the enforced expression of MET inhibited the effect of miR-34c on the function of Bad (Figure 5B). These results indicated that miR-34c can regulate the PI3K/AKT/Bad pathway in ovarian cancer cells by suppressing MET.
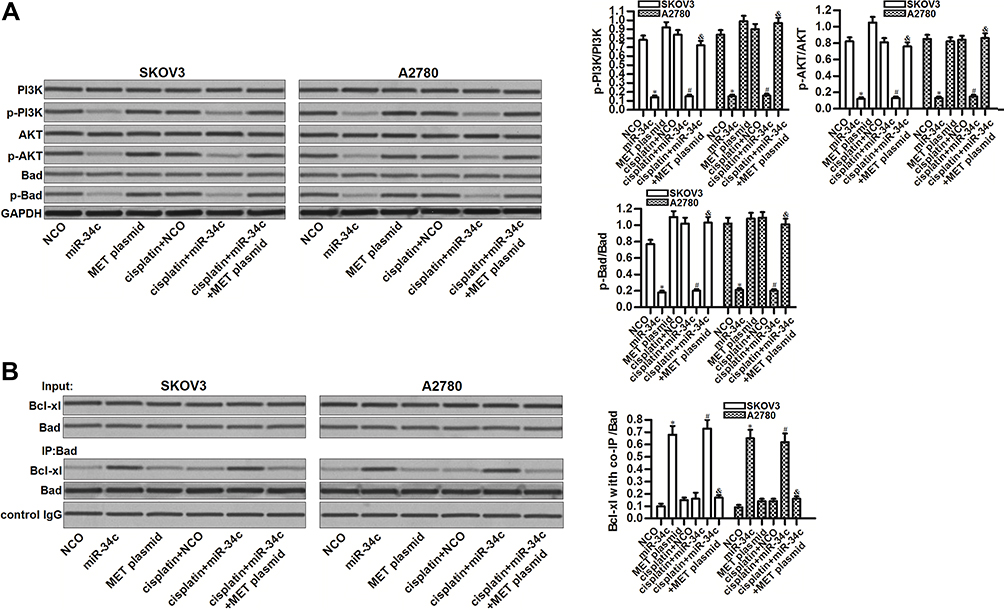 |
Figure 5 The miR-34c/MET axis regulates the PI3K/AKT/Bad pathway in SKOV3 and A2780 cells. (A) Effect of miR-34c (50 pmol/mL), MET plasmid (2 μg/mL), and cisplatin (1 μM) on the phosphorylation of PI3K, AKT, and Bad in SKOV3 and A2780 cells. (B) A co-immunoprecipitation assay was performed to evaluate the interaction between Bad and Bcl-xl in SKOV3 and A2780 cells after treatment with miR-34c (50 pmol/mL), MET plasmid (2 μg/mL), and cisplatin (1 μM). Notes: Data was expressed as mean±SD. *P<0.05 vs. NCO group, #P<0.05 vs. cisplatin + NCO group, &P<0.05 vs. cisplatin + miR-34c group. Abbreviations: MET, hepatocyte growth factor receptor; PI3K, phosphatidylinositol 3-kinase; AKT, threonine protein kinase; Bad, BCL2 associated agonist of cell death; Bcl-xl, B-cell lymphoma-extra large. |
miR-34c Sensitizes Ovarian Cancer Cells to Cisplatin-Induced Apoptosis Through the Mitochondrial Pathway
The preceding results showed that miR-34c increased the levels of free Bcl-xl through the MET/PI3K/AKT/Bad pathway. As Bcl-xl functions as a suppressor of mitochondrial apoptosis and as a stabilizer of mitochondrial inner membrane potential.24,25 We investigated the role of miR-34c in regulating the cisplatin-induced apoptosis pathway. As shown in Figure 6A, miR-34c overexpression enhanced the collapse of MMP induced by cisplatin. However, the enforced expression of MET inhibited the enhancing effect of miR-34c on the cisplatin-dependent collapse of MMP. Due to MMP collapse, co-treatment with miR-34c increased the level of cytoplasmic cytochrome c released from mitochondria in cisplatin-treated SKOV3 and A2780 cells (Figure 6B). Furthermore, we found the clear activation of caspase-9 and −3 (Figure 6C) and cell apoptosis (Figure 6D) in cisplatin and miR-34c co-treated ovarian cancer cells. However, transfection with the MET plasmid partially inhibited the mitochondrial apoptosis pathway. Thus, we concluded that miR-34c sensitized ovarian cancer cells to mitochondrial apoptosis through the MET/PI3K/AKT/Bad pathway.
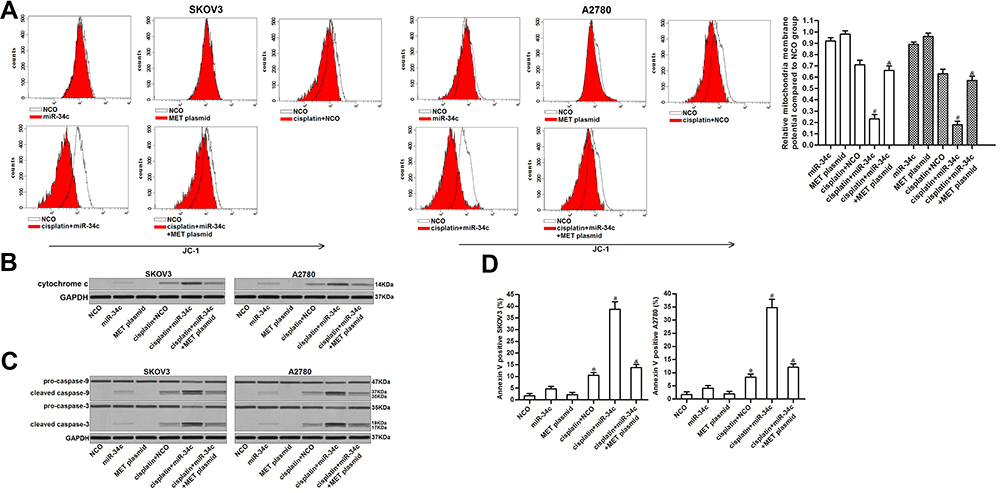 |
Figure 6 miR-34c sensitizes ovarian cancer cells to the cisplatin-induced apoptosis pathway. (A) Effect of miR-34c (50 pmol/mL), MET plasmid (2 μg/mL), and cisplatin (1 μM) on MMP in SKOV3 and A2780 cells. (B) Cytochrome c in the cytoplasmic fraction was detected by Western blot analysis after the removal of mitochondria. (C) Effect of miR-34c (50 pmol/mL), MET plasmid (2 μg/mL), and cisplatin (1 μM) on caspase-9 and caspase-3 in SKOV3 and A2780 cells. (D) Rate of apoptosis in SKOV3 and A2780 cells after treatment with miR-34c (50 pmol/mL), MET plasmid (2 μg/mL), and cisplatin (1 μM). Notes: Data was expressed as mean±SD. *P<0.05 vs. NCO group, #P<0.05 vs. cisplatin + NCO group, &P<0.05 vs. cisplatin + miR-34c group. Abbreviations: MET, hepatocyte growth factor receptor; MMP, mitochondrial membrane potential. |
miR-34c Enhances Anti-Tumor Effect of Cisplatin on Ovarian Cancer in vivo
To investigate the effect of miR-34c on regulating the anti-tumor effect of cisplatin on ovarian cancer in vivo, miR-34c-overexpressing SKOV3 cells were inoculated subcutaneously into nude mice. We found that cisplatin treatment inhibited the growth of SKOV3 tumors. However, the miR-34c-overexpressing SKOV3 tumors exhibited higher sensitivity to cisplatin treatment than control SKOV3 tumors (Figure 7A). This indicated the role of miR-34c in enhancing the anti-tumor effect of cisplatin on ovarian cancer in vivo. Subsequently, we separated and purified the tumor tissues to detect the expression of miR-34c and MET. We confirmed miR-34c overexpression in the miR-34c-LV-transfected tumors (Figure 7B). In contrast, MET expression was decreased in the miR-34c-overexpressing ovarian cancer tumors compared to the control ovarian cancer tumors (Figure 7C). We thus demonstrated that miR-34c targeted MET in ovarian cancer cells in vitro and in vivo.
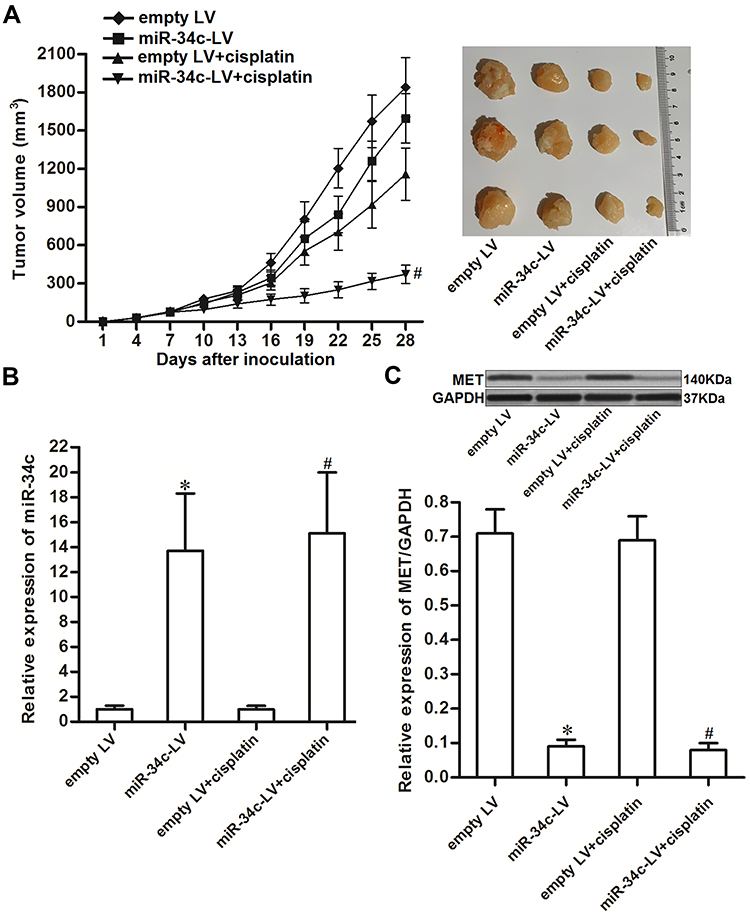 |
Figure 7 miR-34c enhances the anti-tumor effect of cisplatin on ovarian cancer in vivo. (A) Nude mice were inoculated subcutaneously with miR-34c-LV-transfected SKOV3 cells followed by treatment with cisplatin i.p. twice a week (2 mg/kg). Tumor growth was measured every three days. (B) Expression level of miR-34c in purified tumors was detected by qRT-PCR analysis. (C) MET expression in purified tumors was detected by Western blot analysis. Notes: Data was expressed as mean±SD. *P < 0.05 vs. empty LV group, #P < 0.05 vs. empty LV + cisplatin group. Abbreviations: MET, hepatocyte growth factor receptor; LV, lentivirus; qRT-PCR, quantitative Real-Time Polymerase Chain Reaction. |
Discussion
Cisplatin-based chemotherapy is an important approach for the conservative treatment of ovarian cancer.26,27 However, resistance to cisplatin is still a considerable obstacle to achieve a satisfactory therapeutic effect in patients with ovarian cancer.28,29 Strategies are required to sensitize ovarian cancer cells to cisplatin treatment.30,31 Recent studies have demonstrated that cisplatin resistance is usually accompanied with dysregulation of miRNAs in cancers including ovarian cancer.13,32,33 MiR-34c, which has been reported to act as a tumor suppressor in hepatocellular carcinoma and breast cancer,34,35 improves the sensitivity of osteosarcoma to cisplatin.36 These reports suggested the potential role of miR-34c as an anti-tumor treatment.
In this study, we found a significant decrease of miR-34c expression in ovarian cancer cell lines and tumor tissues from patients compared to paracancerous normal tissues. Moreover, despite the fact that the restoration of miR-34c expression did not induce obvious cytotoxicity in ovarian cancer cells, it significantly sensitized these cells to cisplatin-induced cell death. According to the results of in vivo experiments, cisplatin alone treatment exhibited some anti-tumor effect on ovarian cancer despite cisplatin alone cannot affect the MET expression. It is due to the direct cytotoxicity of cisplatin against cancers. Moreover, we found that miR-34c significantly enhanced the effect of cisplatin on reducing the tumor volume. Our finding indicated that miR-34c overexpression enhanced the anti-tumor effect of cisplatin on ovarian cancer in vitro and in vivo.
MET, a receptor of hepatocyte growth factor, acts as an oncogene by promoting tumorigenicity.37 Studies have demonstrated that the activation of MET signaling triggers multiple oncogenic pathways, including the PI3K/AKT pathway, to inhibit apoptosis and promote tumor proliferation, metastasis, and drug resistance.22,38-40 Therefore, MET overexpression usually predicts poor prognosis for cancer patients, and MET has been considered as a potential target in cancer therapy.
In this study, we found that MET was significantly upregulated in ovarian cancer cells. However, change of MET expression alone can not obviously affect the viability of ovarian cancer cells. On the other hand, we found that MET overexpression can decrease the sensitivity of ovarian cancer cells to cisplatin treatment. On the contrary, knockdown of MET sensitized ovarian cancer cells to cisplatin-induced cytotoxicity. These results indicated that MET was an important regulator that regulated the chemosensitivity of ovarian cancer cells. Furthermore, we proved that the effect of miR-34c on enhancing the anti-tumor effect of cisplatin on ovarian cancer was dependent on the inhibition of MET. These findings emphasized the role of the miR-34c/MET axis in changing cisplatin sensitivity in ovarian cancer.
Bad (BCL2-associated agonist of cell death) is a substrate of AKT and functions as a pro-apoptotic protein. However, the effect of Bad is dependent on its phosphorylation status. Non-phosphorylated Bad conjugates with the anti-apoptotic protein Bcl-xl and inactivates it; however, phosphorylated Bad exhibits no function.41,42 In this study, we proved that miR-34c targeted the MET/PI3K/AKT pathway to inhibit the phosphorylation of Bad. Due to the increased levels of dephosphorylated Bad, ovarian cancer cells were facilitated to the apoptosis signals. As a result, we found that the restoration of miR-34c expression sensitized ovarian cancer cells to cisplatin-induced mitochondrial apoptosis.
In conclusion, we have revealed a role for the miR-34c/MET axis in enhancing cisplatin-induced cytotoxicity in ovarian cancer. Despite the need for further studies to determine the clinical prospect of targeting this axis, it may be a potential strategy to increase sensitivity to cisplatin through a combination of certain miRNA sensitizers such as miR-34c.
Acknowledgments
Thanks due to the whole contributors who assisted with this study.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi:10.3322/caac.21551
2. Rooth C. Ovarian cancer: risk factors, treatment and management. Br J Nurs. 2013;22(Sup17):S23–S30. doi:10.12968/bjon.2013.22.Sup17.S23
3. Rosen B, Laframboise S, Ferguson S, et al. The impacts of neoadjuvant chemotherapy and of debulking surgery on survival from advanced ovarian cancer. Gynecol Oncol. 2014;134:462–467. doi:10.1016/j.ygyno.2014.07.004
4. Shylasree TS, Bryant A, Athavale R. Chemotherapy and/or radiotherapy in combination with surgery for ovarian carcinosarcoma. Cochrane Database Syst Rev. 2013;2:CD006246. doi:10.1002/14651858.CD006246
5. Xiong X, Arvizo RR, Saha S, et al. Sensitization of ovarian cancer cells to cisplatin by gold nanoparticles. Oncotarget. 2014;5:6453–6465. doi:10.18632/oncotarget.2203
6. Zhou J, Wang Y, Wang Y, et al. FOXM1 modulates cisplatin sensitivity by regulating EXO1 in ovarian cancer. PLoS One. 2014;9(5):e96989. doi:10.1371/journal.pone.0096989
7. Zhao JX, Liu H, Lv J, Yang XJ. Wortmannin enhances cisplatin-induced apoptosis in human ovarian cancer cells in vitro. Eur Rev Med Pharmacol Sci. 2014;18:2428–2434.
8. Xie Q, Wang S, Zhao Y, Zhang Z, Qin C, Yang X. MiR-519d impedes cisplatin-resistance in breast cancer stem cells by down-regulating the expression of MCL-1. Oncotarget. 2017;8(13):22003–22013. doi:10.18632/oncotarget.15781
9. Xu YY, Wu TT, Zhou SH, et al. Apigenin suppresses GLUT-1 and p-AKT expression to enhance the chemosensitivity to cisplatin of laryngeal carcinoma Hep-2 cells: an in vitro study. Int J Clin Exp Pathol. 2014;7:3938–3947.
10. Hui C, Lan Z, Yue-li L, Li-lin H, Li-lin H. Knockdown of Eag1 expression by RNA interference increases chemosensitivity to cisplatin in ovarian cancer cells. Reprod Sci. 2015;22(12):1618–1626. doi:10.1177/1933719115590665
11. Eljack ND, Ma H-YM, Drucker J, et al. Mechanisms of cell uptake and toxicity of the anticancer drug cisplatin. Metallomics. 2014;6(11):2126–2133. doi:10.1039/c4mt00238e
12. Barabas K, Milner R, Lurie D, Adin C. Cisplatin: a review of toxicities and therapeutic applications. Vet Comp Oncol. 2008;6(1):1–18. doi:10.1111/j.1476-5829.2007.00142.x
13. Yu PN, Yan MD, Lai HC, et al. Downregulation of miR-29 contributes to cisplatin resistance of ovarian cancer cells. Int J Cancer. 2014;134:542–551. doi:10.1002/ijc.28399
14. Wu H, Wang K, Liu W, Hao Q. SPTEN overexpression improves cisplatin-resistance of human ovarian cancer cells through upregulating KRT10 expression. Biochem Biophys Res Commun. 2014;444(2):141–146. doi:10.1016/j.bbrc.2014.01.014
15. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi:10.1016/j.cell.2009.01.002
16. Gargalionis AN, Basdra EK. Insights in microRNAs biology. Curr Top Med Chem. 2013;13(13):1493–1502. doi:10.2174/15680266113139990098
17. Li C, Hashimi SM, Good DA, et al. Apoptosis and microRNA aberrations in cancer. Clin Exp Pharmacol Physiol. 2012;39(8):739–746. doi:10.1111/j.1440-1681.2012.05700.x
18. Hwang HW, Mendell JT. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br J Cancer. 2006;94(6):776–780. doi:10.1038/sj.bjc.6603023
19. Bouyssou JM, Manier S, Huynh D, Issa S, Roccaro AM, Ghobrial IM. Regulation of microRNAs in cancer metastasis. Biochim Biophys Acta. 2014;1845(2):255–265. doi:10.1016/j.bbcan.2014.02.002
20. Ma J, Dong C, Ji C. MicroRNA and drug resistance. Cancer Gene Ther. 2010;17(8):523–531. doi:10.1038/cgt.2010.18
21. Xu S, Fu GB, Tao Z, et al. MiR-497 decreases cisplatin resistance in ovarian cancer cells by targeting mTOR/P70S6K1. Oncotarget. 2015;6(28):26457–26471. doi:10.18632/oncotarget.4762
22. Trovato M, Torre ML, Ragonese M, et al. HGF/c-met system targeting PI3K/AKT and STAT3/phosphorylated-STAT3 pathways in pituitary adenomas: an immunohistochemical characterization in view of targeted therapies. Endocrine. 2013;44(3):735–743. doi:10.1007/s12020-013-9950-x
23. Feng X, Jiang J, Shi S, Xie H, Zhou L, Zheng S. Knockdown of miR-25 increases the sensitivity of liver cancer stem cells to TRAIL-induced apoptosis via PTEN/PI3K/Akt/Bad signaling pathway. Int J Oncol. 2016;49(6):2600–2610. doi:10.3892/ijo.2016.3751
24. Chen YB, Aon MA, Hsu YT, et al. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol. 2011;195(2):263–276. doi:10.1083/jcb.201108059
25. McNally MA, Soane L, Roelofs BA, Hartman AL, Hardwick JM. The N-terminal helix of Bcl-xL targets mitochondria. Mitochondrion. 2013;13(2):119–124. doi:10.1016/j.mito.2013.01.004
26. Gonera A, Wawryka J, Sobkowicz A, et al. SKOV-3 and Me45 cell response to cisplatin-based chemotherapy: an in vitro study. Folia Biol (Praha). 2014;60(5):213–219.
27. Del Prete SA, Ryan SP, Nelson JH. Intraperitoneal cisplatin-based chemotherapy for primary treatment of epithelial ovarian cancer. Conn Med. 2001;65(2):71–75.
28. Sharma S, Santiskulvong C, Rao J, Gimzewski JK, Dorigo O. The role of Rho GTPase in cell stiffness and cisplatin resistance in ovarian cancer cells. Integr Biol (Camb). 2014;134(6):611–617. doi:10.1002/ijc.28399
29. Xia M, Yu H, Gu S, et al. p62/SQSTM1 is involved in cisplatin resistance in human ovarian cancer cells via the Keap1-Nrf2-ARE system. Int J Oncol. 2014;45(6):2341–2348. doi:10.3892/ijo.2014.2669
30. de Almeida Chuffa LG, de Moura Ferreira G, Lupi LA, da Silva Nunes I, Fávaro WJ. P-MAPA immunotherapy potentiates the effect of cisplatin on serous ovarian carcinoma through targeting TLR4 signaling. J Ovarian Res. 2018;11(1):8. doi:10.1186/s13048-018-0380-5
31. RajBhandary UL. Genetic code. Modified bases and aminoacylation. Nature. 1988;336(6195):112–113. doi:10.1038/336112a0
32. Chen Y, Gao Y, Zhang K, et al. MicroRNAs as regulators of cisplatin resistance in lung cancer. Cell Physiol Biochem. 2015;37(5):1869–1880. doi:10.1159/000438548
33. Li B, Chen H, Wu N, Zhang WJ, Shang LX. Deregulation of miR-128 in ovarian cancer promotes cisplatin resistance. Int J Gynecol Cancer. 2014;24:1381–1388. doi:10.1097/IGC.0000000000000252
34. Song J, Wang Q, Luo Y, et al. miR-34c-3p inhibits cell proliferation, migration and invasion of hepatocellular carcinoma by targeting MARCKS. Int J Clin Exp Pathol. 2015;8(10):12728–12737.
35. Tao W-Y, Wang C-Y, Sun Y-H, Su Y-H, Pang D, Zhang G-Q. MicroRNA-34c suppresses breast cancer migration and invasion by targeting GIT1. J Cancer. 2016;7(12):1653–1662. doi:10.7150/jca.14762
36. Hu Y, Yang Q, Wang L, et al. Knockdown of the oncogene lncRNA NEAT1 restores the availability of miR-34c and improves the sensitivity to cisplatin in osteosarcoma. Biosci Rep. 2018;38(3):BSR20180375. doi:10.1042/BSR20180375
37. Ho-Yen CM, Jones JL, Kermorgant S. The clinical and functional significance of c-Met in breast cancer: a review. Breast Cancer Res. 2015;17(1):52. doi:10.1186/s13058-015-0547-6
38. Yao Y, Dou C, Lu Z, Zheng X, Liu Q. MACC1 suppresses cell apoptosis in hepatocellular carcinoma by targeting the HGF/c-MET/AKT pathway. Cell Physiol Biochem. 2015;35(3):983–996. doi:10.1159/000369754
39. Cui Y-M, Jiao H-L, Ye Y-P, et al. FOXC2 promotes colorectal cancer metastasis by directly targeting MET. Oncogene. 2015;34(33):4379–4390. doi:10.1038/onc.2014.368
40. Maroun CR, Rowlands T. The met receptor tyrosine kinase: a key player in oncogenesis and drug resistance. Pharmacol Ther. 2014;142:316–338. doi:10.1016/j.pharmthera.2013.12.014
41. Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13(22):2905–2927. doi:10.1101/gad.13.22.2905
42. Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91(2):231–241. doi:10.1016/S0092-8674(00)80405-5
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.