")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
MiR-344b-1-3p targets TLR2 and negatively regulates TLR2 signaling pathway
Authors Xu H, Wu Y, Li L, Yuan W, Zhang D, Yan Q, Guo Z, Huang W
Received 23 August 2016
Accepted for publication 7 November 2016
Published 14 February 2017 Volume 2017:12 Pages 627—638
DOI https://doi.org/10.2147/COPD.S120415
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Richard Russell
Hong Xu,1–3,* Yuting Wu,2–4,* Li Li,1–3 Weifeng Yuan,1–3 Deming Zhang,2–4 Qitao Yan,2,3 Zhenhui Guo,2,3 Wenjie Huang1–3
1Department of Respiratory Medicine, 2Guangdong Provincial Key Laboratory of Geriatric Infection and Organ Function Support, 3Guangzhou Key Laboratory of Geriatric Infection and Organ Function Support, 4Department of Geriatric Respiratory Medicine, General Hospital of Guangzhou Command of PLA, Guangzhou, Guangdong, People’s Republic of China
*These authors contributed equally to this work
Objectives: COPD is an abnormal inflammatory response characterized by decreased expression of TLR2 in patients, which is suggested to induce invasive pulmonary aspergillosis (IPA). MicroRNAs (miRNAs) have been shown to play important roles in the pathogenesis of human respiratory system disorders. Therefore, the aim of this study was to identify the miRNAs involved in the regulation of TLR2 signaling in COPD.
Materials and methods: miRNA microarray analysis was performed to screen for the dysregulated miRNAs in alveolar macrophages (AMs) isolated from COPD rats. The interaction between these miRNAs and TLR2 gene was predicted using miRBase and validated using dual luciferase assay. Based on the analysis, a novel miR-344b-1-3p was identified as a novel modulator of TLR2 gene. Then, the mechanism through which miR-344b-1-3p regulated TLR2 expression was explored using cigarette smoke extract (CSE)-pretreated NR8383 cells. Moreover, by subjecting CSE-pretreated NR8383 cells to Pam3CSK4, the effect of miR-344b-1-3p on NF-κB activity and other important mediators of COPD, including IRAK-1, ERK, TNF-α, IL-1β, and MIP-2, was also assessed.
Results: COPD rat model was successfully induced by smoke inhalation. Among the 11 upregulated miRNAs in AMs from COPD rats, miR-344b-1-3p was predicted to be a novel miRNA targeting TLR2 gene. In the CSE pretreated NR8383 cells exposed to Pam3CSK4, miR-344b-1-3p inhibition increased the expression levels of TLR2, TNF-α, and IL-1β and decreased the expression levels of MIP-2. In addition, the phosphorylation of IRAK-1, IκBα, and IRK was augmented by miR-344b-1-3p inhibition.
Conclusion: Findings outlined in this study suggest that miR-344b-1-3p was an effective modulator of TLR2 gene, which can be employed as a promising therapeutic and preventive target of IPA in COPD patients.
Keywords: COPD, miR-344b-1-3p, TLR2, invasive pulmonary aspergillosis
Introduction
COPD ranks the fourth of all the causes of morbidity and mortality in the third world countries and is predicted to be the third leading cause of death in the third world countries by the year 2020.1 The disease is characterized by T-cell chronic inflammatory response and increased susceptibility to microbial infection, which markedly contributes to exacerbations and decline of lung function.2,3 As a widely distributed infectious microbe species, Aspergillus spp., have become a severe threat for COPD patients who are impaired by abnormal immune function, resulting in invasive pulmonary aspergillosis (IPA).4,5 Worse still, COPD patients attacked by IPA are always exposed to a higher chance of mortality. Therefore, early intervention of the disorder is critical for better prognosis of IPA in patients with COPD.
Generally, macrophages function as key defense against pathogens, which contribute to inflammation and tissue injury as well as control and resolution of inflammation. In COPD, the number of alveolar macrophages (AMs) increases with the disease severity, but these newly generated cells are always functionally defective.6–8 It has been demonstrated that the phagocytic activity was significantly reduced in AMs from COPD. This is suggested to be attributed to the impaired action of pathogen recognition and anti-inflammation in AMs. The innate immune recognition of pathogens by macrophages is mediated by pathogen recognition receptors. Among these receptors, TLR2 plays important roles in the recognition and removal of Aspergillus species by activating immune responses and phagocytosis of macrophages. As previously reported, AMs from smokers with and without COPD express less TLR2 than nonsmokers,9 which will result in the change of the subsequent intracellular signaling. The diminished TLR2 expression may lead to reduced identification and removal of pathogens and facilitate microbial colonization as well as increased risk of IPA.10
Previous study in our laboratory suggested that after Aspergillus spore transplantation, the upregulated extent of TLR2 in AMs from COPD rats was dramatically smaller than that in normal rats, whereas the increased extent of TLR2 mRNA in AMs from COPD rats was larger than that in normal rats.11 Taken together, this might indicate that transcription of TLR2 was not impaired even with the attack of COPD in rat models. However, the mechanism affecting the protein expression of TLR2 needs further elucidation.
MicroRNAs (miRNAs) are a class of evolutionarily conserved small RNAs, which regulate target gene expression by binding to the 3′-UTR end of target mRNA.12 Hundreds of miRNAs have been identified in plants, animals, and viruses. For Homo sapiens, >700 miRNA genes have been reported and comprehensively studied, including those involved in immune responses and COPD.13 Moreover, several miRNAs have shown their potential as regulators of TLR2 signaling transduction. In human promonocyte THP-1 cells, miR-146a is induced by lipopolysaccharide (LPS) stimulation and negatively regulates TLR2 signaling pathway, contributing to tolerance during microbial infection and sepsis. However, the regulation of TLR2 expression by miRNA in COPD patients still remains unknown. Thus, in this study, the functional link between TLR2 and miRNAs in COPD was selected as the research focus. With a series of in vivo and in vitro assays, the results first demonstrated that miR-344b-1-3p was an important regulator of TLR2. Regulation of the miRNA would not only influence the TLR2 signaling activation but also affect the expression of inflammatory mediators.
Materials and methods
Chemicals and instruments
Coco cigarettes were purchased from Guangdong Tobacco Industry Co. Ltd., (Guangdong, People’s Republic of China). Culture medium (RPMI-1640), fetal bovine serum (FBS), and TRIzol were purchased from Thermo Fisher Scientific Corporation (Carlsbad, CA, USA). The 2× polymerase chain reaction (PCR) master mix was purchased from Arraystar (Rockville, MD, USA). Other chemicals were commercially available and purchased as reagent grade from Sinopharm (Shanghai, People’s Republic of China).
Animals and induction of COPD model by smoke exposure
Sixty male Wistar rats (12 weeks old, weighing 230–250 g) were obtained from Southern Medical University (Guangzhou, People’s Republic of China) and fed in the specific pathogen free (SPF) animal room of Southern Medical University and housed in box cages (<6 rats in every cage) at 23°C±3°C and 55%±10% humidity with a 12-hour light/12-hour dark cycle. All animal experiments were approved by the ethic committee of Southern Medical University (Certificate no 440020100002199). All animal-handling procedures were performed according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and followed the guidelines of the Animal Welfare Act.
After a 3-day adapting period, all animals were randomly divided into two groups (n=30 in each group) and COPD condition was induced as follows: 1) COPD model group: in a home-made smoking box (110×80×75 cm), the rats were exposed to cigarette smoke generated from Yeshu cigarettes twice daily (30 minutes each time) with an interval of 8 hours for 90 days, and 2) control group: rats were subjected to saline tracheal instillation twice daily for 90 days.
COPD pulmonary function detection, CT examination, and histopathology assays
Assessment of pulmonary function is essential for the diagnosis and characterization of COPD. Spirometry was used to detect airway obstruction. The primary parameters of diagnostic assessment are minute ventilation volume (MVV), inspiratory resistance (Ri), peak expiratory flow (PEF), and forced expiratory volume in 0.3 second (FEV0.3).
After anesthesia by intraperitoneal injection of pentobarbital sodium (30 mg/kg body weight), the rats were examined by CT test (Aloka LaTheta LCT-200; Hitachi-Aloka Medical, Ltd, Tokyo, Japan) to determine COPD.
After infusion for macrophage isolation, the left lungs were sampled and fixed in 10% neutral formalin solution for 48 hours. Following gradient dehydration with ethanol, the lungs were embedded in paraffin for the preparation of 4 μm-thick sections. Sections were stained with hematoxylin and eosin (H&E) using standard pathology procedures and evaluated by a pathologist blinded to the study as described previously.14,15
AM isolation and identification
Rats were anesthetized by intraperitoneal injection of pentobarbital sodium (30 mg/kg body weight). The chest of each rat was opened, and the bronchi lobaris inferior sinister was clipped. The lungs were infused with 8 mL of phosphate buffer saline (PBS) at 37°C four times via the main trachea. The alveolar lavage fluid was centrifuged at 1,000 rpm and 4°C for 10 minutes. The supernatant was subsequently discarded, and the pellet was washed twice with PBS, for 10 minutes each time. The cells were then incubated in six-well plates (1 mL/well; 1×106 cells/mL) with cover glasses in 1 mL RPMI-1640 with 10% FBS. After 2-hour culture under 5% CO2 at 37°C, the unattached cells were removed by washing with PBS three times. Using Wright–Giemsa staining and immunofluorescence staining of CD68, the macrophages were identified.
Total RNA isolation and reverse transcription
The preparation and evaluation of AMs was conducted routinely. Total RNA was extracted using TRIzol and miRNeasy Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol, except that isopropanol was replaced with ethanol for RNA precipitation. RNA quality was ascertained using 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Then 1 μg of total RNA was reverse transcribed, and the product (11 μL) was preamplified using Megaplex PreAmp Primers and DBI Bestar® qPCR RT Kit (Applied Biosystems, Foster City, CA, USA) in a 20 μL PCR. The preamplification cycling conditions were 37°C for 60 minutes and 98°C for 10 minutes. The preamplified cDNA was diluted with 0.1× Tris-EDTA buffer (pH 8.0) to 10 μL, and then 1 μL of diluted cDNA was used in each plate for real-time PCRs (RT-PCRs).
miRNA microarray analysis
RNA samples from three randomly selected rats in each group were used for miRNA microarray analysis. After RNA quantification using a NanoDrop spectrophotometer, the samples were labeled using the miRCURY™ Hy3™/Hy5™ Power Labeling Kit and hybridized to the miRCURY LNA™ Array (v.18.0). The samples were hybridized using a hybridization station, and the arrays were scanned with the Axon GenePix 4000B Microarray Scanner. The raw intensity of the image was read using GenePix Pro V6.0. The intensity of the green signal was calculated after background subtraction, and four replicated spots for each probe on the same slide were averaged. MiRNAs with intensity ≥30 in all samples after background correction were used to calculate the normalized factor. The median normalization method was used to normalize the expressed data. After normalization, significantly differentially expressed miRNAs were identified via volcano spot filtering. Subsequently, clustering analysis was performed with the MeV software (v4.6, J.Craig Venter Institute, Rockville, MD, USA), and two-dimensional tree-shaped heat map was acquired.
Bioinformatics analysis
The target genes of the target miRNA were crossly predicted by TargetScan (http://www.targetscan.org/), miRBase (http://www.mirbase.org/index.shtml), and miRanda (http://www.microrna.org/microrna/home.do). Then, the functional gene ontology (GO) biological processes, in terms of the putative targets of candidate miR-34b-1-3p and its target gene TLR2, as well as the Kyoto Encyclopedia of Genes and Genomes pathway analysis were performed by the Database for Annotation, Visualization and Integrated Discovery. For each GO score, the P-value of the function enrichment and the P-value of Benjamini correction were calculated.
Quantitative RT-PCR analysis of miRNAs
Real-time quantification was performed using an Applied Biosystems 7500 Sequence Detection System. The 20 μL PCR included 1 μL RT product (1:5 dilution), 0.5 μL universal reverse primer, 0.5 μL of sense primer, and 10 μL mix buffer (DBI Bestar® SybrGreen qPCR mastermix). The reactions were incubated in a 96-well optical plate at 94°C for 2 minutes, followed by 40 cycles of 94°C for 20 seconds, 58°C for 20 seconds, and 72°C for 20 seconds. All reactions were performed in triplicate. All primers used are listed in Table 1.
 | Table 1 Primer pairs of miRNA used for the RT-PCR |
Plasmid construction
The constructed plasmids (psiCHECK™-2-TLR2-3′UTR/psiCHECK™-2-TLR2-3′UTR-R) contained 3′-UTR promoter of TLR2 gene and were purchased from Sangon (Shanghai, People’s Republic of China).
Luciferase assay
Human 293 cell line (ATCC, Manassas, VA, USA) was maintained in RPMI-1640 with 10% heat inactivated FBS, supplemented with 1% antibiotic–antimycotic solution (Thermo Fisher Scientific Corporation) at 37°C in a humidified atmosphere of 95% air and 5% CO2.
Then, cells were seeded at a density of 2.0×104 cells/mL in 24-well plates to achieve ~70% confluence and were transfected with psiCHECK™-2-TLR2-3′UTR/psiCHECK™-2-TLR2-3′UTR-R plasmids and miR-344b-1-3p inhibitors using Lipofectamine 2000 (Thermo Fisher Scientific Corporation). 293 cells were incubated for an additional 36 hours after transfection. Thereafter, cells were collected and further assayed for firefly luciferase activity using the Dual-Luciferase® Reporter Assay System (Promega, Madison, WI, USA) according the manufacturer’s instruction. The results were recorded using a Synergy™ 4 Microplate Reader (BioTek, Winooski, VT, USA).
Coco cigarette smoke extract (CSE)
CSE was prepared by combusting two coco cigarettes purchased from Guangdong Tobacco Industry Co., Ltd (filters removed) using a peristaltic pump (Watson Marlow 323 E/D) and passing the smoke through 25 mL of FBS-free Dulbecco’s Modified Eagle Medium supplemented with penicillin, streptomycin, and amphotericin B at a rate of 5 minutes/cigarette. The obtained solution is referred to as 100% strength.
Cell treatment
Rat NR8383 cell line (Type Culture Collection of Chinese Academy of Sciences, Beijing, People’s Republic of China) was maintained in RPMI-1640 with 10% heat-inactivated FBS and 1% antibiotic–antimycotic solution (Thermo Fisher Scientific Corporation) at 37°C in a humidified atmosphere of 95% air and 5% CO2. NR8383 cells were seeded in a 24-well plate at the density of 1.0×104 cells/mL and then treated with 0%, 5%, 10%, and 15% CSE.
To select the optimal concentration of CSE for the following exposure, NR8383 cells were exposed to 0%, 5%, 10%, and 15% CSE for 24 hours. Subsequently, the expression level of miRNA-344b-3p and the apoptosis of NR8383 cells under different CSE treatments were determined by RT-PCR and flow cytometry, respectively.
After treatment with 10% CSE for 24 hours, miR-344b-1-3p inhibitor and null inhibitor were transfected into NR8383 cells through lentiviral transfection. Subsequently, the transfected cells were exposed to 1 μg/mL of Pam3CSK4 for an additional 24 hours before harvesting. We also seeded NR8383 cells with CSE and Pam3CSK4 treatment as control.
Flow cytometry assay
Using flow cytometry (Epics XL; Beckman Coulter, Brea, CA, USA), the apoptosis of NR8383 cells after exposure to different concentrations of CSE was analyzed using Annexin V and propidium iodide (PI) staining (eBioscience, Hatfield, UK).
TLR2 expression on the NR8383 cells was determined using flow cytometry by staining with fluorescence-conjugated anti-TLR2. Data acquisition and analysis were performed using CellQuest software.
Western blotting analysis
To evaluate the effect of miR-344b-1-3p on NF-κB signaling, the protein levels of IκBα were determined in NR8383 cells exposed to Pam3CSK4. The concentration of protein extracts from NR8383 was determined using a BCA Kit (Pierce, Rockford, IL, USA), and 20 μg of protein lysates were separated on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis followed by transfer to nitrocellulose membranes. Western blotting analysis was performed regularly, and the signal was detected using an ECL system (Millipore, Billerica, MA, USA). Antibodies used in this study were anti-rat p-IRAK-1 (1:5,000; Abcam, Cambridge, UK), anti-rat p-IκBα (1:20,000; Millipore), and anti-rat p-ERK (1:10,000; Millipore).
Enzyme-linked immunosorbent assay (ELISA)
The concentrations of TNF-α, IL-1β, and MIP-2 in the culture medium were quantified using a commercial rat inflammatory factor heterodimer ELISA kit (BioLegend, San Diego, CA, USA) according to the manufacturer’s protocol. All samples were assayed in duplicate. The mean concentration was determined for each sample. The sensitivity of the assay is 0.13±0.01 ng/mL. The color reaction was stopped with 50 μL of 1 M HCl per well, and the optical density was measured after 1 hour at 492 nm (OD492) on an Infinite® M200 microtiter plate reader (Tecan, Maennedorf, Switzerland).
Statistical analysis
All the data were expressed in the form of mean ± SD, and each assay was represented by three replicates (n=3). Post doc multiple comparisons after ANOVA were performed by Duncan method to control type I error. Difference between two groups was analyzed using Student’s t-test. Significance was accepted when two-tailed P-value was <0.05. All the statistical analyses and graph manipulation were conducted using the Statistical Package for the Social Sciences Version 19.0 (IBM, Armonk, NY, USA).
Results
Morphology analysis of COPD rats
As listed in Table 2, the levels of expiratory resistance and inspiratory resistance were increased in the COPD group when compared with the control group, and the difference was statistically significant (P<0.05). On the contrary, FEV0.3/FVC in the COPD group was decreased when compared with those in the control group (P<0.05). These results indicated that the pulmonary function was impaired in the COPD rats. Lungs from rats in the COPD group displayed more lesions. The CT analysis results showed that the transparency was increased in the rats exposed to the cigarette smoke when compared with that in the healthy rats (Figure 1A). In addition, hyperplasia of ciliated columnar epithelial cells, loss of cilia, thinning of alveolar walls, bronchial effusion, infiltration of inflammatory cells, enlargement of alveolus cavity, and generation of pulmonary bulla were observed in the rats exposed to cigarette smoke (Figure 1B). All these results indicated that the COPD model was successfully developed.
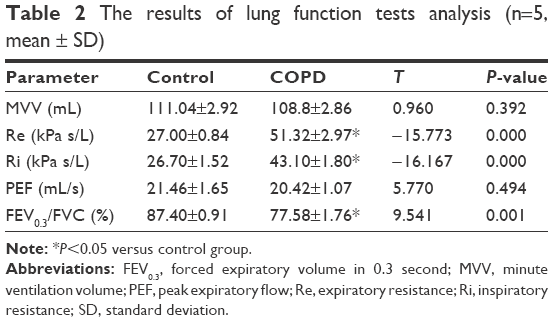 | Table 2 The results of lung function tests analysis (n=5, mean ± SD) |
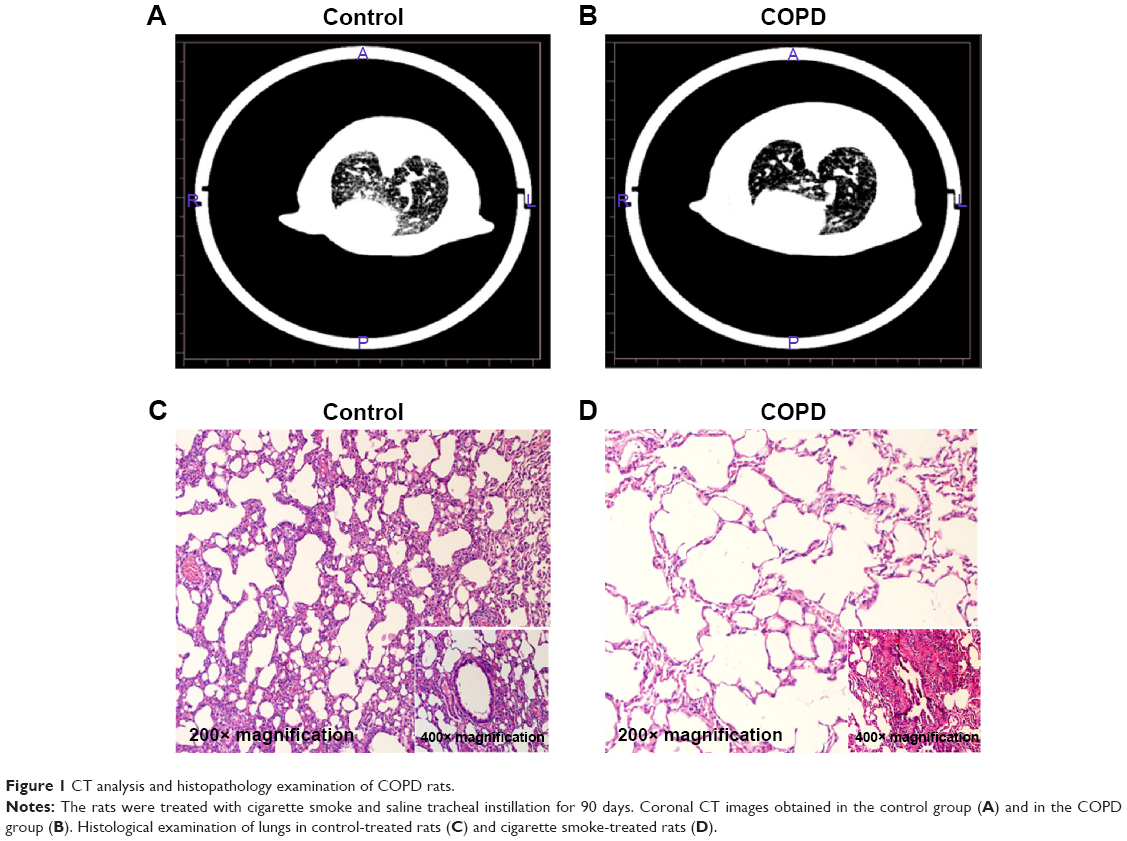 | Figure 1 CT analysis and histopathology examination of COPD rats. |
Differentially expressed miRNAs in AMs of COPD rats
MiRNA expression profiling studies were conducted in AM samples using the miRCURY LNA™ microRNA Array (v.8.0). A total of 13 miRNAs in AMs were identified with a more than twofold change in the levels between the COPD and control groups (Figure 2A). Among all detected miRNAs with miRNA microarray, seven members were predicted to target TLR2 analyzed by miRBase, miRanda, and miRDB databases.
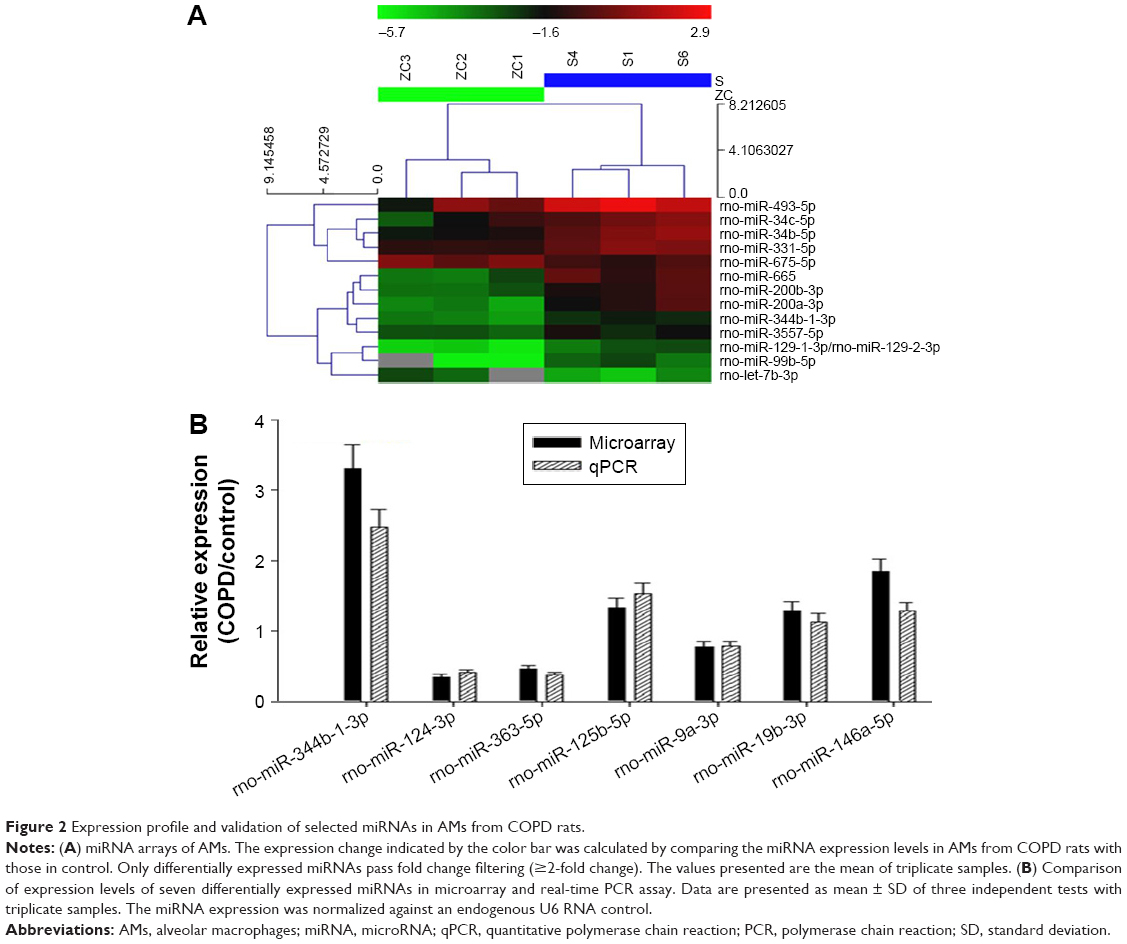 | Figure 2 Expression profile and validation of selected miRNAs in AMs from COPD rats. |
In order to confirm the results obtained from the miRNA microarray, the expression levels of seven miRNAs were analyzed by RT-PCR in the samples analyzed on the microarray. Consistent with the results from the miRNA microarray, the miRNAs, including miR-344b-1-3p, were differentially expressed between the COPD and control groups (Figure 2B). Furthermore, miR-344b-1-3p, miR-125b-5p, and miR-146a-5p levels in COPD AMs were significantly higher than those in control.
TLR2 is a direct target of miR-344b-1-3p
In reference to the significantly induced expression in COPD rats, there are no reports about the relationship between miR-344b-1-3p and TLR2; therefore, this miRNA was selected as the candidate for further studies. Based on the result of luciferase assay, only treatment with both miR-344b-1-3p inhibitor and psiCHECK™-2-TLR2-3′UTR decreased the luciferase activity in 293 cells (Figure 3), representing the direct interaction of miR-344b-1-3p on TLR2 gene transcription.
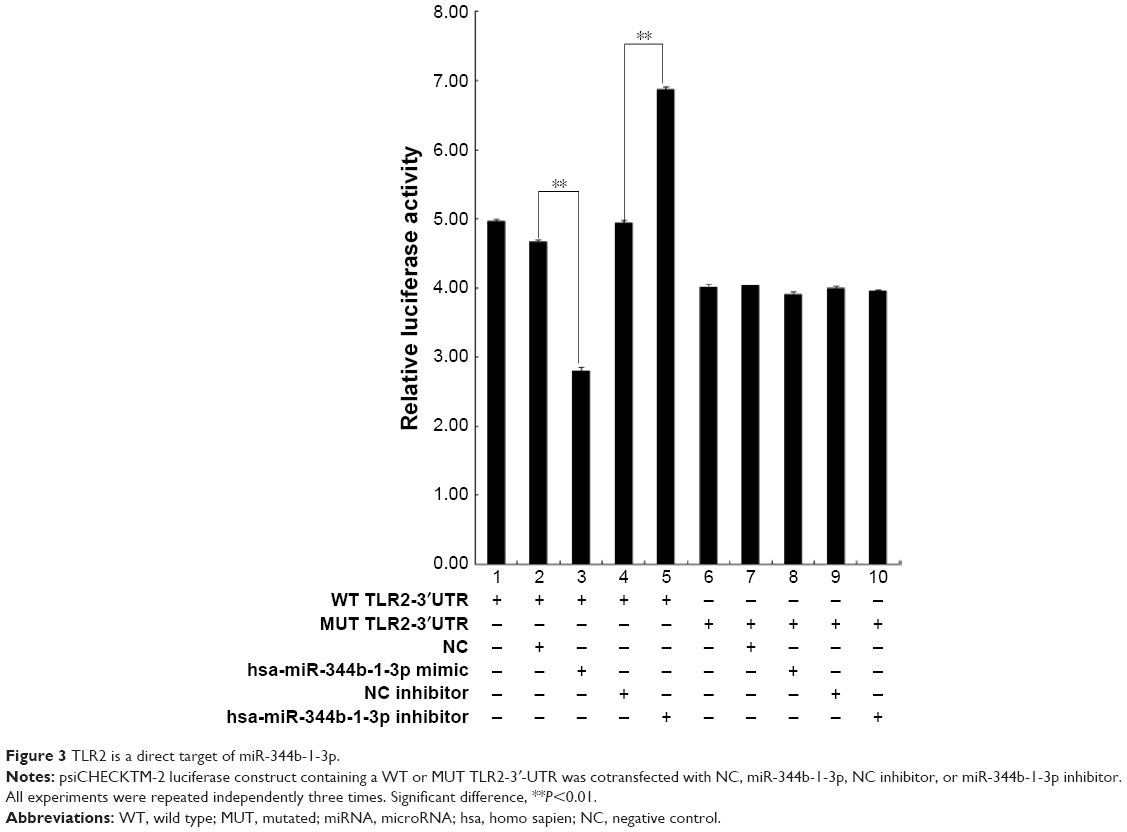 | Figure 3 TLR2 is a direct target of miR-344b-1-3p. |
Detection of miR-344b-1-3p in vitro by RT-PCR
To select the optimal CSE concentration of subsequent assays, the apoptosis of NR8383 cells after treatment with CSE of different concentrations was detected. As shown in Figure 4, NR8383 cell apoptosis rate in all the three CSE treated groups was higher than that in the control group, and the difference was statistically significant (P<0.05). The strongest effect to induce cell apoptosis was observed in 10% CSE group, which was selected for further experiments. Moreover, the expression of miR-344b-1-3p was analyzed by RT-PCR in NR8383 cells to explore the effect of CSE on miR-344b-1-3p expression. It was found that in NR8383 cells subjected to 5% and 10% CSE treatments, the levels of miR-344b-1-3p were upregulated when compared with the control group, and the differences were statistically significant (P<0.01) (Figure 5). However, when the concentration of CSE increased to 15%, the level of miR-344b-1-3p was dramatically decreased when compared with that in the 10% CSE group (Figure 5). Therefore, it was concluded by these findings that CSE could induce the activity of miR-344b-1-3p in NR8383 cells, which could be employed for the establishment of COPD condition in vitro. The strongest effect was observed when the concentration of CSE was 10%, which confirmed the results of flow cytometry.
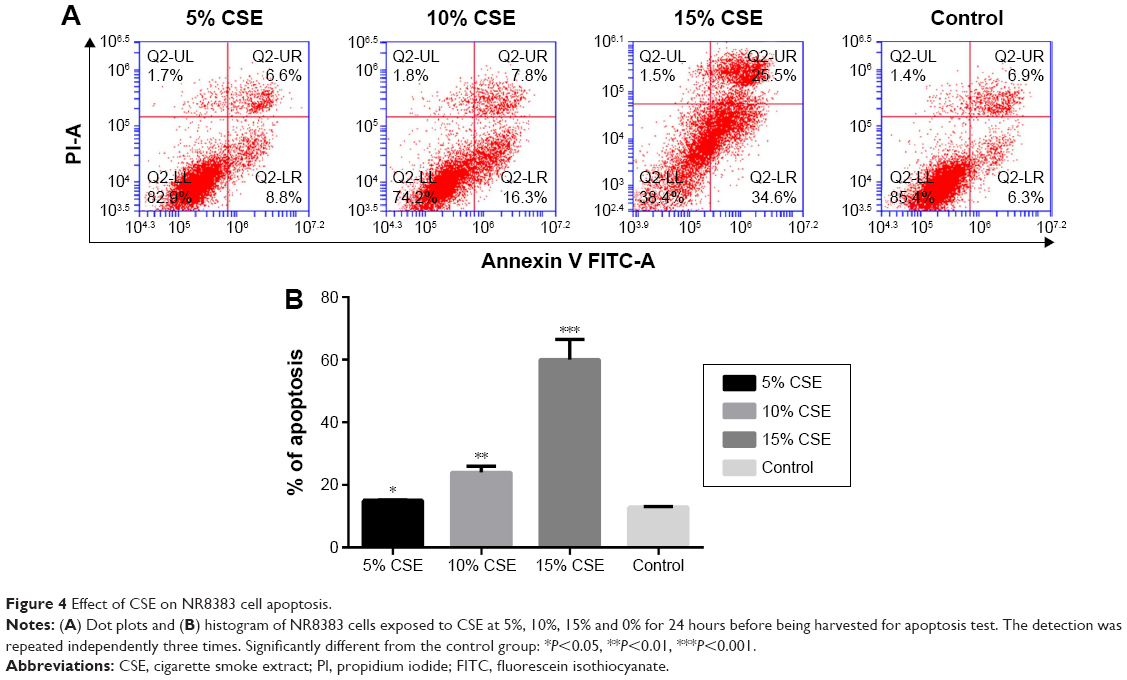 | Figure 4 Effect of CSE on NR8383 cell apoptosis. |
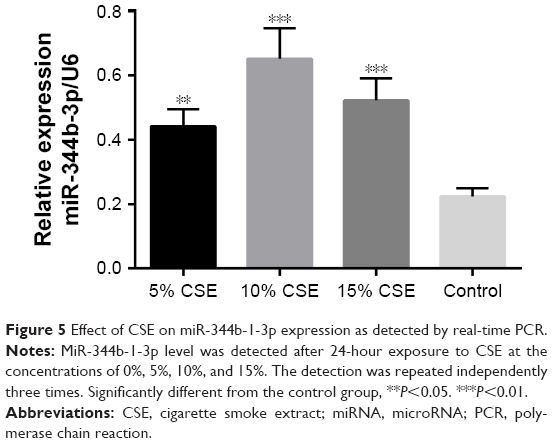 | Figure 5 Effect of CSE on miR-344b-1-3p expression as detected by real-time PCR. |
Effect of miR-344b-1-3p on TLR2 and downstream signaling transduction
As shown in Figure 6, the TLR2 level was increased after inhibiting miR-344b-1-3p. To determine the effect of miR-344b-1-3p on inflammatory responses of NR8383 cells induced by TLR2, the levels of TNF-α, IL-1β, and MIP-2 in supernatant were evaluated. Results of ELISA are shown in Figure 7A–C. The levels of TNF-α, IL-1β, and MIP-2 were significantly increased after inhibiting miR-344b-1-3p.
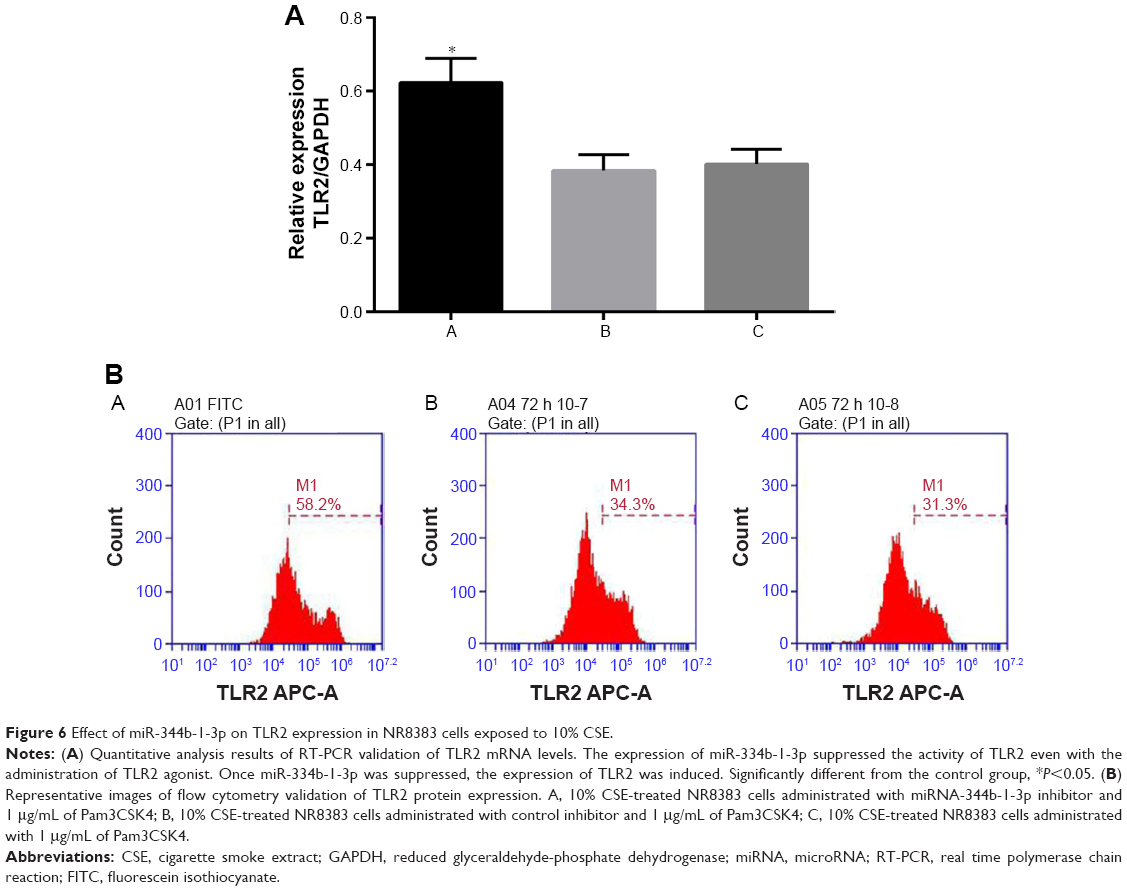 | Figure 6 Effect of miR-344b-1-3p on TLR2 expression in NR8383 cells exposed to 10% CSE. |
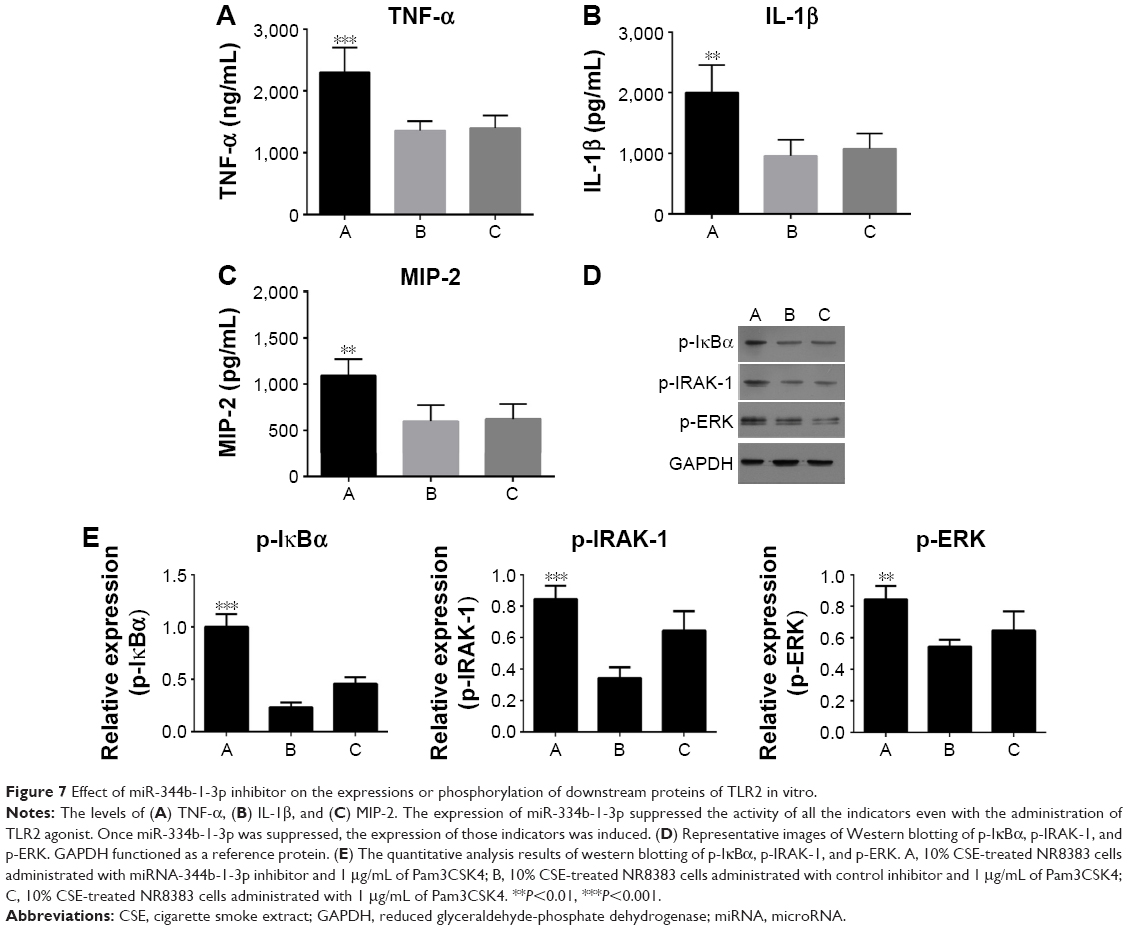 | Figure 7 Effect of miR-344b-1-3p inhibitor on the expressions or phosphorylation of downstream proteins of TLR2 in vitro. |
Furthermore, the phosphorylation levels of IκBα, IRAK-1, and ERK in NR8383 cell line pretreated with CSE were augmented following transfection of miR-344b-1-3p inhibitor (Figure 7D and E).
Discussion
It is well known that patients with severe COPD are at higher risk of developing IPA.16 This has been increasingly accepted to be associated with the decreased activity of AMs. TLR2 is the first pattern recognition receptor identified as the cell-activating receptor for Gram-negative lipopolysaccharides, as well as the receptor for both Gram-positive and Gram-negative bacteria, mycobacteria, Mycoplasma, and yeast.17 In addition to the functions mentioned earlier, recent study has shown that TLR2 deficiency induced a high risk for succumbing to IPA,9 which further confirms the key role of TLR2 in the innate immune system of mammalians. In this study, for the first time, we report that miRNA-344b-1-3p plays a pivotal role in the downregulation of TLR2 in lung AMs, indicating that decreased TLR2 expression induced by miRNAs might result in the high sensitivity of patients with COPD to Aspergillus species.
Emerging reports elaborate that the abnormal expression of some miRNAs is an important regulator of inflammatory responses and lung diseases.18–20 As it has been proved that TLR2 is the target of several miRNAs, including miR-105, miR-19, miR-1225-5P, miRNA-143, miRNA-154, and miR-UL112-3P, in a variety of cell lines,21–25 it is reasonable to observe the association of miRNAs with TLRs in COPD AMs. To fulfill such a purpose, in this study, COPD was induced in rats by smoke exposure. It was found that cigarette smoke exposure decreased FEV0.3/FVC and increased pulmonary transparency. In addition, rats exposed to cigarette smoke showed typical changes of COPD, including hyperplasia of ciliated columnar epithelial cells, thinning of alveolar walls, enlargement of alveolus cavity, and generation of pulmonary bulla. Using the analysis of microarray chips and the validation of RT-PCR in this study, we confirmed that production of miR-344b-1-3p in COPD AMs was significantly enhanced compared to that in the control group. Using dual luciferase assay, the targeting relationship between miR-344b-1-3p and TLR2 was confirmed as well. The above findings showed that miR-344b-1-3p played an important role in the post-transcriptional regulation of TLR2 and might be involved in the onset of IPA in COPD. As previously reported, miR-344b-1-3p is capable of activating Wnt/β-catenin signaling and inhibiting adipocyte differentiation, which is associated with renal disease.26 In this study, its involvement in immunity was observed for the first time.
To observe the functional effect of miR-344b-1-3p on the regulation of TLR2-related signaling, we examined whether TLR2 agonist, Pam3CSK4, could affect the function of miR-344b-1-3p in the activation of NF-κB pathway. NF-κB is usually in the inactive state by its inhibitory protein, IκB. The phosphorylation of IκBα allows NF-κB to initiate the functionality. It has been reported that NF-κB plays an important role in macrophage phagocytic activity and upregulates the production of proinflammatory cytokines.27 In this study, the data showed that the level of p-IκBα in the NR8383 cells exposed to CSE was upregulated after inhibition of miR-344b-1-3p, indicating that this miRNA was involved in the decreased NF-κB function by suppressing TLR2 expression. All the results suggested that miR-344b-1-3p decreased the phagocytic activity of AMs by suppressing NF-κB signaling pathway.
TNF-α, IL-1β, and MIP-2 are important proinflammatory factors expressed by AMs and play an important role in the defense of the body against foreign pathogens, such as viruses, bacteria, and fungi. It has been reported that TNF-α plays a critical role in defending against Aspergillus by promoting the production of some inflammatory cytokines to activate immune responses, inducing the activation and recruitment of neutrophils, as well as increasing the level of superoxide dismutase and lysosomal enzymes, to facilitate clearance of Aspergillus. IL-1β is critically involved in the differentiation, maturation, and recruitment of inflammation cells, contributing to the host response against Aspergillus. MIP-2 could recruit neutrophils in the airway to protect from Aspergillus infection. Therefore, production of TNF-α, IL-1β, and MIP-2 was examined in this study to monitor the effects of miR-344b-1-3p on the TLR2-mediated inflammation. After transfection with miR-344b-1-3p inhibitor and exposure to Pam3CSK4, the levels of TNF-α, IL-1β, and MIP-2 secreted by NR8383 cells were determined. The data revealed that miR-344b-1-3p inhibitor significantly increased TNF-α, IL-1β, and MIP-2 expressions, which suggested that the downexpression of TLR2 in AMs contributed to the reduced TNF-α, IL-1β, and MIP-2 expressions in COPD AMs. Based on the study of Yang et al,28 TLR2 is capable of initiating the generation of reactive oxygen species (ROS) via an ASK1/p38 MAPK/p47phox cascade. Once activated, downstream signaling pathways will finally lead to the production of proinflammatory factors, including TNF-α, IL-1β, and MIP-2,29,30 which might explain the mechanism through which miR-344b-1-3p decreased the defense activity of AMs.
A previous study suggested the important role of activated ERK in the defense against Aspergillus fumigatus by inducing the production of TNF-α through a TLR2-independent manner.31 However, we observed that the increased expression of TLR2 by miR-344b-1-3p inhibitor induced the activation of ERK. The inconsistency may be due to the different experiment systems between studies. In this study, NR8383 cells were exposed to CSE, while TLR2 and Myd88 knock out mice were used in the previous study. COPD induced by smoking not only suppressed the expression of TLR2 but also altered the biological property permanently. In this pathological status, the expression of TLR2 might play an important role in the defense against IPA. IRAK-1 is an important cytokine inducing the activation of NF-κB and proinflammatory responses.32 The increased p-IRAK-1 was observed after miR-344b-1-3p inhibition in this study. Taken together, it indicated that inhibition of TLR2 by the miRNA might play an important role in the pathogenesis of IPA by decreasing the activation of IRAK-1 and ERK. In addition, the impairments of smoking on the normal function of smokers are irreversible if proper treatments are not conducted. Therefore, patients with COPD suffer from inflammation and immune abnormalities even years after they quit smoking. However, whether these abnormalities are attributed to the dysexpression of miRNAs induced by smoking needs to be further explored with a long-term experiment.
Conclusion
Our data revealed that miR-344b-1-3p plays an important role in suppressing TLR2 expression and signaling, indicating that this miRNA functionally decreases the defense activity of AMs in COPD rats. TLR2 inhibition can impair host immune defenses against respiratory infections in smokers. This study suggested that miRNA-modulated TLR2 may be a preventive and therapeutic target for the treatment of IPA. However, further studies with COPD human participants will be required to comprehensively delineate the relative contribution of miRNAs to the sensitivity of COPD patients to Aspergillus.
Acknowledgment
This study was funded by the National Natural Science Foundation of People’s Republic of China (grant nos 81200025 and 81370713), the Sci-Tech Research Projects of Guangdong Province (grant no 2012A061400010), and the Natural Science Foundation of Guangdong Province (grant no 2014A030313597).
Disclosure
The authors report no conflicts of interest in this work.
References
Caramori G, Kirkham P, Barczyk A, Di Stefano A, Adcock I. Molecular pathogenesis of cigarette smoking-induced stable COPD. Ann N Y Acad Sci. 2015;1340:55–64. | ||
Sethi S, Muscarella K, Evans N, Klingman KL, Grant BJ, Murphy TF. Airway inflammation and etiology of acute exacerbations of chronic bronchitis. Chest. 2000;118(6):1557–1565. | ||
Hiemstra PS. Altered macrophage function in chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2013;10(suppl):S180–S185. | ||
Ader F, Nseir S, Le Berre R, et al. Invasive pulmonary aspergillosis in chronic obstructive pulmonary disease: an emerging fungal pathogen. Clin Microbiol Infect. 2005;11(6):427–429. | ||
Bulpa PA, Dive AM, Garrino MG, et al. Chronic obstructive pulmonary disease patients with invasive pulmonary aspergillosis: benefits of intensive care? Intensive Care Med. 2001;27(1):59–67. | ||
Green GM, Carolin D. The depressant effect of cigarette smoke on the in vitro antibacterial activity of alveolar macrophages. N Engl J Med. 1967;276(8):421–427. | ||
Hodge S, Hodge G, Scicchitano R, Reynolds PN, Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol. 2003;81(4):289–296. | ||
Kirkham PA, Spooner G, Rahman I, Rossi AG. Macrophage phagocytosis of apoptotic neutrophils is compromised by matrix proteins modified by cigarette smoke and lipid peroxidation products. Biochem Biophys Res Commun. 2004;318(1):32–37. | ||
Droemann D, Goldmann T, Tiedje T, Zabel P, Dalhoff K, Schaaf B. Toll-like receptor 2 expression is decreased on alveolar macrophages in cigarette smokers and COPD patients. Respir Res. 2005;6:68. | ||
von Scheele I, Larsson K, Dahlen B, et al. Toll-like receptor expression in smokers with and without COPD. Respir Med. 2011;105(8):1222–1230. | ||
Wu Y, Xu H, Yuan W, Zhang D, Guo Z, Huang W. The mechanism of up regulation of TLR2 in COPD rats. COPD. In press 2017. | ||
Wang G, Wang R, Strulovici-Barel Y, et al. Persistence of smoking-induced dysregulation of miRNA expression in the small airway epithelium despite smoking cessation. PLoS One. 2015;10(4):e0120824. | ||
De Smet EG, Mestdagh P, Vandesompele J, Brusselle GG, Bracke KR. Non-coding RNAs in the pathogenesis of COPD. Thorax. 2015;70(8):782–791. | ||
Naserzadeh P, Hosseini MJ, Mohamadzadeh Asl B, Pourahmad J. Toxicity mechanisms of cigarette smoke on mouse fetus mitochondria. Iran J Pharm Res. 2015;14(suppl):131–138. | ||
Prasad S, Sajja RK, Park JH, Naik P, Kaisar MA, Cucullo L. Impact of cigarette smoke extract and hyperglycemic conditions on blood-brain barrier endothelial cells. Fluids Barriers CNS. 2015;12:18. | ||
Meersseman W, Lagrou K, Maertens J, Van Wijngaerden E. Invasive aspergillosis in the intensive care unit. Clin Infect Dis. 2007;45(2):205–216. | ||
Dziarski R, Wang Q, Miyake K, Kirschning CJ, Gupta D. MD-2 enables toll-like receptor 2 (TLR2)-mediated responses to lipopolysaccharide and enhances TLR2-mediated responses to Gram-positive and Gram-negative bacteria and their cell wall components. J Immunol. 2001; 166(3):1938–1944. | ||
Zhang Y, Wang F, Lan Y, et al. Roles of microRNA-146a and microRNA-181b in regulating the secretion of tumor necrosis factor-alpha and interleukin-1beta in silicon dioxide-induced NR8383 rat macrophages. Mol Med Rep. 2015;12(4):5587–5593. | ||
Tomankova T, Petrek M, Kriegova E. Involvement of microRNAs in physiological and pathological processes in the lung. Respir Res. 2010;11:159. | ||
Oglesby IK, McElvaney NG, Greene CM. MicroRNAs in inflammatory lung disease – master regulators or target practice? Respir Res. 2010;11:148. | ||
Landais I, Pelton C, Streblow D, DeFilippis V, McWeeney S, Nelson JA. Human cytomegalovirus miR-UL112-3p targets TLR2 and modulates the TLR2/IRAK1/NFkappaB signaling pathway. PLoS Pathog. 2015;11(5):e1004881. | ||
Benakanakere MR, Li Q, Eskan MA, et al. Modulation of TLR2 protein expression by miR-105 in human oral keratinocytes. J Biol Chem. 2009;284(34):23107–23115. | ||
Philippe L, Alsaleh G, Suffert G, et al. TLR2 expression is regulated by microRNA miR-19 in rheumatoid fibroblast-like synoviocytes. J Immunol. 2012;188(1):454–461. | ||
Guo H, Chen Y, Hu X, Qian G, Ge S, Zhang J. The regulation of toll-like receptor 2 by miR-143 suppresses the invasion and migration of a subset of human colorectal carcinoma cells. Mol Cancer. 2013;12:77. | ||
Sallustio F, Serino G, Costantino V, et al. miR-1915 and miR-1225-5p regulate the expression of CD133, PAX2 and TLR2 in adult renal progenitor cells. PLoS One. 2013;8(7):e68296. | ||
Jiang CB, Wei MG, Tu Y, et al. Triptolide attenuates podocyte injury by regulating expression of miRNA-344b-3p and miRNA-30b-3p in rats with adriamycin-induced nephropathy. Evid Based Complement Alternat Med. 2015;2015:107814. | ||
Zhang DM, Bao YL, Yu CL, Wang YM, Song ZB. Cripto-1 modulates macrophage cytokine secretion and phagocytic activity via NF-kappaB signaling. Immunol Res. 2016;64(1):104–114. | ||
Yang CS, Shin DM, Lee HM, et al. ASK1-p38 MAPK-p47phox activation is essential for inflammatory responses during tuberculosis via TLR2-ROS signalling. Cell Microbiol. 2008;10(3):741–754. | ||
Barreiro E, de la Puente B, Minguella J, et al. Oxidative stress and respiratory muscle dysfunction in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;171(10):1116–1124. | ||
Zuo L, Lucas K, Fortuna CA, Chuang CC, Best TM. Molecular regulation of toll-like receptors in asthma and COPD. Front Physiol. 2015;6:312. | ||
Dubourdeau M, Athman R, Balloy V, et al. Aspergillus fumigatus induces innate immune responses in alveolar macrophages through the MAPK pathway independently of TLR2 and TLR4. J Immunol. 2006;177(6):3994–4001. | ||
Meier A, Kirschning CJ, Nikolaus T, Wagner H, Heesemann J, Ebel F. Toll-like receptor (TLR) 2 and TLR4 are essential for Aspergillus-induced activation of murine macrophages. Cell Microbiol. 2003;5(8):561–570. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.