")
Back to Journals » OncoTargets and Therapy » Volume 9
MiR-29c inhibits cell growth, invasion, and migration of pancreatic cancer by targeting ITGB1
Authors Lu Y, Hu J, Sun W, Li S, Deng S, Li M
Received 20 July 2015
Accepted for publication 9 November 2015
Published 30 December 2015 Volume 2016:9 Pages 99—109
DOI https://doi.org/10.2147/OTT.S92758
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Yebin Lu, Juanjuan Hu, Weijia Sun, Shengyu Li, Shuangya Deng, Ming Li
Department of General Surgery, Xiangya Hospital, Central South University, Changsha, People’s Republic of China
Abstract: MiR-29c is frequently dysregulated in many cancers; however, the roles of miR-29c in pancreatic cancer (PC) and underlying mechanisms remain poorly understood. In this study, we investigated the role of miR-29c in PC. Using quantitative real-time polymerase chain reaction, we demonstrated that miR-29c was frequently downregulated in clinical PC tissues and cell lines. Overexpression of miR-29c significantly inhibited the proliferation, migration, and invasion of PC cells in vitro, which demonstrated that miR-29c acts as a tumor suppressor in PC cells. Further analysis revealed that ITGB1 is one of the functional target genes of miR-29c, and knockdown of ITGB1 inhibited the proliferation, migration, and invasion of PC cells, which was similar to the effects of overexpression of miR-29c. Taken together, our results highlight the significance of miR-29c–ITGB1 interaction in the development and progression of PC.
Keywords: miR-29c, tumor suppressor, pancreatic cancer, ITGB1
Introduction
Pancreatic cancer (PC) is one of the most aggressive cancers and the fourth-leading cause of cancer-related deaths.1,2 The 5-year survival rate for PC patients is only 5%, mainly due to late diagnosis and no effective therapies.3–5 Better understanding of the molecular mechanisms underlying PC development will be useful to identify novel treatment strategies and improve current therapeutics.
MicroRNAs (miRNAs) are a class of small noncoding regulatory RNAs, which can broadly regulate target genes by binding to a complementary sequence in their 3′UTR6,7 and have important roles in the regulation of principal cellular processes, including cell proliferation, differentiation, and migration.8 Increasing evidence has revealed that dysregulation of miRNAs occurs in a variety of cancers, including PC.9 MiR-29c is a member of the miR-29 family, and has been reported to be deregulated in many different types of cancer.10–12 MiR-29c is frequently silenced or downregulated in many kinds of cancers, including hepatocellular cancer,13,14 nasopharyngeal cancer,15 glioma,16,17 gastric cancer,18,19 bladder cancer,20 and colorectal cancer,21 and in those cancers, miR-29c usually has tumor-suppressive effects, while the expression levels of miR-29c in osteosarcoma tissues and patients’ sera are all significantly higher than those in normal controls.22 In PC, recent reports have shown that miR-29c suppresses PC liver metastasis in a nude mouse model and affects patient survival,23 and reduction of miR-29c enhances PC cell migration and stem cell-like phenotype.24 However, the roles of miR-29c in PC and underlying mechanisms remain poorly understood.
In the present study, we found that miR-29c was significantly downregulated in clinical PC samples and cell lines. Ectopic expression of miR-29c inhibited cell proliferation, migration, and invasion in PC cells. Furthermore, we found that ITGB1 is one of the direct target genes of miR-29c, and confirmed that miR-29c might exerts its effect on the inhibition of cell growth, migration, and invasion by downregulating ITGB1 in PC cells. Our findings suggest that miR-29c may function as a tumor suppressor in PC.
Materials and methods
Patient samples and cell lines
A total of 23 human PC tissues and their matched adjacent pancreatic tissues were obtained during surgery at Xiangya Hospital between July 2012 and December 2014. All tissues were immediately snap-frozen in liquid nitrogen and stored at −80°C until used. None of the patients received chemotherapy or radiotherapy before the surgical excision, and specimen information is provided in Table 1. The use of these samples was approved by the Clinical Research Ethics Committee of Central South University (Changsha, People’s Republic of China [PRC]). Written informed consent was obtained from all subjects. Four PC cell lines (SW1990, AsPC-1, BxPC-3, and PANC-1) and normal human pancreatic duct epithelial (HPDE) cells were purchased from the Chinese Center for Type Culture Collection (Shanghai, PRC) and cultured in Roswell Park Memorial Institute 1640 medium supplemented with 10% fetal bovine serum and antibiotics (100 U/mL penicillin and 100 mg/mL streptomycin sulfate).
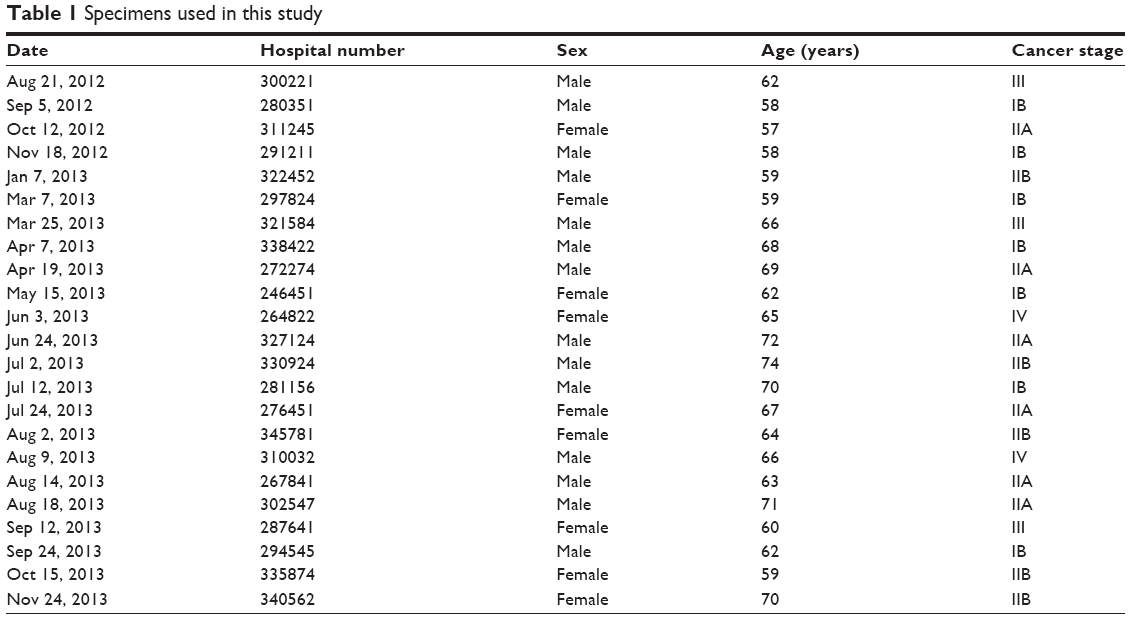 | Table 1 Specimens used in this study |
RNA isolation and quantitative real-time PCR
Total RNA from cultured cells or fresh surgical tissues was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Quantitative real-time reverse-transcription polymerase chain reaction (qRT-PCR) was performed using the All-in-One™ miRNA qRT-PCR detection kit (GeneCopoeia, Rockville, MD, USA) for miR-29c and small nuclear RNA U6, which was used as an endogenous control. The relative expressions of mRNA were detected by SYBR green qPCR assay (Bio-Rad Laboratories Inc, Hercules, CA, USA), and β-actin was used as an endogenous control. All qRT-PCR was performed on ABI 7500 thermocycler (Thermo Fisher Scientific, Waltham, MA, USA). Primers used were as follows: β-actin, 5′-AGTGTGACGTGGACATCCGCAAAG-3′ (forward), 5′-ATCCACATCTGCTGGAAGGTGGAC-3′ (reverse); ITGB1, 5′-AATGTAACCAACCGTAGC-3′ (forward), 5′-CAGGTCCATAAGGTAGTAGA-3′ (reverse). The specific primers for miR-29c (HmiRQP0376) and U6 (HmiRQP9001) were purchased from RiboBio (Guangzhou, PRC). The relative expression levels were calculated using the 2−ΔΔct method.
Western blot
Cells were lysed using the protein-extraction reagent RIPA (Beyotime Biotechnology, Nantong, PRC) supplemented with protease inhibitors, and approximately 50 μg protein extractions were separated by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes (GE Healthcare, Little Chalfont, UK), and incubated with specific primary antibodies against ITGB1 (18638; Sigma-Aldrich Co, St Louis, MO, USA), MMP2 (ab110186; Abcam PLC, Cambridge, UK), MMP9 (ab76003; Abcam), E-cadherin (ab76055; Abcam), and β-actin (Santa Cruz Biotechnology Inc, Dallas, TX, USA), then probed with horseradish peroxidase-conjugated secondary antibodies. Signals were visualized with an Enhanced Chemiluminescence Plus kit (GE Healthcare).
miRNA mimics, siRNAs, plasmid construction, and transfection
MiR-29c mimics, scrambled mimics, and corresponding inhibitors were purchased from RiboBio. ITGB1-specific small interfering RNAs (siRNA-ITGB1) and the control small interfering RNAs (siRNA-control) were purchased from GeneChem (Shanghai, PRC). The coding sequence of ITGB1 was amplified and cloned into a pcDNA3.1 vector to generate ITGB1-expression vectors, and the empty pCDNA3.1 vector was used as control. Primers used for the ITGB1 coding sequence were 5′-GGATCCATGAATTTACAACCAATTTTC-3′ (forward) and 5′-GAATTCTTAGAGACCAGCTTTACGTC-3′ (reverse). Cells were cultured to approximately 80% of the plates, then transfection of miRNA mimics (50 nM) and siRNAs was performed using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocol.
Luciferase-reporter assay
The wild-type 3′UTR of ITGB1 was amplified with the primers 5′-CCGCTCGAGCTTAGATAAAAGAACCGAG (Xho I)-3′ (forward) and 5′-GTTGCGGCCGCACTAAAGGCACTTAAAACA (Not I)-3′ (reverse), and ligated into the psiCheck-2 reporter vector (Promega Corporation, Fitchburg, WI, USA). Site-directed mutagenesis of the miR-29c-binding sequence in the 3′UTR of ITGB1 was performed using the QuikChange™ Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA). Cells were seeded into 24-well plates for 24 hours, then cotransfected with 0.3 μg of the reporter vector and 40 nM miR-29c mimics or scrambled mimics using Lipofectamine 2000. Cells were harvested 48 hours after transfection, and luciferase activity was examined using a dual-luciferase reporter assay system (Promega) according to the manufacturer’s protocol. All experiments were performed in triplicate.
Cell-proliferation assays
MTT assays were performed to evaluate cell proliferation. Briefly, the PC cells (5,000 per well) were plated in 96-well plates and allowed to grow for 24, 48, and 72 hours, then were assessed by a colorimetric assay using MTT solution (10 mg/mL) at 570 nm. All the experiments were performed three times.
Cell-cycle analysis by flow cytometry
The cells were harvested by trypsinization, washed twice with cold phosphate-buffered saline (PBS), and fixed in 70% ethanol overnight at 4°C. The fixed cells were then washed with cold PBS, resuspended in staining solution (50 μg/mL of propidium iodide, 1 mg/mL of RNase A, 0.1% Triton X-100 in PBS), and incubated for 30 minutes at 4°C, then analyzed with a flow cytometer (Beckman Coulter Inc, Brea, CA, USA).
Cell-migration and -invasion assays
Cell migration was assessed by wound-healing assays. In brief, cells were seeded in six-well plates and cultured to 100% confluence. Using a sterile pipette tip, wounds were generated and the cells cultured for 48 hours. Then, wound closure was assessed by Scion Image software (Scion Corporation, Frederick, MD, USA). For cell-invasion assays, Matrigel invasion chambers (BD Biosciences, San Jose, CA, USA) were used to assess cell-invasion ability. Briefly, 1×105 cells were seeded in the upper chamber with media containing 0.1% fetal bovine serum, while the lower chamber was filled with media with 10% fetal bovine serum. After incubation for 48 hours, noninvading cells were removed with cotton swabs, and cells that migrated through the membrane were fixed with 70% alcohol, stained with crystal violet imaged, and counted under a microscope (Olympus, Tokyo, Japan).
Statistical analysis
All statistical analyses were carried out with SPSS 19.0 software. Data are presented as means ± standard deviation. Statistical significance between two groups was analyzed using Student’s t-test. The correlation between miR-29c and ITGB1 expression was analyzed using Spearman’s correlation analysis. P<0.05 was considered significant.
Results
MiR-29c is significantly downregulated in PC tissues and cell lines
We first evaluated the expression levels of miR-29c in pancreatic tumors and paired adjacent tissues (n=23). qRT-PCR showed that the expression levels of miR-29c were significantly decreased in tumor tissues compared with those in adjacent tissues (Figure 1A). Moreover, we examined miR-29c-expression levels in four PC cell lines (SW1990, AsPC-1, BxPC-3, and PANC-1), and found the relative expression levels of miR-29c in these PC cells were all significantly lower than in the HPDE cells (Figure 1B). Therefore, these data suggest that decreased miR-29c may be associated with pancreatic carcinogenesis.
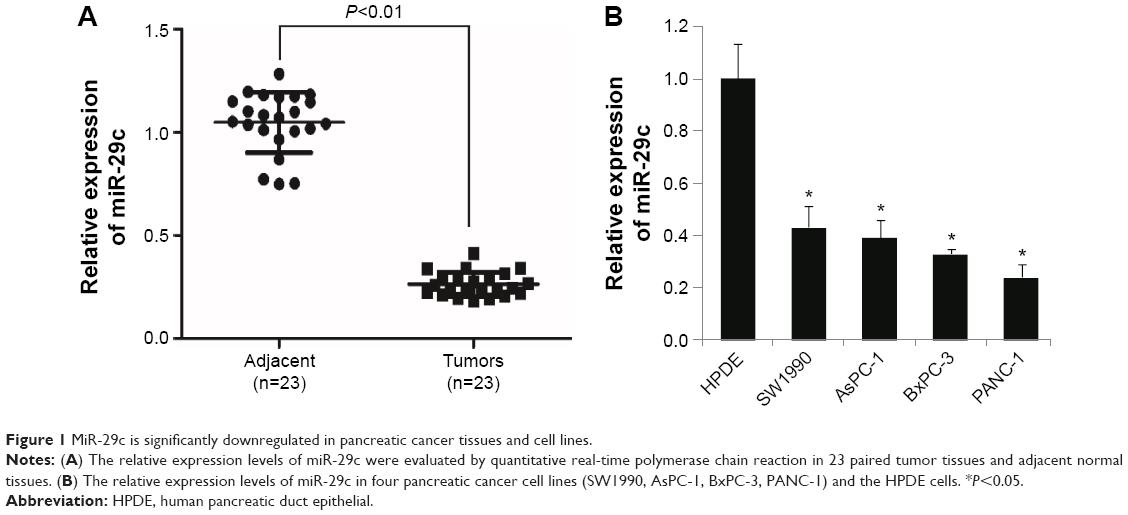 | Figure 1 MiR-29c is significantly downregulated in pancreatic cancer tissues and cell lines. |
MiR-29 inhibits proliferation by inducing cell-cycle arrest in PC cells
In order to identify the effects of miR-29c on PC cells, miR-29c mimics, anti-miR-29c mimics, and corresponding negative controls were transfected to BxPC-3 and PANC-1 cells, qRT-PCR revealed that miR-29c mimics significantly increased, while anti-miR-29c mimics decreased the miR-29c expression in BxPC-3 and PANC-1 cells (Figure 2A). MTT assays then revealed that overexpression of miR-29c inhibited the proliferation of BxPC-3 and PANC-1 cells, while downregulation of miR-29c promoted the proliferation of BxPC-3 and PANC-1 cells compared to the controls (Figure 2B). To investigate the mechanisms of miR-29c inhibition on the proliferation of PC cells, we analyzed the cell cycle using flow cytometry. As shown in Figure 2C, miR-29c significantly increased the proportion of G1 phases and reduced the proportion of G2 and S phases in BxPC-3 and PANC-1 cells compared to the controls, and the miR-29c inhibitor resulted in the opposite phenomenon.
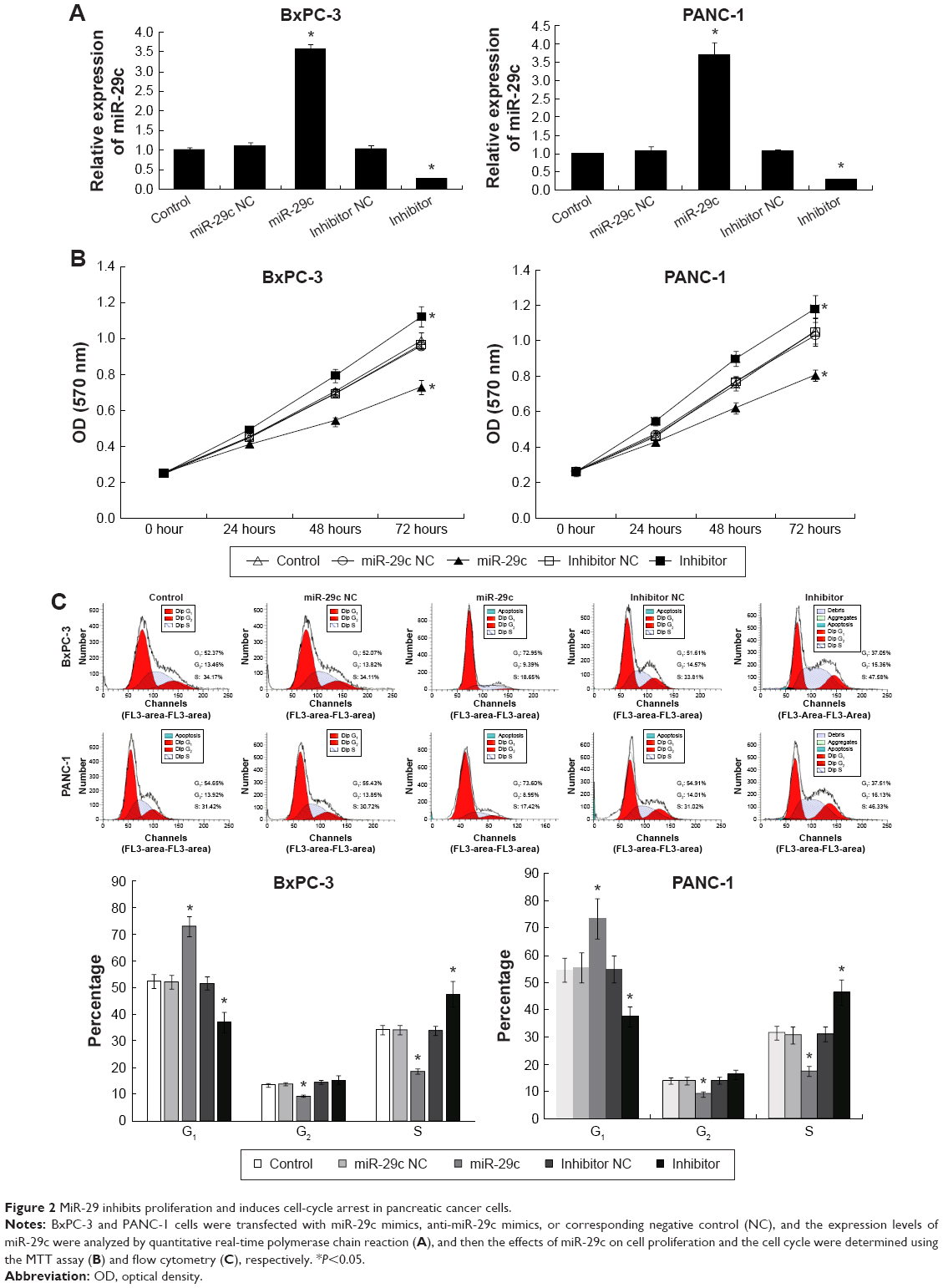 | Figure 2 MiR-29 inhibits proliferation and induces cell-cycle arrest in pancreatic cancer cells. |
MiR-29 inhibits the invasion and migration of PC cells and the expression of MMP2, MMP9, and E-cadherin
The effects of miR-29c on PC cell invasion and migration were investigated by transwell assays with Matrigel and wound-healing assays, respectively. We found that overexpression of miR-29c in BxPC-3 and PANC-1 cells significantly inhibited cell invasion and migration ability, respectively (Figure 3). In contrast, when endogenous miR-29c was inhibited by anti-miR-29c mimics, cell invasion and migration increased (Figure 3). These results revealed that miR-29c mediated the cell invasion and migration of PC cells. To investigate the mechanisms of miR-29c inhibition on the invasion and migration of PC cells, we analyzed the effects of miR-29c on the expressions of migration- and invasion-related genes: MMP2, MMP9, and E-cadherin. This revealed that miR-29c mimic-transfected BxPC-3 and PANC-1 cells had decreased expression levels of MMP2 and MMP9 and increased expression level of E-cadherin compared to the controls (Figure 4).
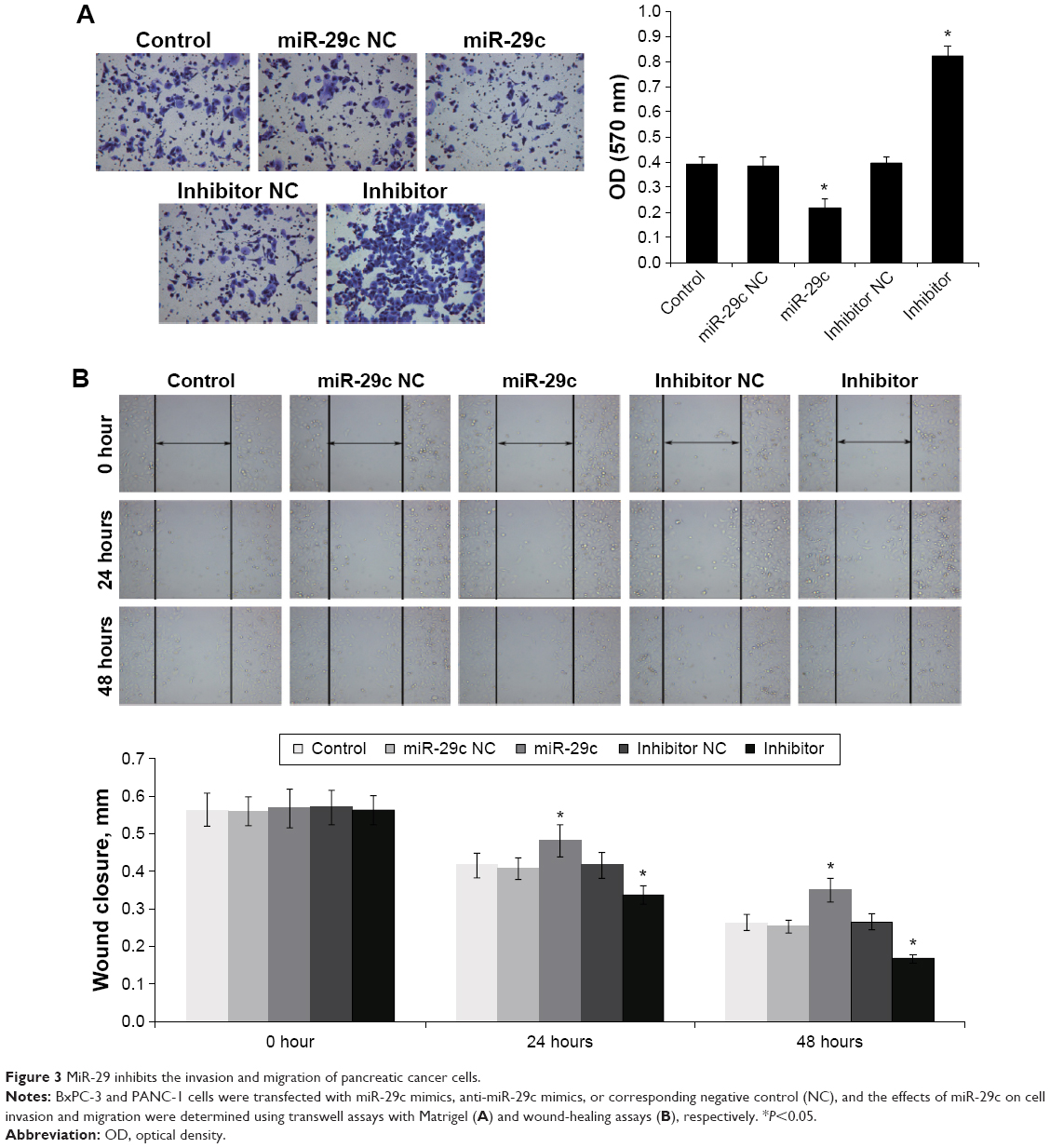 | Figure 3 MiR-29 inhibits the invasion and migration of pancreatic cancer cells. |
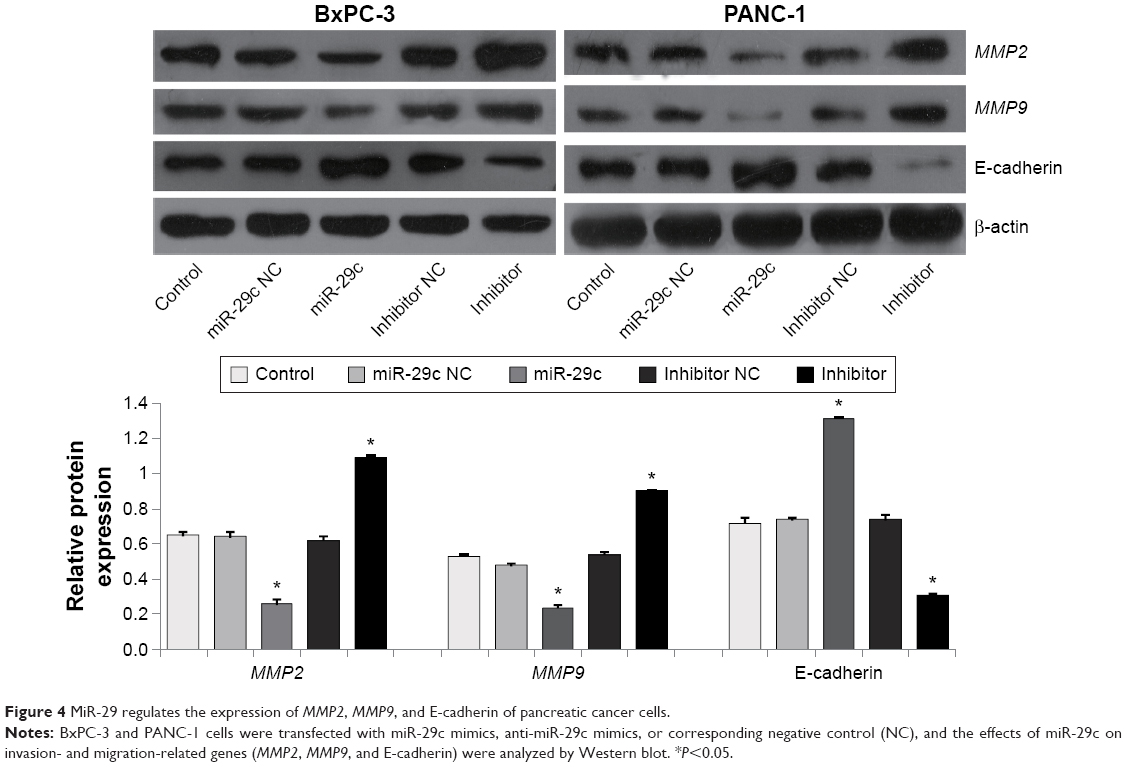 | Figure 4 MiR-29 regulates the expression of MMP2, MMP9, and E-cadherin of pancreatic cancer cells. |
MiR-29c directly inhibits ITGB1 expression
To elucidate the molecular mechanism underlying miR-29c exerting its inhibitory effects on the proliferation, migration, and invasion of PC cells, we predicted potential targets of miR-29c using the TargetScan, miRanda, and PicTar online tools, and identified ITGB1 as one of the potential targets. Luciferase-reporter assays were performed to validate if miR-29c could directly target potential binding sites in the 3′UTR of ITGB1. A fragment of wild-type or mutant 3′UTR of ITGB1 was cloned into the psiCheck-2 reporter vector (Figure 5A). Our results showed that miR-29c mimics significantly decreased the firefly luciferase activity of the vector with the wild-type 3′UTR of ITGB1 cells, but had no significant effect on the vector with the mutant 3′UTR of ITGB1 (Figure 5A). Furthermore, qRT-PCR analysis revealed that miR-29c mimics significantly reduced the expression level of ITGB1, and anti-miR-29c mimics increased the expression level of ITGB1 in BxPC-3 and PANC-1 cells (Figure 5B). Moreover, we also examined the expression levels of ITGB1 in pancreatic tumors and paired adjacent tissues, and found a significant inverse correlation between expression levels of miR-29c and ITGB1 mRNAs in PC tissues (Figure 5C). These results demonstrated that miR-29c can directly inhibit ITGB1 expression in pancreatic cells.
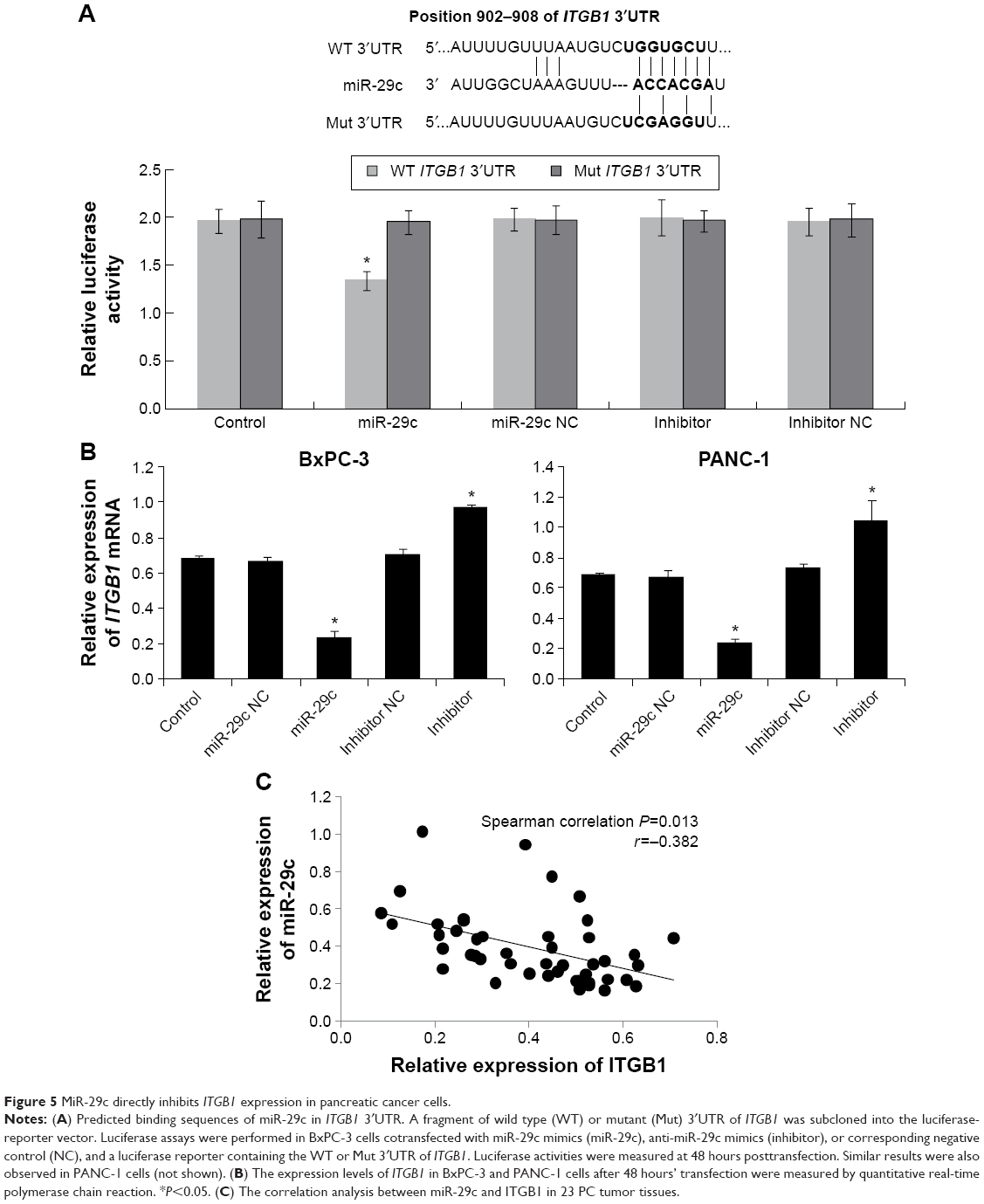 | Figure 5 MiR-29c directly inhibits ITGB1 expression in pancreatic cancer cells. |
To determine further whether miR-29c exerts its inhibitory effect through downregulation of ITGB1, we transfected ITGB1-expression plasmids lacking 3′UTR to rescue ITGB1 expression in miR-29c mimic-treated PANC-1 cells. As shown in Figure 6, ITGB1 overexpression significantly attenuated miR-29c-induced inhibition on cellular migration and invasion in PANC-1 cells. Taken together, these results suggest that ITGB1 is a functional target of miR-29c in PC cells.
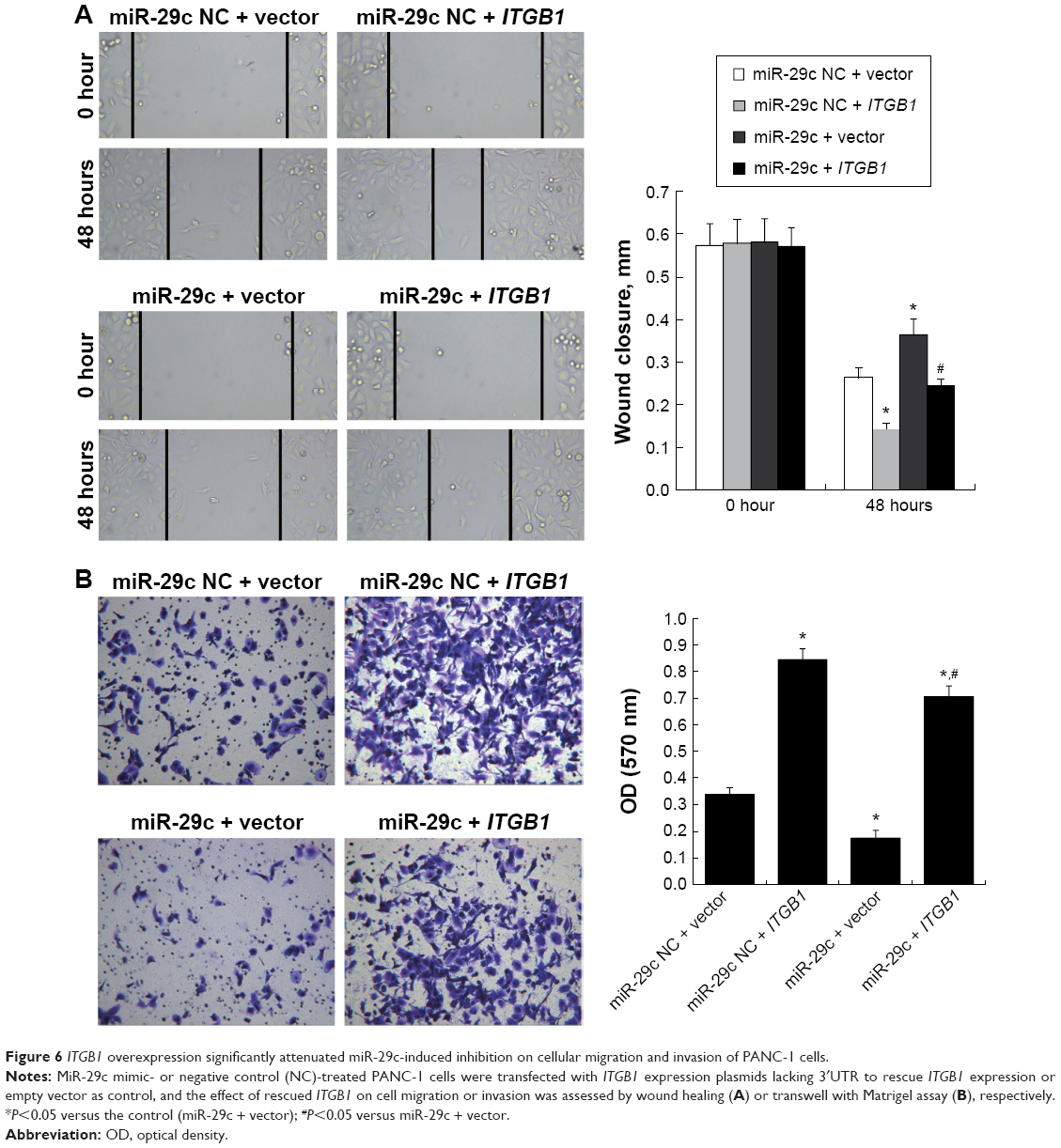 | Figure 6 ITGB1 overexpression significantly attenuated miR-29c-induced inhibition on cellular migration and invasion of PANC-1 cells. |
Knockdown of ITGB1 suppresses the proliferation, invasion, and migration of PANC-1 cells
To determine further the role of ITGB1 in PC cells, we knocked down ITGB1 in PANC-1 cells by ITGB1-specific siRNAs (siRNA-ITGB1) (Figure 7A). We found that ITGB1 knockdown significantly inhibited cell proliferation, invasion, and migration of PANC-1 cells (Figure 7B–E), which was similar to the effects of miR-29c overexpression. Moreover, knockdown of ITGB1 also resulted in decreased expression levels of MMP2 and MMP9 and increased expression of E-cadherin compared to that of the controls (Figure 7A).
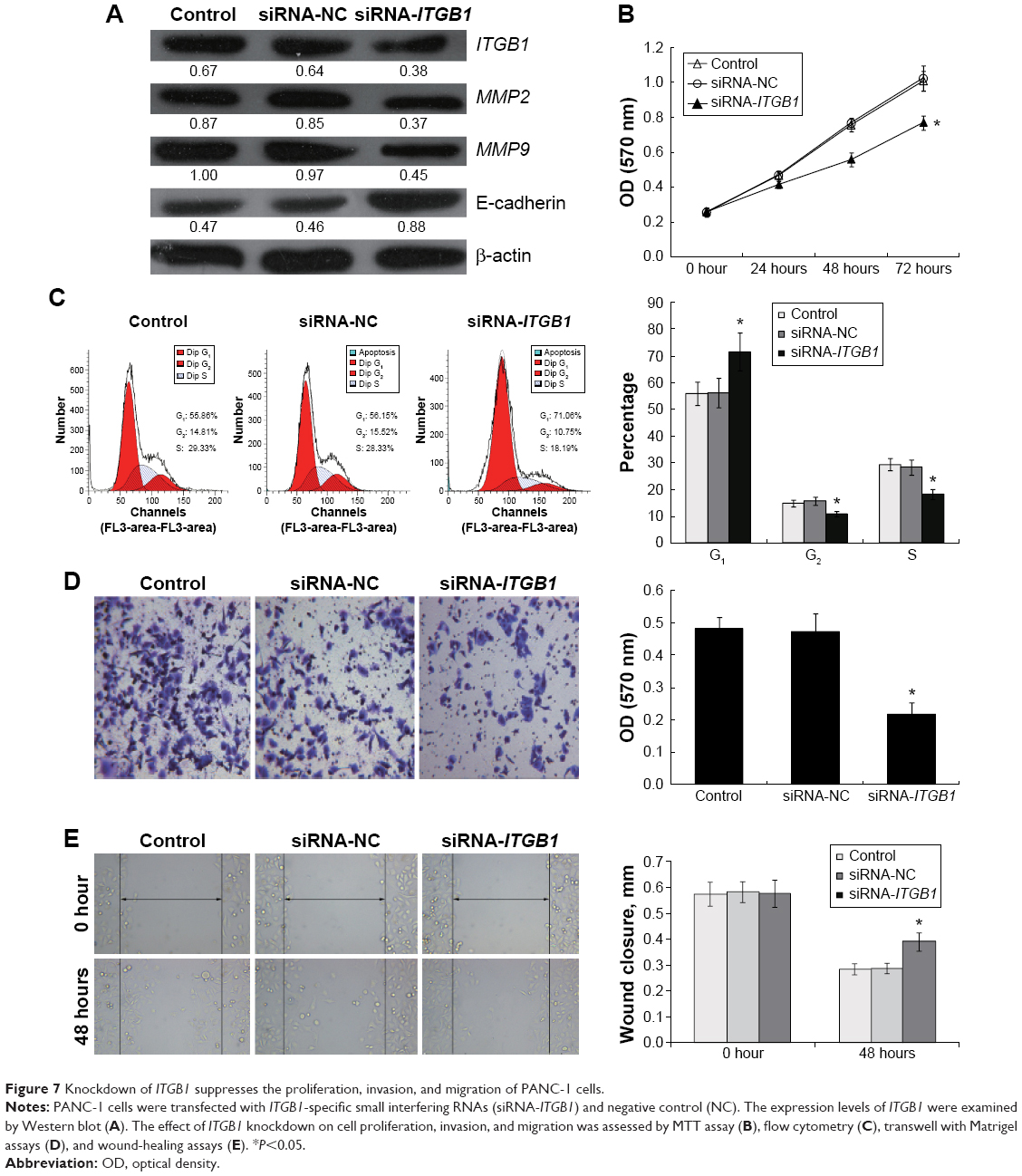 | Figure 7 Knockdown of ITGB1 suppresses the proliferation, invasion, and migration of PANC-1 cells. |
Discussion
In the present study, we demonstrated that miR-29c expression is decreased in clinical PC tumors compared with adjacent normal pancreatic tissues. The expression levels of miR-29c in four PC cell lines were also significantly downregulated. Moreover, we confirmed that ITGB1 is a functional target of miR-29c in PC cells. Therefore, these findings demonstrated that miR-29c might be a tumor-suppressor gene in PC cells, and abnormal alteration of miR-29c–ITGB1 interaction might contribute to PC development.
To validate our hypothesis, we investigated the functional roles of miR-29c in two PC cell lines: BxPC-3 and PANC-1. MTT assays and cell-cycle analysis using flow cytometry demonstrated that miR-29c inhibited cell proliferation, at least partially, by inducing cell-cycle arrest. Wound-healing and Matrigel transwell assays revealed that overexpression of miR-29c could significantly inhibit PC cell migration and invasion ability. These observations were consistent with previous findings that overexpression of miR-29c suppressed cancer cell growth, migration, and invasion in many kinds of cancers,12,13,15,16,19–21 but inconsistent in osteosarcoma, in which the expression levels of miR-29 family members (miR-29a, miR-29b, and miR-29c) in osteosarcoma tissues and patients’ sera were all significantly higher than normal controls, suggesting miR-29 family members as oncogenic miRNAs.22 These data suggest that the roles of miR-29c might vary in different kinds of cancers.
A given miRNA affects cancer development through posttranscriptional regulation of its target genes.25,26 Therefore, the establishment of an miRNA–target interaction may help us to better understand the molecular mechanism underlying cancer progression and provide potential therapeutic targets for the clinical treatment of cancers. In this study, we predicted and further validated that ITGB1 was a direct target of miR-29c by luciferase-reporter assays in PC cells. qRT-PCR and Western blot analysis revealed that ITGB1 was significantly downregulated by miR-29c overexpression at both the mRNA and protein levels. Functional studies also revealed that knockdown of ITGB1 inhibited cell migration and invasion of PANC-1 cells. Although ITGB1 has been demonstrated to be the target gene of miR-29c in gastric cancer cells,18 this is the first validation of the function of miR-29c–ITGB1 interaction in the progression of PC.
Integrins are cell-adhesion molecules, and aberrant upregulation has been associated with tumor progression, particularly during invasion, migration, and metastasis.27,28 As for ITGB1, it has been reported to promote cell growth, migration, and tumor metastasis in many types of human cancers, including PC,29–33 and thus ITGB1 is a therapeutic target for the treatment of these cancers. For example, a monoclonal antibody against ITGB1 was used to block hepatocellular cancer cell invasion,34 and knockdown of ITGB1 by lentivirus-based RNA interference inhibited PC cell growth and migration in vitro, as well as tumor growth and metastasis in vivo.31 Our study showed that targeted inhibition of ITGB1 by miR-29c introduction might be a potential strategy for the treatment of certain ITGB1-related cancers.
In conclusion, our findings revealed that miR-29c was frequently downregulated in PC, and demonstrated the tumor-suppressive roles of miR-29c in inhibiting the proliferation and migration and invasion of PC cells, at least partially by negatively regulating ITGB1, which suggests that miR-29c might be applied as a potential prognostic biomarker and inhibitor in PC.
Disclosure
The authors report no conflicts of interest in this work.
References
Michaud DS. Epidemiology of pancreatic cancer. Minerva Chir. 2004;59(2):99–111. | ||
Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol. 2010;7(3):163–172. | ||
Cameron JL, Riall TS, Coleman J, Belcher KA. One thousand consecutive pancreaticoduodenectomies. Ann Surg. 2006;244(1):10–15. | ||
Ducreux M, Boige V, Malka D. Treatment of advanced pancreatic cancer. Semin Onco. 2007;34(2 Suppl 1):S25–S30. | ||
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59(4):225–249. | ||
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. | ||
Engels BM, Hutvagner G. Principles and effects of microRNA-mediated post-transcriptional gene regulation. Oncogene. 2006;25(46):6163–6169. | ||
Calin GA, Croce CM. MicroRNA-cancer connection: the beginning of a new tale. Cancer Res. 2006;66(15):7390–7394. | ||
Reddy KB. MicroRNA (miRNA) in cancer. Cancer Cell Int. 2015;15:38. | ||
Jiang H, Zhang G, Wu JH, Jiang CP. Diverse roles of miR-29 in cancer (review). Oncol Rep. 2014;31(4):1509–1516. | ||
Schmitt MJ, Margue C, Behrmann I, Kreis S. MiRNA-29: a microRNA family with tumor-suppressing and immune-modulating properties. Curr Mol Med. 2013;13(4):572–585. | ||
Wang Y, Zhang X, Li H, Yu J, Ren X. The role of miRNA-29 family in cancer. Eur J Cell Biol. 2013;92(3):123–128. | ||
Bae HJ, Noh JH, Kim JK, et al. MicroRNA-29c functions as a tumor suppressor by direct targeting oncogenic SIRT1 in hepatocellular carcinoma. Oncogene. 2014;33(20):2557–2567. | ||
Wang CM, Wang Y, Fan CG, et al. miR-29c targets TNFAIP3, inhibits cell proliferation and induces apoptosis in hepatitis B virus-related hepatocellular carcinoma. Biochem Biophys Res Commun. 2011;411(3):586–592. | ||
Liu N, Tang LL, Sun Y, et al. MiR-29c suppresses invasion and metastasis by targeting TIAM1 in nasopharyngeal carcinoma. Cancer Lett. 2013;329(2):181–188. | ||
Fan YC, Mei PJ, Chen C, Miao FA, Zhang H, Li ZL. MiR-29c inhibits glioma cell proliferation, migration, invasion and angiogenesis. J Neurooncol. 2013;115(2):179–188. | ||
Wang Y, Li Y, Sun J, et al. Tumor-suppressive effects of miR-29c on gliomas. Neuroreport. 2013;24(12):637–645. | ||
Han TS, Hur K, Xu G, et al. MicroRNA-29c mediates initiation of gastric carcinogenesis by directly targeting ITGB1. Gut. 2015;64(2):203–214. | ||
Zhang H, Cheng Y, Jia C, Yu S, Xiao Y, Chen J. MicroRNA-29s could target AKT2 to inhibit gastric cancer cells invasion ability. Med Oncol. 2015;32(1):342. | ||
Fan Y, Song X, Du H, et al. Down-regulation of miR-29c in human bladder cancer and the inhibition of proliferation in T24 cell via PI3K-AKT pathway. Med Oncol. 2014;31(7):65. | ||
Zhang JX, Mai SJ, Huang XX, et al. MiR-29c mediates epithelial-to-mesenchymal transition in human colorectal carcinoma metastasis via PTP4A and GNA13 regulation of β-catenin signaling. Ann Oncol. 2014;25(11):2196–2204. | ||
Hong Q, Fang J, Pang Y, Zheng J. Prognostic value of the microRNA-29 family in patients with primary osteosarcomas. Med Oncol. 2014;31(8):37. | ||
Zou Y, Li J, Chen Z, et al. miR-29c suppresses pancreatic cancer liver metastasis in an orthotopic implantation model in nude mice and affects survival in pancreatic cancer patients. Carcinogenesis. 2015;36(6):676–684. | ||
Jiang J, Yu C, Chen M, Zhang H, Tian S, Sun C. Reduction of miR-29c enhances pancreatic cancer cell migration and stem cell-like phenotype. Oncotarget. 2015;6(5):2767–2778. | ||
Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010;9(10):775–789. | ||
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. | ||
Foubert P, Varner JA. Integrins in tumor angiogenesis and lymphangiogenesis. Methods Mol Biol. 2012;757:471–486. | ||
Gotzmann J, Mikula M, Eger A, et al. Molecular aspects of epithelial cell plasticity: implications for local tumor invasion and metastasis. Mutat Res. 2004;566(1):9–20. | ||
Caccavari F, Valdembri D, Sandri C, Bussolino F, Serini G. Integrin signaling and lung cancer. Cell Adh Migr. 2010;4(1):124–129. | ||
Felding-Habermann B. Integrin adhesion receptors in tumor metastasis. Clin Exp Metastasis. 2003;20(3):203–213. | ||
Grzesiak JJ, Tran Cao HS, Burton DW, et al. Knockdown of the β(1) integrin subunit reduces primary tumor growth and inhibits pancreatic cancer metastasis. Int J Cancer. 2011;129(12):2905–2915. | ||
Song J, Zhang J, Wang J, Wang J, Guo X, Dong W. β1 Integrin mediates colorectal cancer cell proliferation and migration through regulation of the Hedgehog pathway. Tumour Biol. 2015;36(3):2013–2021. | ||
Yang Z, Zhou X, Liu Y, et al. Activation of integrin β1 mediates the increased malignant potential of ovarian cancer cells exerted by inflammatory cytokines. Anticancer Agents Med Chem. 2014;14(7):955–962. | ||
Masumoto A, Arao S, Otsuki M. Role of β1 integrins in adhesion and invasion of hepatocellular carcinoma cells. Hepatology. 1999;29(1):68–74. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.