")
Back to Journals » OncoTargets and Therapy » Volume 11
miR-206 regulates 5-FU resistance by targeting Bcl-2 in colon cancer cells
Received 6 December 2017
Accepted for publication 7 February 2018
Published 29 March 2018 Volume 2018:11 Pages 1757—1765
DOI https://doi.org/10.2147/OTT.S159093
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tohru Yamada
Xiaomin Meng, Rao Fu
Department of Applied Chemistry, Northeast Electric Power University, Jilin, People’s Republic of China
Introduction: Previous studies have found that miRNAs play a key role in drug resistance. Multiple reports show that miRNAs act as regulators in colorectal cancer (CRC) cells, but the role of miR-206 in CRC is still not well understood. The current study aimed to explore the potential function of miR-206 in 5-FU resistance.
Methods: To indentify the role of miR-206 in 5-FU resistance, the expression of miR-206 was examined by real-time polymerase chain reaction (RT-PCR) in 5-FU-resistant (FR) CRC (HCT116/FR and RKO/FR) and their parental cell lines. miR-206 mimic was transfected to 5-FU-FR CRC, and the 5-FU sensitivity was detected by MTS and flow cytometry. Using miRNA target prediction software, we found that miR-206 could target the 3' untranslated region (3'UTR) sequence of Bcl-2.
Results: miR-206 was found to be downregulated in 5-FU-FR CRC in comparison with their parental cell lines, suggesting its crucial relevance for colon cancer biology. Downregulation of miR-206 promoted drug resistance and decreased apoptosis of parental cells, while overexpression of miR-206 promoted drug cytotoxicity and apoptosis of HCT116/FR cells. We also identified miR-206 targeting Bcl-2 directly in CRC, which is required for miR-206 mediated-5-FU resistance.
Conclusion: Our results show that miR-206 targets Bcl-2 to mediate chemoresistance, proliferation, and apoptosis in CRC. This study provides a novel promising candidate for colon cancer therapy.
Keywords: miR-206, 5-FU, resistance, Bcl-2, colorectal cancer
Introduction
Colorectal cancer (CRC) is a leading cause of cancer incidence and death worldwide.1,2 The overall relative 5-year survival of patients with CRC is about 50%. Almost half of the patients with CRC are confronted with liver metastasis either at the time of diagnosis or later during the course of the disease. 5-Fluorouracil (5-FU)-based chemotherapy is the key therapy for advanced disease.3 The researchers used a hydrogen that simulated the location of a pyrimidine ring in the form of a carbon-5 uracil and fluorine, to meet the expected biochemical, pharmacological, and clinical anticancer drug activity.4,5 5-FU resistance in CRC is an important clinical problem.6 There are a number of mechanisms that have been identified to be responsible for 5-FU resistance, such as activation of notch, Wnt, and Hh signaling pathways.7–9
miRNAs are endogenous 21–24-nucleotide single-strand noncoding RNAs that bind to the target mRNAs at 3′-UTR through imperfect complementary match to decrease protein expression by either translation repression or mRNA cleavage.10 A previous report indicated that miRNAs participate in various biological processes, such as tissues homeostasis, organ development, cellular proliferation, apoptosis, and human diseases.11 In cancer cells, miRNAs may function as tumor promoters by targeting tumor suppressor genes as well as tumor suppressors by inhibiting cellular oncogene expression.12,13
Right now, miR-206 is one of the well-studied and excellently characterized miRNAs.14,15 miR-206 downregulation has been shown in many types of cancers.14 A previous study showed that miR-206 suppresses cancer cell invasion and migration via Cdc42.16 Moreover, miR-206 also inhibits VEGF expression and regulates the migration and apoptosis of laryngeal cancer cells.17 However, the role of miR-206 in 5-FU resistance regulation remains unclear.
In this study, the downregulation of miR-206 was found in the 5-FU-resistant CRC. Our results shown that miR-206 regulates 5-FU resistance by changing the protein levels of its target gene Bcl-2. Our findings indicated that miR-206/Bcl-2 as a novel regulator mediates 5-FU resistance in CRC.
Materials and methods
Cell culture and reagents
CRC cell lines including HCT116 and RKO were obtained from American Type Culture Collection (Rockville, MD, USA). The cells were cultured in Dulbecco’s Modified Eagle’s Medium (Gibco, Gaithersburgh, MD, USA) supplemented with 10% fetal bovine serum (Gibco), and 1% penicillin–streptomycin at 37°C in a humidified environment with 5% CO2. miR-206 inhibitor, miR-206 mimic, or the appropriate negative controls (NCs) of miRNA inhibitor and miRNA mimic were obtained from GenePharma (Shanghai, People’s Republic of China).
5-FU-resistant cell lines, HCT116/FR and RKO/FR, were derived by incubation with stepwise increasing concentrations of 5-FU. Briefly, HCT116 and RKO cells were treated with increasing dose of 5-FU from 10 to 100 μg/mL for more than 12 months. During the treatment, after each round, when the surviving cells reached >70% confluence they were passaged by trypsinization, and the dose of 5-FU was increased.
MTS assay
Indicated cells were transfected with miR-206 mimic/inhibitor. After 24 hours, cells were cultured in 96-well plates at a density of 4×103 cells/well. Then the cells were treated with increasing concentration of 5-FU for 72 hours. Subsequently, MTS assay was performed using the MTS assay kit (Promega, Fitchburg, WI, USA) according to the manufacturer’s instructions. Luminescence was measured with a Wallac Victor 1420 Multilabel Counter (Perkin Elmer, Bridgeville, PA, USA). Each assay was conducted in triplicate and repeated three times.
siRNA transfection
Bcl-2 siRNA and control siRNA were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). HCT116 cells were seeded in 12-well plates for 24 hours to reach 30%–40% confluence and transiently transfected cells with Bcl-2 siRNA or control siRNA with Lipofectamine 2000 (Invitrogen, Carsbad, CA, USA) according to the manufacturer’s instructions.
RNA extraction and real-time reverse transcription-polymerase chain reaction (PCR)
Total RNA was extracted using the TRIzol RNA Kit (Invitrogen) according to the manufacturer’s protocol. Briefly, total RNA was used to generate cDNA using SuperScript II reverse transcriptase (Invitrogen). Polymerase chain reaction (PCR) was performed in triplicate using SsoFasrTM Probes Supermix (Bio-Rad, Hercules, CA, USA) in a final reaction volume of 20 μL with gene-specific primer/probe sets and a standard thermal cycling procedure (35 cycles) on a Bio-Rad CFX96™ Real-time PCR System. Relative gene mRNA levels were assessed using TaqMan Gene Expression Real-Time PCR assays. Result was expressed as the threshold cycle (Ct). The relative quantification of the target transcripts was determined by the comparative Ct method (ΔΔCt) according to the manufacturer’s protocol. The 2−ΔΔCt method was used to analyze the relative changes in gene expression. Control experiments were conducted without reverse transcription to confirm that the total RNA was not contaminated with genomic DNA. β-Actin expression served as the internal reference for Bcl-2. U6 RNA served as an internal control for miRNA. The primers used were as follows: Bcl-2: forward: 5′-CTGCACCTGACGCCCTTCACC-3′; reverse: 5′-CACATGACCCCACCGAACTCAAAGA-3′; β-actin: forward: 5′-GACCTGACACACTACCTCAT-3′, reverse: 5′-AGACAGCACTGTGTTGGCTA-3′.
Western blotting
Western blotting was performed as previously described,18,19 with antibodies for Bcl-2 (ab692; Abcam, Cambridge, MA, USA) and β-actin (#4970; Cell signaling, Danvers, MA, USA).
Apoptosis assays
Annexin V/propidium iodide staining was performed using annexin–Alexa 488 (Invitrogen) and PI.20
Luciferase reporter assay
To evaluate the function of miR-206, the 3′-UTR of Bcl-2 was amplified and inserted downstream of the luciferase reporter gene in the luciferase reporter pMIR-REPORT luciferase reporter vector. The mutant 3′-UTR of Bcl-2 was amplified using wild-type (WT) Bcl-2 3′-UTR as the template, and the mutant plasmid was created using a Site-Directed Mutagenesis Kit. For luciferase reporter assays, the cells were cotransfected with miR-206 mimics and WT or mutant Bcl-2 3′-UTR, along with Renilla luciferase pRL-TK vector (Promega) as an internal control. Following transfection for 24 hours, the cells were collected and lysed using RIPA buffer. Luciferase activity was then measured by using Dual Luciferase Assay System (Promega) according to the manufacturer’s instructions.
Xenograft study
All procedures and experiments involving animals in this study were approved by the Committee on the Ethics of Animal Experiments of Northeast Electric Power University and were performed in compliance with the institutional ethical guidelines for animal experimentation according to the guide of the Committee on the Ethics of Animal Experiments of Northeast Electric Power University. HCT116 and HCT116/FR cells were suspended in 100 μL of phosphate-buffered saline at a concentration of 4×106 cells/mL and injected into either flank of the same NOD/SCID mice at 5–6 weeks of age (n=6). 5-FU (20 mg/kg), polyplex containing 500 nM miR-206 mimic in 50 μL of phosphate-buffered saline, or their combination was administrated by intraperitoneal injection twice a week. Tumor growth was monitored using calipers, and tumor volumes were calculated according to the formula: 1/2× length × width2. Mice were euthanized when tumors reached ~1.0 cm3 in size. Tumors were dissected and fixed in 10% formalin and embedded in paraffin. TUNEL immunostaining was performed on 5 μM paraffin-embedded tumor sections by using an AlexaFluor 488-conjugated secondary antibody (Invitrogen) for signal detection.
Statistical analysis
All data are represented as the mean of at least triplicate samples ± standard deviation. Statistical analysis included a one-way analysis of variance or Student’s t-test using GraphPad Prism V software (GraphPad Software, La Jolla, CA, USA). P<0.05 was considered statistically significant.
Results
Generation of 5-FU-resistant CRC
To investigate the mechanism of 5-FU resistance in CRC, we generated HCT116 and RKO 5-FU-resistant cell lines (HCT116/FR and RKO/FR cells). To test the resistance properties of HCT116/FR and RKO/FR cells and the parental cells, we exposed these cells to 5-FU. As expected, 5-FU treatment could remarkably inhibit the proliferation and induce apoptosis of parental cells but not resistant cells (Figure 1A and B). Moreover, significant changes were observed in the distribution of cell cycle phases in HCT116/FR and RKO/FR after 5-FU treatment. As shown in Figure 1C, 5-FU treatment could induce G1 phase arrest strikingly in HCT116 and RKO cells, but not in HCT116/FR and RKO/FR. These data demonstrate that 5-FU-resistant cells (HCT116/FR and RKO/FR) were generated successfully.
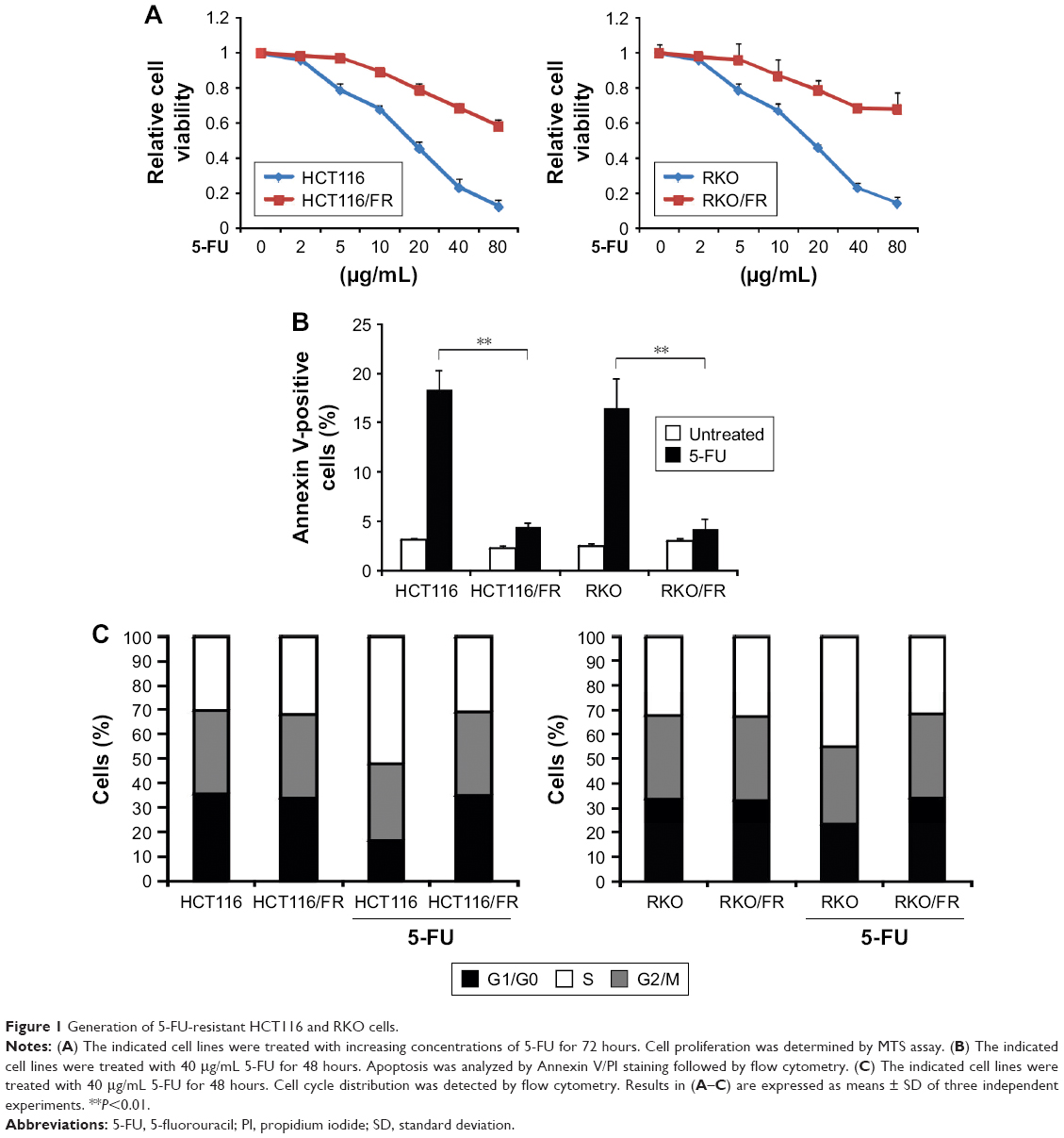 | Figure 1 Generation of 5-FU-resistant HCT116 and RKO cells. |
miR-206 was downregulated in HCT116/FR and RKO/FR cells
To determine the involvement of miR-206 in 5-FU-resistant HCT116/FR and RKO/FR cells, we used qRT-PCR to examine miR-206 expression in these cells. Compared to parental cells, the expression level of miR-206 was significantly decreased in 5-FU-resistant cells (Figure 2A and B). These results indicate that miR-206 downregulation may contribute to 5-FU resistance in CRC.
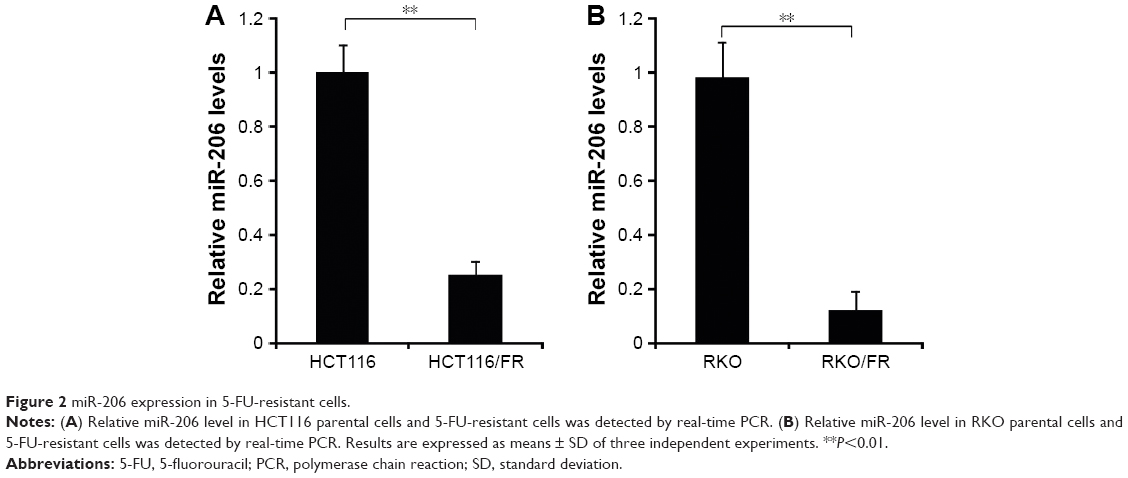 | Figure 2 miR-206 expression in 5-FU-resistant cells. |
5-FU resistance can be overcome by miR-206 overexpression in CRC
Next, we investigated the role of miR-206 in regulating resistance to 5-FU in CRC. miR-206 was overexpressed by miR-206 mimic in HCT116/FR and RKO/FR cell lines (Figure 3A). Compared to miR-mimic-NC cells, MTS of the miR-206-overexpression (miR-206 mimic) HCT116/FR and RKO/FR cell lines was inhibited to a greater extent by 5-FU (Figure 3B and C). Furthermore, miR-206 mimic recovered apoptosis induced by 5-FU in 5-FU-resistant cells (Figure 3D). Thus, the results demonstrate that miR-206 mimic overcame 5-FU resistance in 5-FU-resistant cells.
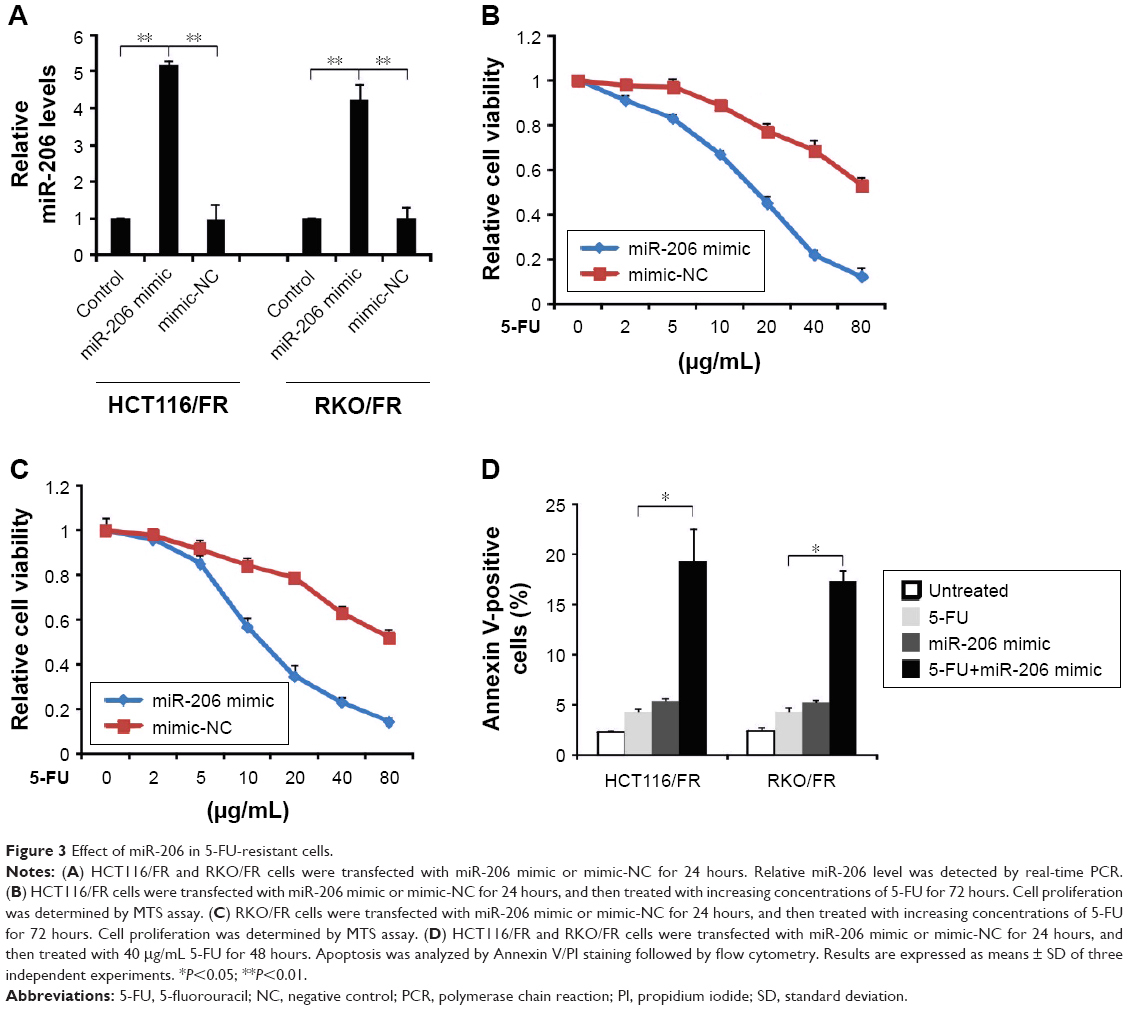 | Figure 3 Effect of miR-206 in 5-FU-resistant cells. |
Inhibition of miR-206 promotes resistance to 5-FU in CRC
Then, to determine the association of 5-FU resistance and miR-206 in HCT116 and RKO cells, we examined the role of inhibition of miR-206 in the 5-FU-sensitive cells. As shown in Figure 4A, qRT-PCR results indicated that miR-206 inhibitor decreased levels of miR-206, suggesting that miR-206 is efficiently the downregulated in HCT116 and RKO cells. HCT116 and RKO cells transfected with miR-206 inhibitor demonstrated significant resistance to 5-FU compared with the NC group (Figure 4B). Moreover, HCT116 and RKO cells treated with miR-206 inhibitor had a lower apoptosis rate than the NC group in response to 5-FU treatment (Figure 4C). Collectively, these data indicate that miR-206 downregulation contributes to the resistance to 5-FU in CRC.
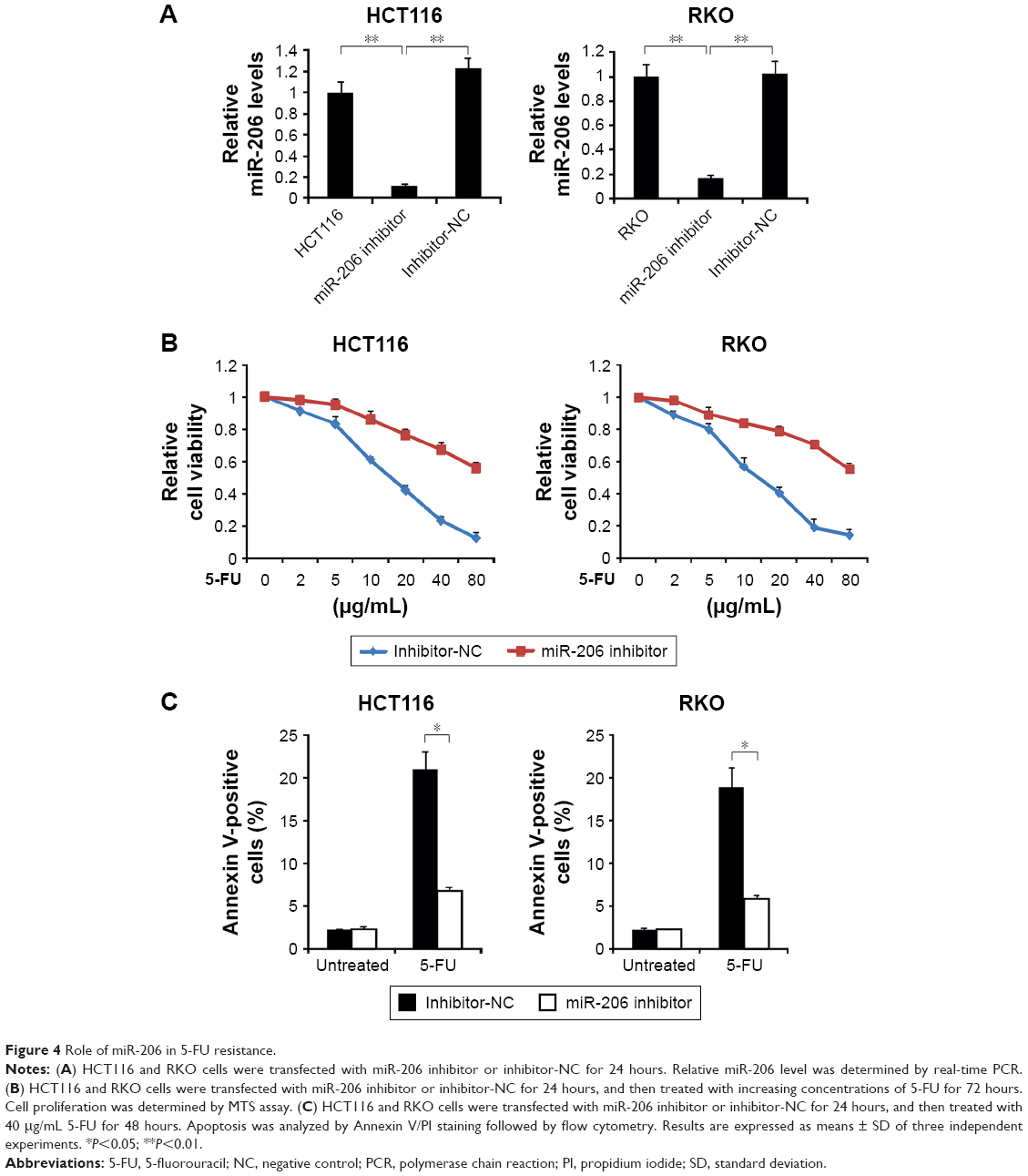 | Figure 4 Role of miR-206 in 5-FU resistance. |
Bcl-2 regulates miR-206-mediated 5-FU resistance
Next, we used TargetScanHuman 6.2 (http://www.targetscan.org) to predict the targeting gene and found that Bcl-2 is a target gene of miR-206 (Figure 5A). To confirm whether Bcl-2 is targeted by miR-206, a luciferase reporter vector with the target site or the mutant target site downstream of the luciferase gene (Bcl-2-3′-UTR-WT and Bcl-2-3′-UTR-mut) was generated. Compared with the Bcl-2-3′-UTR-mut and miR-206 mimic cotransfected group, a significant decrease was observed in Bcl-2-3′-UTR-WT and miR-206 mimic cotransfected group in HCT116 cells (Figure 5B). These data indicate that Bcl-2 is a target gene of miR-206.
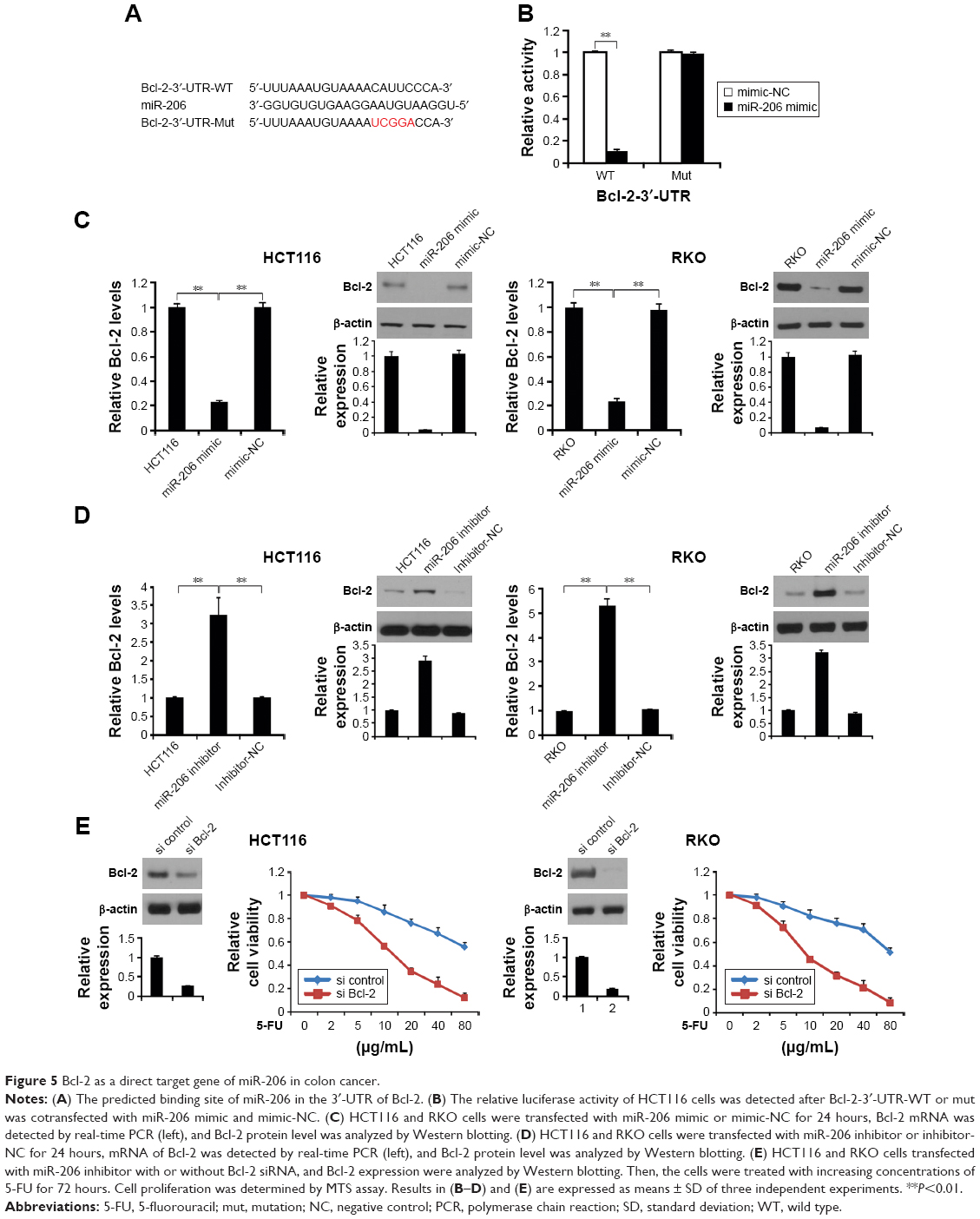 | Figure 5 Bcl-2 as a direct target gene of miR-206 in colon cancer. |
Next, we analyzed whether miR-206 regulates Bcl-2 protein level in HCT116 and RKO cells. HCT116 and RKO cells transfected with miR-206 mimic downregulated mRNA and protein level of Bcl-2 in the cells (Figure 5C). Moreover, transfected miR-206 inhibitor upregulated mRNA and protein levels of Bcl-2 in HCT116 and RKO cells (Figure 5D). These results indicate a strong inverse and consistent correlation between the miR-206 levels and Bcl-2.
To investigate if Bcl-2 plays an important role in miR-206-mediated 5-FU resistance, we transfected the Bcl-2 or control siRNA into the miR-206 inhibitor-treated 5-FU-resistant HCT116 and RKO cells, and cell survival rate was assessed. The results confirmed that Bcl-2 level was reduced in HCT116 and RKO cells, and Bcl-2 siRNA significantly decreased the survival rate of the 5-FU-resistant HCT116 and RKO cells compared with the control group (Figure 5E), suggesting that miR-206 downregulation modulates 5-FU resistance in HCT116 cells by upregulating Bcl-2.
Inhibition of miR-206 overcomes 5-FU resistance in vivo
Based on our findings that 5-FU combined with miR-206 mimic synergistically inhibited the growth of 5-FU-resistant cells in vitro, we confirmed the combination effect in vivo in mice. HCT116 and HCT116/FR tumors were treated with miR-206 mimic, 5-FU, or their combination. The combination of 5-FU with miR-206 mimic inhibited the growth of tumors more efficiently than the 5-FU single treatment (Figure 6A), which indicates that miR-206 mimic might be a potential method for therapy in combination 5-FU in CRC. The apoptosis of the tumor cell also showed a similar trend (Figure 6B). These results further support the finding that miR-206 contributed to overcome 5-FU resistance in vivo.
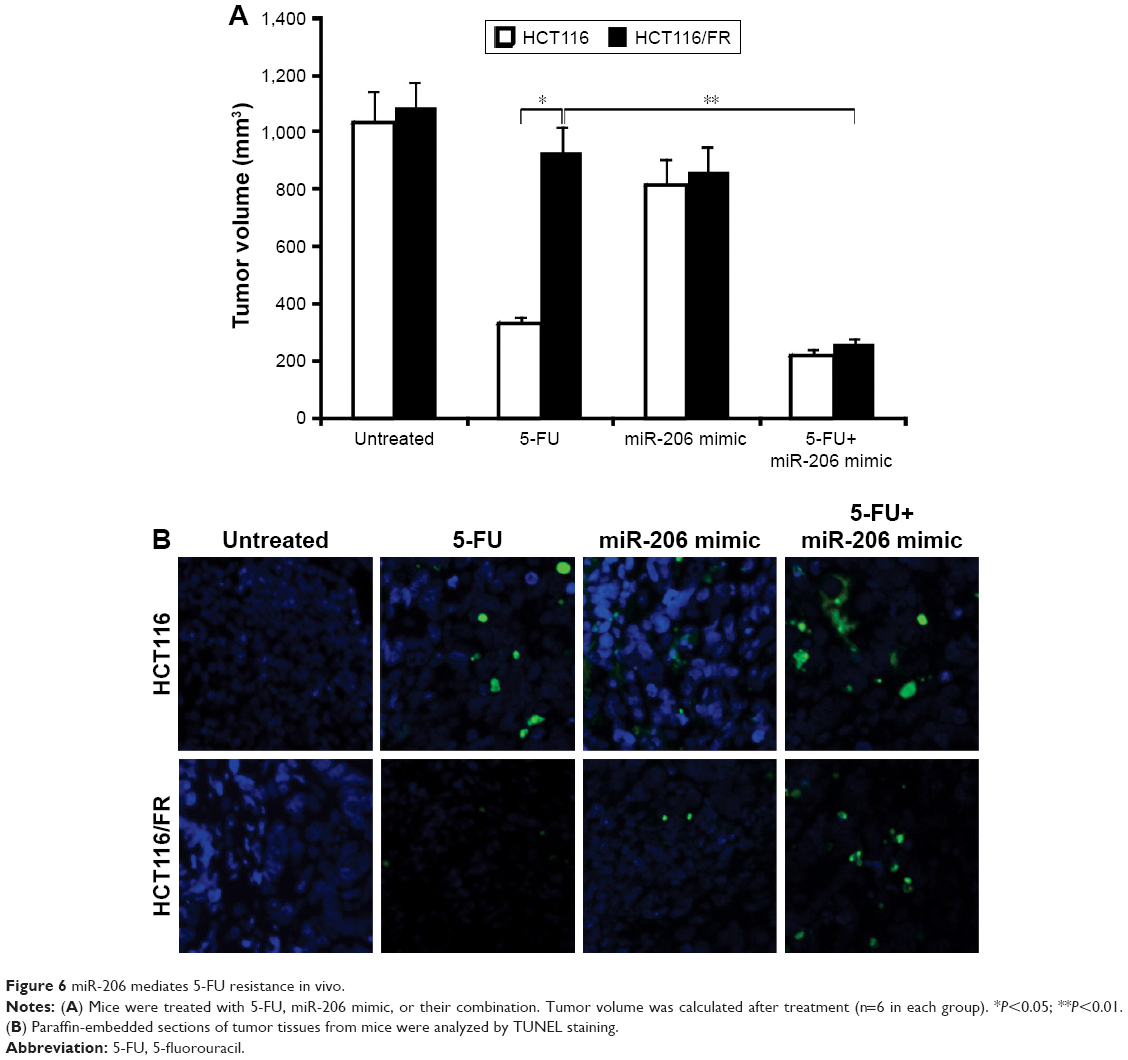 | Figure 6 miR-206 mediates 5-FU resistance in vivo. |
Discussion
Usually, drug resistance is caused by the sequential addition of genetic mutations that reroute key signaling pathways promoting cancer cell infiltration and invasive phenotype.21 Although the cause of drug resistance has been investigated for many years, there are still no remedies to improve drug resistance in clinical outcomes.21 In a previous study, miRNAs have been stratified into mediators that regulate the response of cancer cells to therapeutic cells.22
It has been shown that the response of patients to chemotherapy is closely related to the functional status of miRNAs.23,24 Although the mechanism of miRNA regulation of drug resistance is unclear, current evidence suggests several roles of miRNAs, including changes in drug targets that affect treatment-induced cell death, regulation of multiple drug resistance (MDR)-related proteins, the use of drug concentrations, and the promotion angiogenesis.23,25 In the current study, we found that miR-206 was downregulated in HCT116/FR and RKO/FR cells compared with the parental cells.
In addition, our results also demonstrated that downregulation of miR-206 promotes resistance to 5-FU in CRC, but miR-206 overexpression reverses 5-FU resistance. Target prediction tools identified miR-206 targeting Bcl-2. It has been studied that Bcl-2 protein plays a key role in various cell processes, including apoptosis, invasion, cell proliferation, and metastases in many types of cancers.26 Thus, the function of miR-206 in targeting Bcl-2 is in agreement with the known biological effects of miR-206. Actually, other miRNAs are able to confer resistance by targeting Bcl-2 in many other types of cancer. Previous studies have shown that Bcl-2 dysfunction has prognostic significance in several malignancies, such as colorectal cancer.27,28 These reports demonstrated a mechanism in which reduced expression of miR-206 should be associated with a reduced survival of colon cancer patients and also supports the development of miR-206 mimic as a potential target for reversing drug resistance. The overexpression of miR-206 can successfully induce cancer cell sensitivity to 5-FU. In the future, therapeutic strategies can be developed based on the predicted level of miR-206. In addition, miR-206 may potentially bind to 5-FU to prolong the drug susceptibility in CRC.
Conclusion
Our results uncovered a unique role of miR-206 in 5-FU resistance in CRC. The drug resistance is a major challenge for the 5-FU treatment, but the strategy of upregulating the miR-206 may be a potential way to increase the sensitivity of CRC to 5-FU.
Disclosure
The authors report no conflicts of interest in this work.
References
De Rosa M, Pace U, Rega D, et al. Genetics, diagnosis and management of colorectal cancer (review). Oncol Rep. 2015;34:1087–1096. | ||
Tong J, Wang P, Tan S, et al. Mcl-1 Degradation is required for targeted therapeutics to eradicate colon cancer cells. Cancer Res. 2017;77:2512–2521. | ||
He K, Chen D, Ruan H, et al. BRAFV600E-dependent Mcl-1 stabilization leads to everolimus resistance in colon cancer cells. Oncotarget. 2016;7:47699–47710. | ||
Lee JJ, Beumer JH, Chu E. Therapeutic drug monitoring of 5-fluorouracil. Cancer Chemother Pharmacol. 2016;78:447–464. | ||
van Kuilenburg AB, Maring JG. Evaluation of 5-fluorouracil pharmacokinetic models and therapeutic drug monitoring in cancer patients. Pharmacogenomics. 2013;14:799–811. | ||
Wilhelm M, Mueller L, Miller MC, et al. Prospective, multicenter study of 5-fluorouracil therapeutic drug monitoring in metastatic colorectal cancer treated in routine clinical practice. Clin Colorectal Cancer. 2016;15:381–388. | ||
Liu J, Fan H, Ma Y, et al. Notch1 is a 5-fluorouracil resistant and poor survival marker in human esophagus squamous cell carcinomas. PLoS One. 2013;8:e56141. | ||
Wu X, Luo F, Li J, Zhong X, Liu K. Tankyrase 1 inhibitior XAV939 increases chemosensitivity in colon cancer cell lines via inhibition of the Wnt signaling pathway. Int J Oncol. 2016;48:1333–1340. | ||
Liu Y, Du F, Zhao Q, Jin J, Ma X, Li H. Acquisition of 5-fluorouracil resistance induces epithelial-mesenchymal transitions through the Hedgehog signaling pathway in HCT-8 colon cancer cells. Oncol Lett. 2015;9:2675–2679. | ||
Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15:509–524. | ||
Gonzalez-Duarte RJ, Cazares-Ordonez V, Avila-Chavez E. The microRNA biogenesis machinery: regulation by steroid hormones and alterations in cancer. Rev Invest Clin. 2014;66:460–464. | ||
Zhang W, Liu J, Wang G. The role of microRNAs in human breast cancer progression. Tumour Biol. 2014;35:6235–6344. | ||
Zheng Q, Chen C, Guan H, Kang W, Yu C. Prognostic role of microRNAs in human gastrointestinal cancer: a systematic review and meta-analysis. Oncotarget. 2017;8(28):46611–46623. | ||
Ma G, Wang Y, Li Y, et al. MiR-206, a key modulator of skeletal muscle development and disease. Int Biol Sci. 2015;11:345–352. | ||
Sun W, Zhang L, Li R. Overexpression of miR-206 ameliorates chronic constriction injury-induced neuropathic pain in rats via the MEK/ERK pathway by targeting brain-derived neurotrophic factor. Neurosci Lett. 2017;646:68–74. | ||
Cui Y, Xie S, Luan J, Zhou X, Han J. Quantitative proteomics and protein network analysis of A549 lung cancer cells affected by miR-206. Biosci Trends. 2013;7:259–263. | ||
Wang CQ, Huang YW, Wang SW, et al. Amphiregulin enhances VEGF-A production in human chondrosarcoma cells and promotes angiogenesis by inhibiting miR-206 via FAK/c-Src/PKCdelta pathway. Cancer Lett. 2017;385:261–270. | ||
Tong JS, Zhang QH, Huang X, et al. Icaritin causes sustained ERK1/2 activation and induces apoptosis in human endometrial cancer cells. PLoS One. 2011;6:e16781. | ||
Tong JS, Zhang QH, Wang ZB, et al. ER-alpha36, a novel variant of ER-alpha, mediates estrogen-stimulated proliferation of endometrial carcinoma cells via the PKCdelta/ERK pathway. PLoS One. 2010;5:e15408. | ||
Tong J, Tan S, Zou F, Yu J, Zhang L. FBW7 mutations mediate resistance of colorectal cancer to targeted therapies by blocking Mcl-1 degradation. Oncogene. 2017;36:787–796. | ||
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. | ||
Zheng T, Wang J, Chen X, Liu L. Role of microRNA in anticancer drug resistance. Int J Cancer. 2010;126:2–10. | ||
Li H, Yang BB. MicroRNA-in drug resistance. Oncoscience. 2014;1:3–4. | ||
Wang J, Yang M, Li Y, Han B. The role of MicroRNAs in the chemoresistance of breast cancer. Drug Dev Res. 2015;76:368–374. | ||
Magee P, Shi L, Garofalo M. Role of microRNAs in chemoresistance. Ann Transl Med. 2015;3:332. | ||
Delbridge AR, Strasser A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015;22:1071–1080. | ||
Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. | ||
Delbridge AR, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016;16:99–109. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.