")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
miR-190a-5p participates in the regulation of hypoxia-induced pulmonary hypertension by targeting KLF15 and can serve as a biomarker of diagnosis and prognosis in chronic obstructive pulmonary disease complicated with pulmonary hypertension
Authors Jiang J, Xia Y , Liang Y, Yang M, Zeng W, Zeng X
Received 7 August 2018
Accepted for publication 11 October 2018
Published 20 November 2018 Volume 2018:13 Pages 3777—3790
DOI https://doi.org/10.2147/COPD.S182504
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chunxue Bai
Jing Jiang,1 Yimeng Xia,2 Yi Liang,1 Meiling Yang,1 Wen Zeng,1 Xiaocong Zeng3
1Department of Respiratory and Critical Care Medicine, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi 530021, People’s Republic of China; 2Department of Anesthesiology, Ruijin Hospital, Shanghai Jiaotong University School of Medicine, Shanghai 200025, People’s Republic of China; 3Department of Cardiology, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi 530021, People’s Republic of China
Purpose: miR-190a-5p expression alters dynamically in response to hypoxia. However, the role of miR-190a-5p expression in hypoxia-induced pulmonary hypertension (PH) remains unclear. We sought to correlate the miR-190a-5p expression levels with the severity, diagnosis, and prognosis of PH in relation to chronic obstructive pulmonary disease (COPD-PH). Additionally, we evaluated the effect of miR-190a-5p through in vitro experiments on human pulmonary endothelial cells (HPECs) that were exposed to hypoxia and in vivo experiments using an animal model of hypoxia-induced PH.
Methods: Circulating miR-190a-5p levels were measured from 73 patients with PH and 32 healthy controls through quantitative real-time PCR. The levels of miR-190a-5p and the expression of Krüppel-like factor 15 (KLF15) were analyzed in HPECs that were exposed to hypoxia, and the effects of antagomir-190a-5p in mice with chronic hypoxia-induced PH were tested. Target gene analysis was performed by Western blot and luciferase assay.
Results: The miR-190a-5p level was significantly higher in patients with COPD-PH than in the healthy controls. Higher miR-190a-5p levels were associated with a greater severity of COPD-PH. In vitro experiments on HPECs showed that exposure to hypoxia increased the miR-190a-5p levels significantly. KLF15 was validated as a target of miR-190a-5p. Transfection with miR-190a-5p mimicked inhibition of KLF15 expression in HPECs. In the mouse model of PH, antagomir-190a-5p reduced right ventricular systolic pressure and enhanced the KLF15 expression levels in lung tissue.
Conclusion: miR-190a-5p regulates hypoxia-induced PH by targeting KLF15. The circulating levels of miR-190a-5p correlate with the severity of COPD-PH, thereby confirming the diagnostic and prognostic value of this parameter in COPD-PH.
Keywords: hypoxia, pulmonary hypertension, COPD, miR-190a-5p, KLF15
Introduction
Pulmonary hypertension (PH) is a devastating, life-threatening condition characterized by vasoconstriction and vascular remodeling, resulting in a progressive increase in pulmonary arterial pressure (PAP).1,2 Chronic obstructive pulmonary disease (COPD) leads to the development of hypoxia, which in turn causes vascular occlusion and neointimal formation.3,4 The treatment of PH involves the establishment of pulmonary vascular homeostasis, which is mainly achieved through the use of endothelial nitric oxide synthase (eNOS).5,6 Arginase 2 (Arg2) is an important isoenzyme that is present in the endothelial cells (ECs) of humans and other species.7 Arg2 competes with eNOS to bind with L-arginine, which has been implicated in the development of vascular dysfunction.8 The Krüppel-like factor (KLF) family – a subclass of the zinc finger family of DNA-binding transcriptional factors – regulates a broad range of cellular processes, including cell differentiation, angiogenesis, and erythropoiesis.9,10 Recent studies have shown that KLF15 plays a critical role in protection against cell proliferation and migration, heart failure, aortic aneurysm formation, and activation of proinflammatory processes in vascular smooth muscle and atherogenesis.11,12 Furthermore, KLF15 has been reported to contribute to pulmonary endothelial homeostasis by regulating the expression of endothelial Arg2 and eNOS. This suggests that modulation of KLF15 overexpression may represent a novel therapeutic target in PH.13
Several circulating biomarkers have been identified as having the potential to be noninvasive, objective, and repeatable parameters that may be useful for the diagnostic and prognostic assessment and the evaluation of the treatment response in various diseases.14 In the last few years, circulating miRNAs have emerged as promising biomarkers in multiple conditions.15,16 The expression levels of circulating miRNAs have been correlated with the onset and development of multiple diseases, including PH.17–19 This opens new avenues for the screening and monitoring of PH. Recent studies have indicated that miRNAs may play key roles in regulating the physiological and pathophysiological processes that are triggered on exposure to hypoxic conditions.20 The miRNAs that show dynamic alterations in expression on exposure to hypoxic conditions are called “hypoxamirs.”21 Hypoxamirs are believed to contribute to the pathogenesis and development of hypoxia-induced PH – a major complication of COPD – by regulating the target gene expression.22,23 miR-190 has been reported to be a hypoxamir.24 It has also been shown to be a positive regulator of Ca2+ influx and to play an important role in hypoxic pulmonary vasoconstriction.25 A previous study reported that progressive hypobaric hypoxia significantly increases the levels of circulating miR-190 and that these increases correlate significantly with increased systolic PAP, independent of the extent of hypoxemia.26 Recently, miR-190 was officially listed after miR-190a-5p in the miRBase (http://www.mirbase.org/, Accession number: MIMAT0000458). Moreover, using a target prediction algorithm TargetScan database (TargetScan 7.2; http://www.targetscan.org) and microRNA.org database (http://www.microrna.org), the mRNA sequence of KLF15 was predicted to contain a conserved “seed” sequence complementary to miR-190a-5p in the 3′-UTR. The above findings prompted us to further evaluate whether miR-190a-5p exerts its effects via KLF15 in hypoxia-induced PH.
The present study aimed to investigate the potential diagnostic and prognostic role of circulating miR-190a-5p in patients with COPD that is complicated with PH. In addition, we investigated the effect of miR-190a-5p on human pulmonary endothelial cells (HPECs) that were exposed to hypoxia and in an animal model of hypoxic PH. Additionally, we sought to determine whether miR-190a-5p interacted with the Arg2 and eNOS pathways by targeting KLF15.
Material and methods
Patients and samples
This was a prospective, observational study conducted on 73 consecutive patients with PH and 32 healthy controls recruited from the department of Respiratory and Critical Care Medicine, The First Affiliated Hospital of Guangxi Medical University, between January 1, 2015, and December 31, 2015. The blood samples of all the patients were collected to evaluate the miR-190a-5p levels. The inclusion criteria for this study were as follows: 1) Patient underwent right-sided heart catheterization for measurement of PAP. 2) Mean PAP (mPAP) was elevated to more than 25 mm Hg, and pulmonary capillary wedge pressure was <15 mm Hg (and pulmonary vascular resistance [PVR]>3 Wood units).27 3) The diagnosis and classification of PH were made according to the standard criteria defined in the 2015 guidelines for PH.28 Patients were excluded if they had malignancy, impaired level of consciousness, obstructive sleep apnea syndrome, or sepsis; if they had undergone renal replacement therapy; if they required mechanical ventilation, intensive care unit admission, or treatment with vasopressors; or if they had a history of arrhythmia, valvular or coronary heart disease, or left ventricle dysfunction. We also excluded patients who had PH secondary to left-sided heart disease because the World Health Organization classifies these patients as group 2 PH or pulmonary venous hypertension.28 The group of healthy controls included 32 healthy volunteers who underwent ultrasonography and showed no evidence of PH. Patient follow-ups were performed via telephone interviews conducted by the study investigators every 3 months for 2 years. The primary endpoint was defined as all-cause mortality. None of the subjects in this study were lost to follow-up.
The demographic characteristics of the patients, including age, sex, and body mass index (BMI), were recorded. Blood samples were collected from each patient at the time of admission. The samples were used for the measurement of the arterial blood gases (partial pressure of arterial oxygen [PaO2] and partial pressure of arterial carbon dioxide [PaCO2]), estimated glomerular filtration rate (eGFR), C-reactive protein (CRP), and laboratory variables. Peripheral blood samples were collected and centrifuged at 3,000× g for 15 minutes at 4°C; the supernatant was then acquired and further centrifuged at 12,000× g for 10 minutes at 4°C. Next, the supernatants were transferred into fresh tubes and stored at −80°C before quantitative reverse transcriptase-PCR (qRT-PCR) analysis. Respiratory function data and echocardiographic data were obtained within 48 hours of collecting the blood samples. In addition, invasive hemodynamic data were included if right heart catheterization was performed within 2 weeks of the blood sample collection. Depending on the mPAP and cardiac index (CI) values, patients with COPD-PH were classified into severe and non-severe COPD-PH subgroups. The severe COPD-PH group was defined as patients with an mPAP of ≥35 mm Hg or mPAP of ≥25 mm Hg with a low CI (<2.0 L/min/m2). The non-severe COPD-PH subgroup was defined as patients with an mPAP of 25–35 mm Hg with a normal CI.29,30
The study protocol was approved by the Ethics Committee of The First Affiliated Hospital of Guangxi Medical University, and the study was carried out in accordance with the Declaration of Helsinki. All of the participants enrolled in this study received oral and written information regarding the objectives of the study, following which they provided written consent.
Cell culture experiments
Human pulmonary arterial endothelial cells (HPAECs), human pulmonary microvascular endothelial cells (HPMECs), human pulmonary artery smooth muscle cells (HPASMCs), and human pulmonary artery fibroblasts (HPAFs) (all purchased from Science Cell Research Laboratories, Carlsbad, CA, USA) were maintained in endothelial medium culture medium, according to the supplier’s instructions. For the experiments, HPAECs, HPMECs, HPASMCs, and HPAFs were cultured under normoxic or hypoxic conditions for 24 hours. The hypoxic environment was produced by placing the cells within hypoxic pouches (GasPak™ EZ; BD Biosciences, San Jose, CA, USA) that were equilibrated with 95% N2 and 5% CO2 and maintained at 37°C for 24 hours.31,32
Transfection of HPAECs with miRNAs and siRNA
To induce the overexpression of miR-190a-5p, HPAECs were transfected with 100 nM of has-miR-190a-5p mimic (sense: 5′-UGAUAUGUUUGAUAUAUUAGGU-3′, miR10000458-1-5; purchased from Guangzhou RiboBio Co., Ltd., Guangzhou, China) To inhibit miR-190a-5p expression, HPAECs were transfected with 100 nM of has-miR-190a-5p inhibitor (sense: 5′-ACCUAAUAUAUCAAACAUAUCA-3′, miR20000458-1-5; purchased from Guangzhou RiboBio Co., Ltd.). The control cells were transfected with a negative control (sense: 5′-UCACAACCUCCUAGAAAGAGUAGA-3′, miR01201-1-5; purchased from Guangzhou RiboBio Co., Ltd) under the same experimental conditions. For KLF15 knockdown, HPAECs were transfected with human endoribonuclease-prepared small interfering RNA against KLF15 (sense: 5′-GCAUUUCUGCUUGCCCGAGUUUCCU-3′, human siRNA KLF15, AM16708; purchased from Thermo Fisher Scientific, Waltham, MA, USA). To achieve complete KLF15 knockdown, the transfection was performed twice at intervals of 24 hours, and the experiments were performed 24 hours after the second transfection. According to the manufacturer’s instructions, we used Lipofectamine® 2000 reagent (Thermo Fisher Scientific, Waltham, MA, USA) to perform all of the transfections.
Luciferase reporter assay
The KLF15 3′-UTR containing the miR-190a-5p target sequence, as well as the mutant sequences that were used, were chemically synthesized (GenScript, Nanjing, China) and cloned into the PGL3 control vector (Promega Corporation, Madison, WI, USA) downstream of the luciferase gene using the XbaI site. The resulting plasmids were designated KLF15 3′-UTR-luci-wild type (WT) and KLF15 3′-UTR-luci-mutant (Mut). HPAECs were transfected with either KLF15 3′-UTR-luci-WT or KLF15 3′-UTR-luci-Mut in 24-well plates using the transfection reagent Lipofectamine 2000, according to the manufacturer’s instructions. The cells were also co-transfected with an miR-190a-5p mimic, an miR-190a-5p inhibitor, or the same concentration of the negative control. In addition, each well was co-transfected with the pRL-TK plasmid (Promega Corporation) to assess the transfection efficiency. Subsequently, the Dual-Luciferase® Reporter Assay System (Promega Corporation) was used to measure the firefly and renilla luciferase activity levels in the cells that were harvested 24 hours after transfection. Each transfection was performed in triplicate.
Hypoxic PH animal model and experimental protocols
All animal care practices and animal experimental procedures in this study were approved by the Guangxi Medical University Animal Welfare & Ethics Committee and were conducted in accordance with the guidelines of the National Research Council (US) for the care and use of laboratory animals (2011).
The experimental animals were obtained from the Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and maintained at the Animal Experiment Center of Guangxi Medical University (Guangxi, China). The method for the establishment of an animal model for hypoxia-induced PH has been described extensively in previous studies.33,34 Twenty-four adult (aged between 8 and 10 weeks) male C57BL/6J mice were randomly assigned to three groups in equal number: a normoxia group, a hypoxia-antagomir negative control (hypoxia-antagomir-NC) group, and a hypoxia-antagomir-190a-5p group. The mice were housed either in room air or in a hypoxic chamber with 10%±0.5% O2 for a period of 4 weeks. The antagomir-190a-5p (sense: 5′-ACCUAAUAUAUCAAACAUAUCA-3′, miR30000458-1-10; purchased from Guangzhou RiboBio Co., Ltd.) or the antagomir-NC (sense: 5′-UUUGUACUACACAAAAGUACUG-3′, miR03101-1-10; purchased from Guangzhou RiboBio Co., Ltd.) were dissolved in prewarmed PBS and administered as an intravenous injection (injected into the tail vein at a dose of 8 mg/kg) in the groups exposed to hypoxic conditions on days 14, 17, 20, 23, and 26.33
Measurement of hemodynamic parameters and right-sided heart hypertrophy
The mice were anesthetized by administration of sodium pentobarbital (60 mg/kg), tracheotomized, and placed under artificial ventilation. A catheter was introduced into the right jugular vein and advanced into the right ventricle for measurement of the right ventricular systolic pressure (RVSP). The RVSP was measured and analyzed using a PowerLab system (AD Instruments, Colorado Springs, Australia). After the completion of the hemodynamic measurements, the hearts and lungs were removed. The right ventricle (RV) wall was carefully separated from the left ventricle (LV) and septum (S), and both the separated portions were blotted dry and weighed immediately. The ratio of the free wall of the right ventricle weight to the weight of the left ventricle and septum (RV/(LV+S)) was calculated as an index based on hypertrophy of the RV. Frozen lung tissues were used for Western blotting analysis and real-time (RT)-PCR analysis, as described below.
Real-time quantitative PCR analysis
The levels of miR-190a-5p were quantified by qRT-PCR in human plasma, human pulmonary cell lines, and the lung tissues of mice. The relative expression levels of KLF15 mRNA were quantified by real-time qRT-PCR of human plasma. The total RNA was isolated using the TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions. The RNA was then reverse transcribed into cDNA using a PrimerScript Real-time reagent kit (Takara Bio Inc., Otsu, Shiga, Japan). In accordance with the manufacturer’s instructions, qPCR was performed using SYBR Green PCR Master Mix (Takara Biotechnology Co., Ltd.) and the ABI 7500 Real-time PCR system (Applied Biosystems; Thermo Fisher Scientific, Inc.). The primer sequences used were as follows: miR-190a-5p forward, 5′-GCAGGCCTCTGTGTGATATGT-3′ and reverse, 5′-GGCAAGACACTGTAGGAATATGT-3′; KLF15 forward, 5′-TACACCAAAAGCAGCCACCT-3′ and reverse, 5′-TCTTCTCGCACACAGGACAC-3′ (Shanghai GenePharma Co., Ltd.). The PCR conditions included template denaturation at 95°C for 10 minutes, followed by a total of 40 cycles at 95°C for 15 seconds and 60°C for 1 minute. Cel67 and GAPDH were used as the internal references. Data analyses were performed using the 2-ΔΔCq method for calculating the relative expression levels of miR-190a-5p.
Western blot analysis
Solubilized protein lysates were isolated from the HPAECs, HPMECs, and lung tissue of mice and were used to determine the levels of KLF15, eNOS, and Arg2. The total protein from HPAECs, HPMECs, and lung tissues of mice was extracted by using 1% radioimmunoprecipitation assay lysis buffer (Beyotime Institute of Biotechnology, Jiangsu, China) containing 1 mM phenylmethanesulfonyl fluoride. The supernatants were then collected, and the protein concentration was determined using a Bicinchoninic Acid Protein Assay Kit (Pierce Biotechnology, Inc., Rockford, IL, USA). The isolated proteins (80 μg) were then resolved onto 12% SDS-PAGE gels and transferred to nitrocellulose membranes (Sigma-Aldrich, St. Louis, MO, USA). The membranes were then blocked for 1 hour at room temperature, using 5% non-fat milk dissolved in PBS, and incubated overnight at 4°C with the following polyclonal antibodies (Santa Cruz Biotechnology, Inc., Dallas, TX, USA): mouse anti-KLF15 antibody (1:500; cat. no sc-271675), mouse anti-Arg2 antibody (1:500), mouse anti-eNOS/NOS3 antibody (1:500), and mouse anti-GADPH antibody (1:500). Antibody treatment was followed by three 10-minute washes with Tris-buffered saline solution containing 0.05% Tween-20 (TBST) and incubation with horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G (1:1,000; Santa Cruz Biotechnology, Inc.). Next, the TBST washes were again repeated in the same fashion, and the protein was detected by chemiluminescence with the Pierce-enhanced chemiluminescence Western blotting substrate (Pierce Biotechnology, Inc.). All the data for Western blot analysis were quantified using ImageJ 1.48 software (National Institutes of Health, Bethesda, MD, USA) and data were normalized to GADPH.
Statistical analysis
The data were processed using GraphPad Prism 7 for Windows, version 7.04 (GraphPad Software, Fay Avenue, La Jolla, CA, USA) and STATA 12.0 (Lakeway Drive, College Station, TX, USA). The data are presented as absolute numbers, percentages, mean ± SD, median, interquartile range, or scatter dot plots (each dot represents one patient or an independent experiment). For multiple comparisons, a one-way or two-way analysis of variance was used, followed by Tukey’s test. For unpaired data, the two-tailed Student t-test was used to evaluate single comparisons between the different experimental groups. Data normality was tested by visual inspection of histograms and by using the skewness and kurtosis test for normality. Cox regression analysis included potential confounders, and selected variables with P<0.1 were forced into the model. Kaplan–Meier survival analysis was performed to estimate survival. For all survival analyses, the time from sampling to event/censoring was taken into consideration. Receiver operator characteristic (ROC) curves were plotted to assess the sensitivity and specificity of the biomarkers and to determine the cutoff points.
Results
Demographic and clinical characteristics
In this study, we enrolled a total of 73 patients with PH and 32 healthy subjects, including 27 patients with COPD-PH, 15 patients with idiopathic pulmonary arterial hypertension (IPAH), 13 patients with connective tissue disorder-associated PAH (PAH-CTD), and 18 patients with chronic thromboembolic pulmonary hypertension (CTEPH). The demographic and clinical characteristics of the participants are shown in Table 1. Among the 73 patients with PH in this study, 44 (60%) were women. The median age of all 73 patients with PH was 59 years. The mean BMI of the patients with PH was 27.5±2.8 kg/m2, and the mean eGFR of the patients with PH was 60.9±20.9 mL/min/1.73 m2.
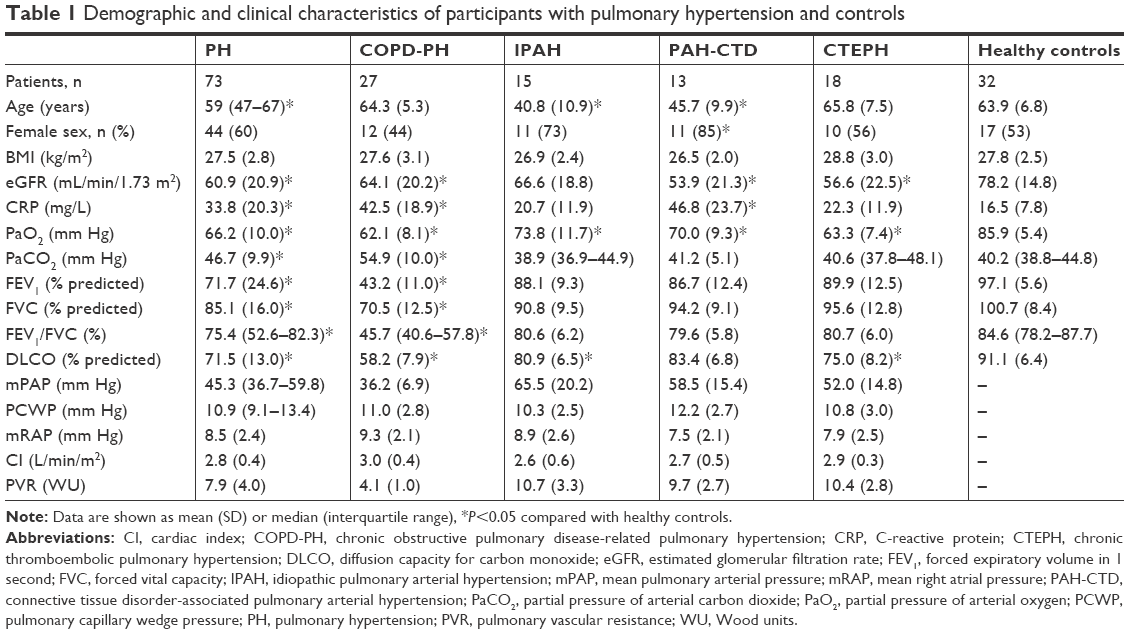 | Table 1 Demographic and clinical characteristics of participants with pulmonary hypertension and controls |
Levels of circulating miR-190a-5p are higher in patients with COPD-PH than in healthy controls, in correlation with disease severity
To determine whether miR-190a-5p was responsible for the development and severity of various types of hypoxia-induced PH, such as PH in patients with COPD, we quantified the expression levels of miR-190a-5p and KLF15 mRNA. The levels of miR-190a-5p were significantly higher in the global cohort of patients with PH and in the subgroup of patients with COPD-PH than in healthy controls (Figure 1A). The relative expression levels of KLF15 mRNA were significantly lower in the global cohort of patients with PH and in the subgroup of patients with COPD-PH than in the healthy controls (Figure 1B). Patients with COPD-PH who had low arterial oxygen tension (PO2 of <60 mm Hg) were found to have significantly higher levels of miR-190a-5p and lower relative expression levels of KLF15 mRNA than the patients with high arterial oxygen tension (PO2 >60 mm Hg; Figure 1C). Furthermore, patients with severe COPD-PH had higher miR-190a-5p levels and lower KLF15 mRNA levels as compared to those in the mild or moderate COPD subgroups (Figure 1D). The miR-190a-5p levels were negatively correlated with the relative expression of KLF15 mRNA levels in the subgroup of patients with COPD-PH (Figure 1E).
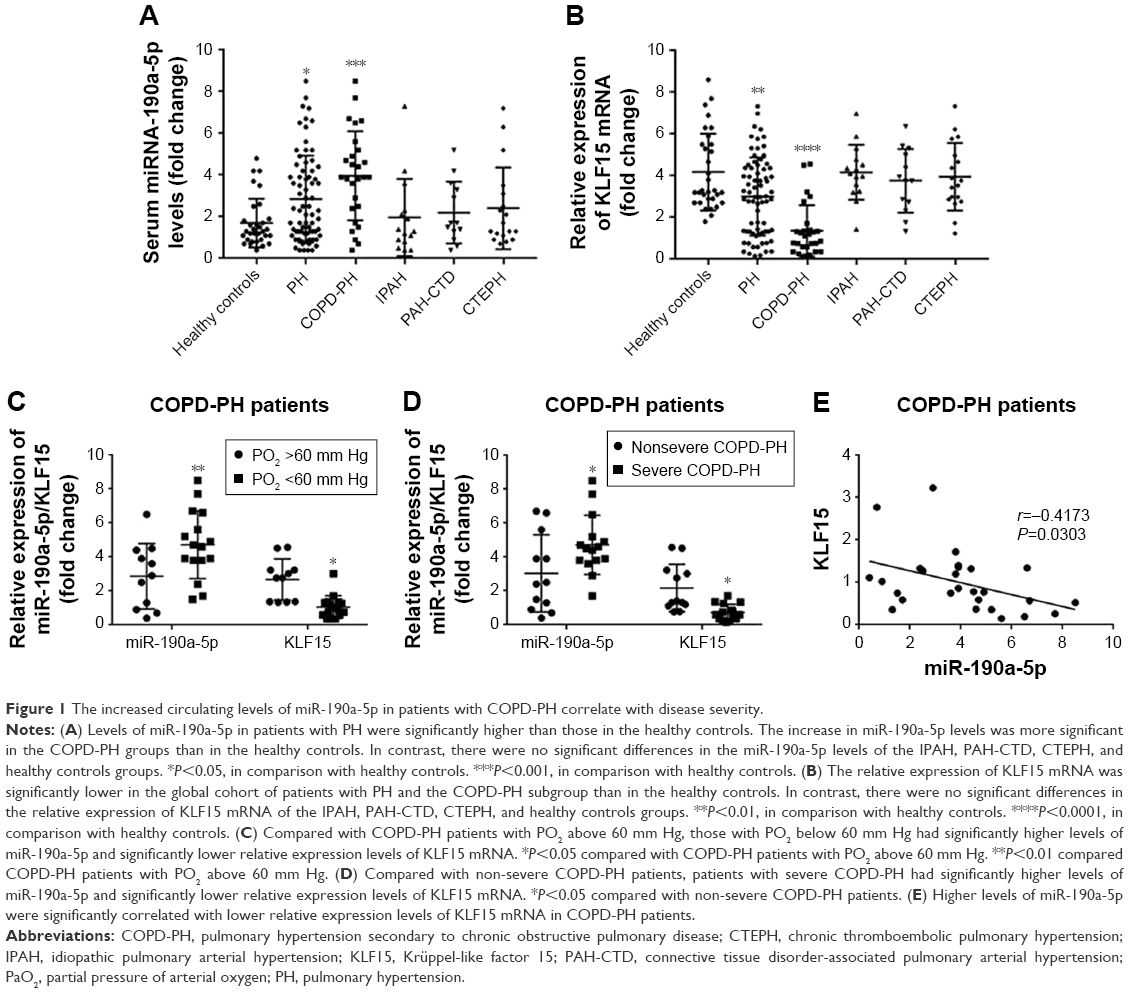 | Figure 1 The increased circulating levels of miR-190a-5p in patients with COPD-PH correlate with disease severity. |
Circulating miR-190a-5p levels as markers for diagnosis and prognosis in patients with COPD-PH
Because the increase in the miR-190a-5p levels correlated with the severity of COPD-PH, we investigated whether miR-190a-5p could be used as a diagnostic and prognostic marker for the condition. The area under the curve (AUC) for the prediction of PH using circulating miR-190a-5p levels was 0.658 (95% CI 0.553–0.764), and the cutoff point was 2.0, yielding a sensitivity of 0.534 and a specificity of 0.781 (Figure 2A). The AUC of circulating miR-190a-5p to predict COPD-PH was 0.811 (95% CI 0.693–0.929) and the cutoff point was 2.75, with a sensitivity of 0.704 and a specificity of 0.844 (Figure 2B). Univariate Cox regression analyses identified PO2 as a risk factor for all-cause mortality (HR 0.873; 95% CI; 0.791–0.963; P=0.007) in patients with COPD-PH (Table 2); this finding remained consistent even in multivariate analysis, indicating that PO2 was an independent risk factor for all-cause mortality (HR 0.847, 95% CI 0.762–0.987; P=0.034) in this patient population. The forced expiratory volume in 1 second (FEV1%) and miR-190a-5p level were the two other parameters identified as independent risk factors of all-cause mortality in patients with COPD-PH (HR 0.904, 95% CI 0.824–0.992; P=0.032 and HR 1.740, 95% CI 1.053–2.875; P=0.031, respectively). Survival analysis showed no relationship between miR-190a-5p levels and all-cause mortality in patients with PH (Figure 2C). However, there was an increased 2-year survival rate in patients with COPD-PH that had low miR-190a-5p levels (<2.75) as compared to those with high miR-190a-5p levels (≥2.75, P=0.049; Figure 2D).
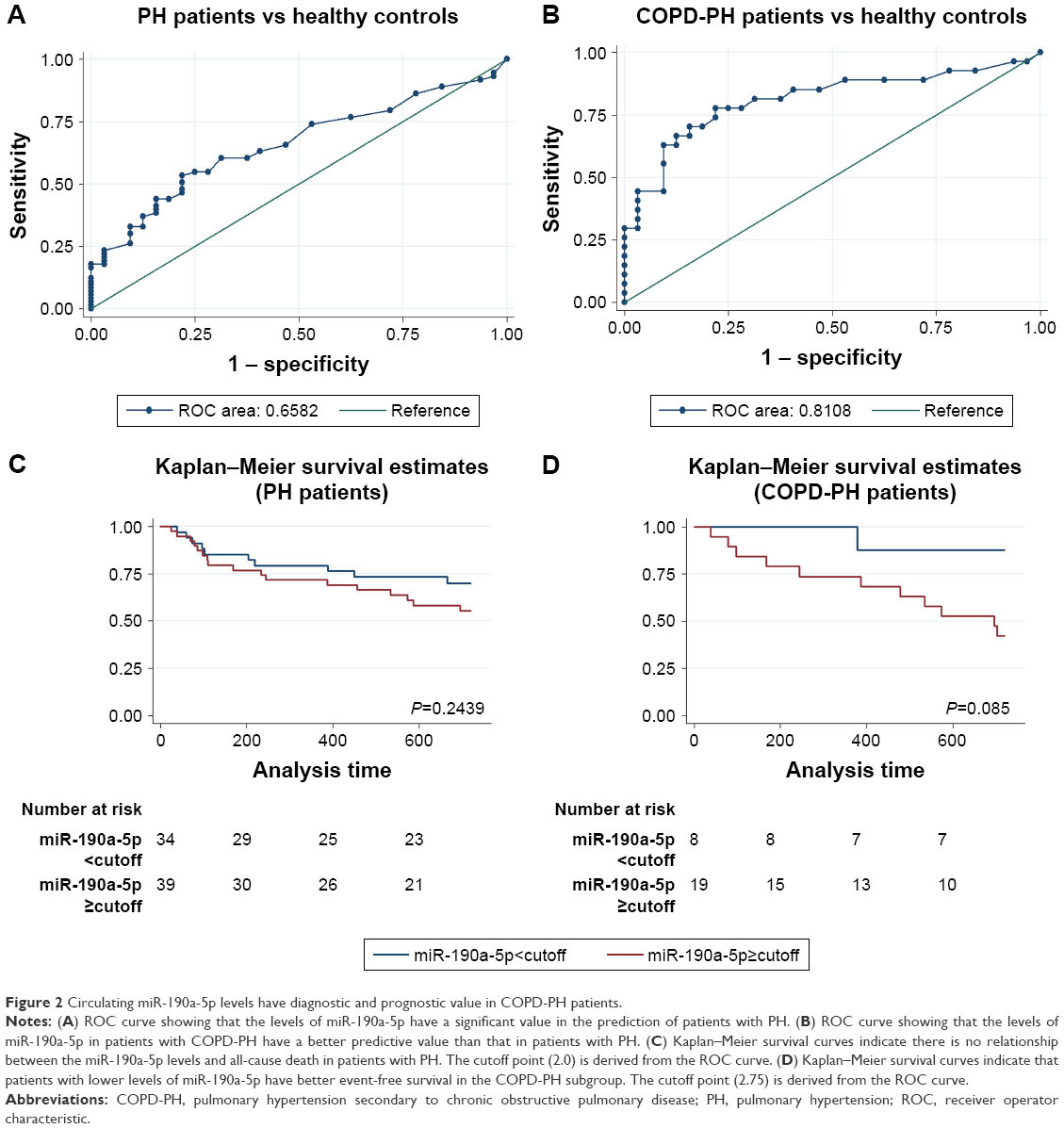 | Figure 2 Circulating miR-190a-5p levels have diagnostic and prognostic value in COPD-PH patients. |
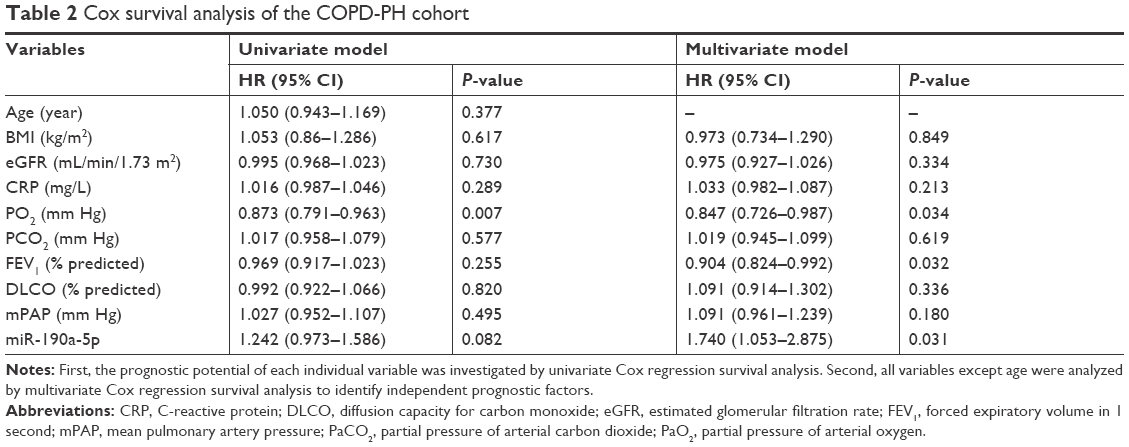 | Table 2 Cox survival analysis of the COPD-PH cohort |
Effects of hypoxia on the expression of miR-190a-5p, KLF15, Arg2, and eNOS in HPECs
Once the clinical relevance of miR-190a-5p was established, we attempted to identify potential miR-190a-5p sources that could mediate the effect of hypoxia on the onset and progression of PH in cell and animal models. Exposure to hypoxia significantly decreased the expression of KLF15 and eNOS protein in HPAECs and HPMECs (Figure 3A–C). However, the expression level of Arg2 protein was significantly increased in HPAECs and HPMECs that were exposed to hypoxia (Figure 3A–C). The results also showed that the levels of miR-190a-5p in HPAECs and HPMECs that were exposed to hypoxia for 24 hours were significantly higher than those in the cells that were maintained under normoxic conditions (Figure 3D). The levels of miR-190a-5p in HPASMCs and HPAFs that were exposed to hypoxia were not significantly different from those in the cells that were maintained under normoxic conditions (Figure 3D).
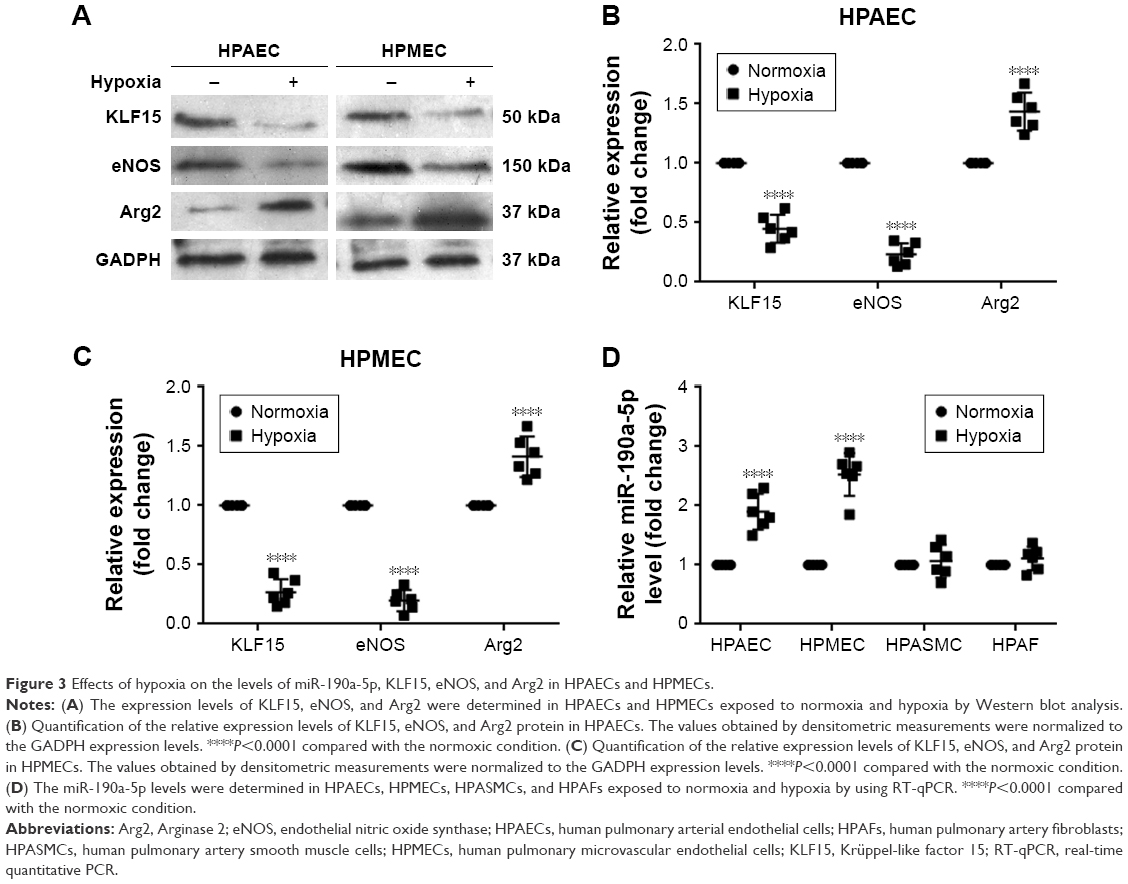 | Figure 3 Effects of hypoxia on the levels of miR-190a-5p, KLF15, eNOS, and Arg2 in HPAECs and HPMECs. |
miR-190a-5p expression correlated with the expression levels of Arg2 and eNOS by targeting KLF15
KLF15 has been reported to serve as a critical regulator of pulmonary endothelial homeostasis by modulating the endothelial expression of Arg2 and eNOS. Therefore, we investigated whether miR-190a-5p levels correlated with the expression levels of Arg2 and eNOS by targeting KLF15. The potential binding sites for KLF-15 on miR-190a-5p were predicted using Target Scan (Figure 4A). The luciferase reporter assay was used to confirm these predictions. Data from the luciferase reporter assay showed that HPAECs that were transfected with an miR-190a-5p inhibitor had markedly higher luciferase activity of KLF15 3′-UTR-luci-WT than observed in the control group, without any such obvious changes in the luciferase activity of KLF15 3′-UTR-luci-Mut (Figure 4B). In contrast, the luciferase activity of KLF15 3′-UTR-luci-WT was lower in cells that were transfected by the miR-190a-5p mimic than that observed in the control group (P<0.0001; Figure 4B).
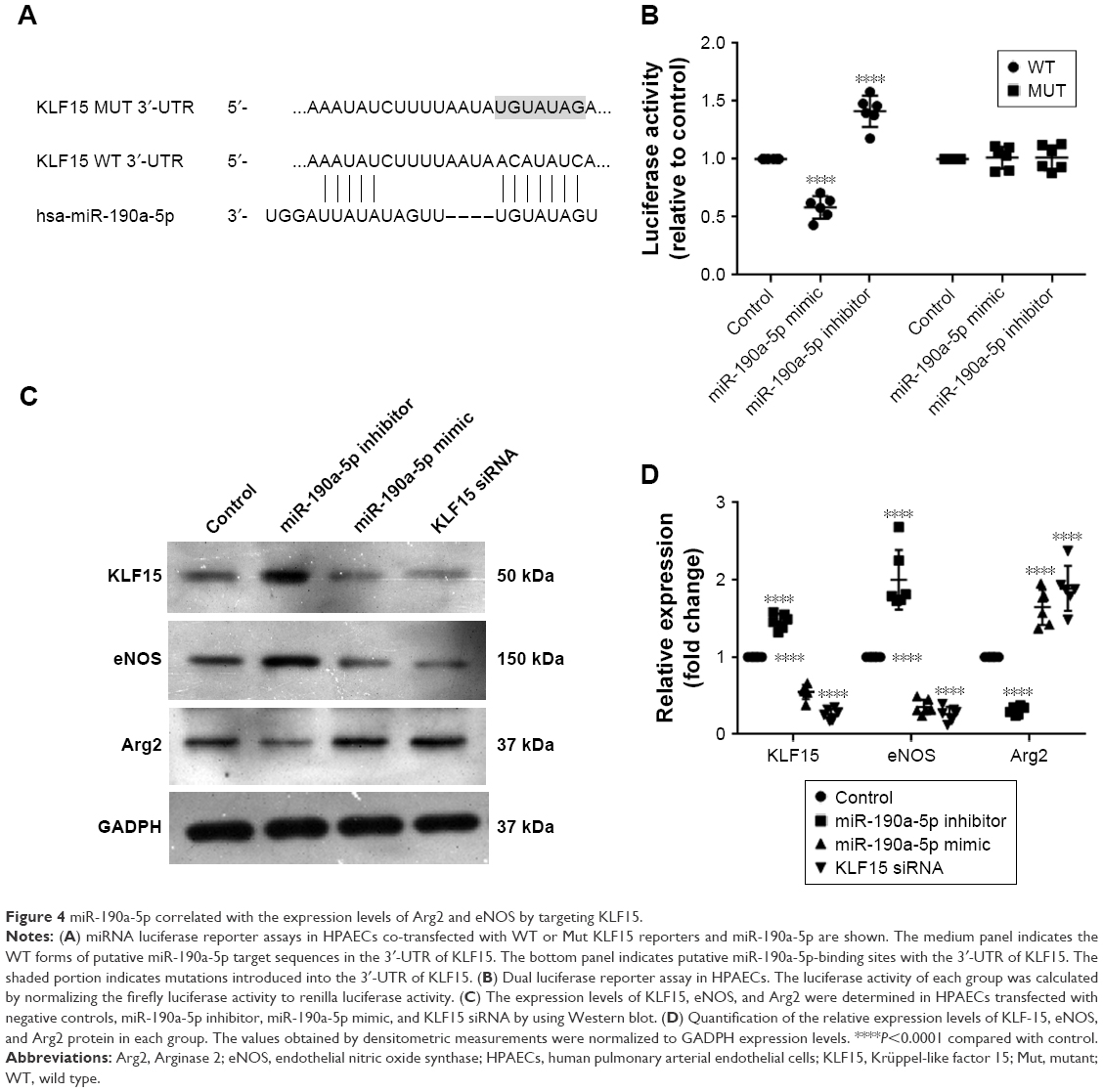 | Figure 4 miR-190a-5p correlated with the expression levels of Arg2 and eNOS by targeting KLF15. |
In the HPAECs that were transfected with the miR-190a-5p mimic and siRNA KLF15, the protein expression levels of KLF15 and eNOS were significantly lower, while the expression levels of Arg2 were significantly higher compared to the values that were observed in the control cells (Figure 4C and D). In contrast, the inhibitor of miR-190a-5p significantly increased the protein levels of KLF15 and eNOS and decreased the expression of Arg2, as compared to levels observed in the control groups (Figure 4C and D).
The in vivo effect of antagomir-190a-5p on the expression of KLF15, Arg2, and eNOS and the development of hypoxic PH
To study the effect of miR-190a-5p inhibition on the expression levels of KLF15, Arg2, and eNOS, as well as the development of hypoxic PH, we established an animal model of PH by exposing mice to hypoxia and intravenously administering antagomir-190a-5p or a scramble negative control. In the hypoxia-antagomir-NC group, RVSP and RV/(LV+S) were significantly higher than those in the normoxia group (Figure 5A–E). The increased RVSP and RV/(LV+S) was not observed in mice exposed to chronic hypoxia following administration of antagomir-190a-5p. The levels of miR-190a-5p in the hypoxia-antagomir-NC group were significantly higher than those in the normoxia group. The levels of miR-190a-5p in the hypoxia-antagomir-190a-5p group were significantly lower than those in the hypoxia-antagomir-NC group (Figure 5F). Compared to the normoxia group, the hypoxia-antagomir-NC group showed significantly lower expression levels of KLF15 protein and significantly higher expression levels of eNOS and Arg2 protein (Figure 5G and H). Compared to the hypoxia-antagomir-NC group, the hypoxia-antagomir-190a-5p group showed significantly higher expression levels of KLF15 and eNOS protein and significantly decreased levels of Arg2 protein.
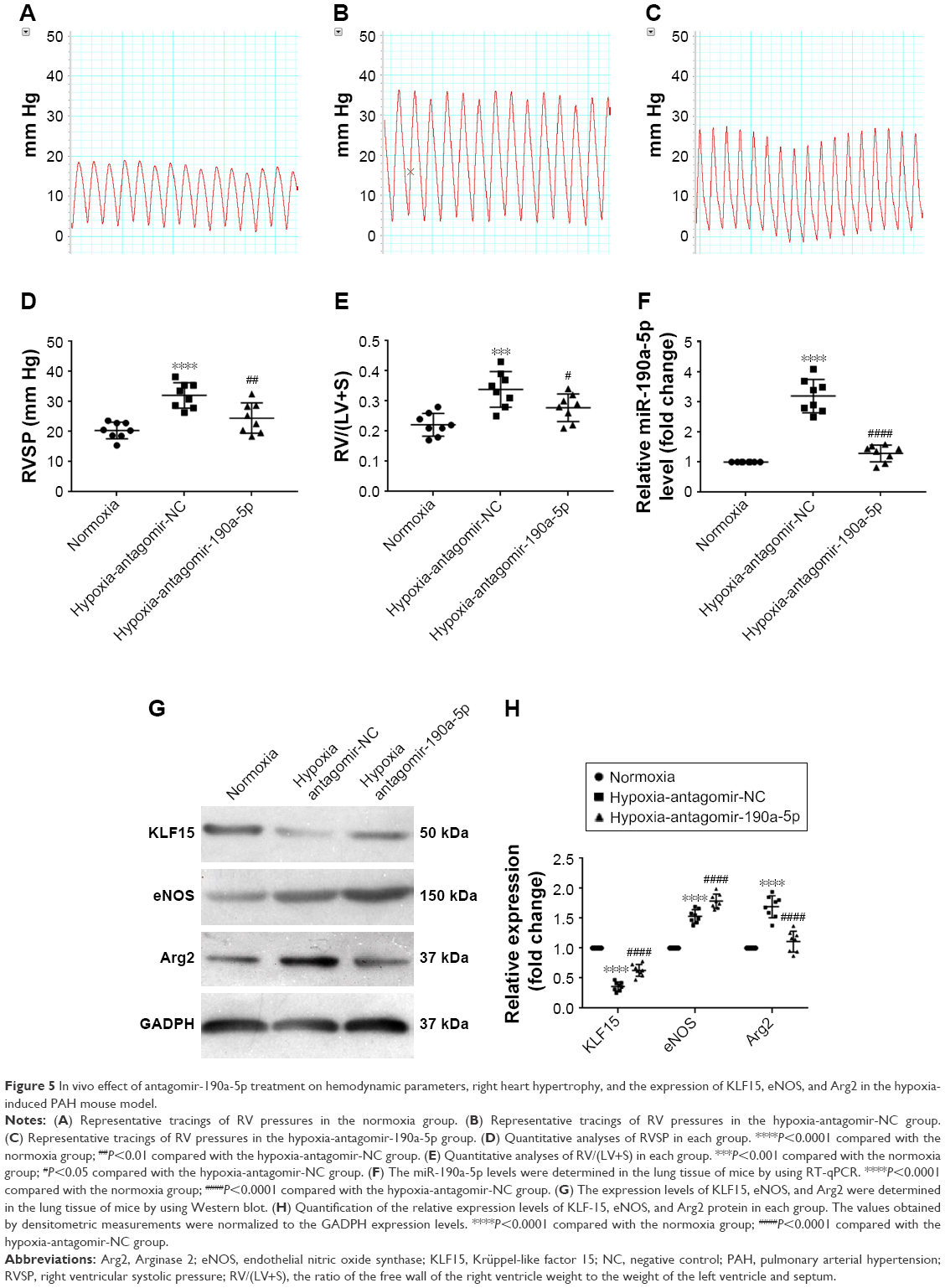 | Figure 5 In vivo effect of antagomir-190a-5p treatment on hemodynamic parameters, right heart hypertrophy, and the expression of KLF15, eNOS, and Arg2 in the hypoxia-induced PAH mouse model. |
Discussion
The findings of the present study show that the levels of circulating miR-190a-5p were increased in patients with PH, especially among those with COPD-PH. Higher levels of miR-190a-5p were associated with a lower PO2 and increased COPD-PH severity. We also found that circulating levels of miR-190a-5p may have the potential to be a new serum biomarker for the diagnosis and prognosis of COPD-PH. Moreover, we observed that hypoxia upregulates the expression of miR-190a-5p in HPAECs and HPMECs. Additionally, we found that the miR-190a-5p expression correlated with the expression levels of Arg2 and eNOS’ by targeting KLF15. Our results suggest that the inhibition of miR-190a-5p modulates the expression of KLF15, Arg2, and eNOS, thereby affecting the development of hypoxia-induced PH in an animal model.
PH can be idiopathic or hereditary or it can occur as a result of thromboembolism, hypoxia caused by vascular or pulmonary parenchymal disease, connective tissue disease, congenital heart disease, heart failure, or a combination of these factors.28 In the case of COPD, hypoxia has been identified as the principal pathophysiological mechanism underlying the development of PH.35,36 Hypoxia leads to a decrease in the expression of eNOS in vascular ECs.37,38 However, several studies have indicated that the gene expression, protein content, and activity of lung eNOS are high in mice with chronic hypoxic PH.39–41 These findings are consistent with our observations. Nitric oxide (NO) is a potent pulmonary artery vasodilator; altered NO production may contribute to the development of PH.42,43 Our study shows that, in response to hypoxia, the expression of KLF15 and eNOS decreased significantly, while the expression of Arg2 protein increased. Studies have shown that the production of eNOS-derived NO and endothelial function is reciprocally regulated by Arg2.44,45 KLF15 has been reported to mediate Arg2 regulation of the eNOS signaling pathway.13 This was also confirmed in the present study. Moreover, we found that miR-190a-5p not only targeted KLF15 but also showed a correlation with the expression levels of Arg2 and eNOS. Hypoxia induces pulmonary endothelial dysfunction46 as well as the resultant changes in the expression of KLF15, Arg2, and eNOS.13 Moreover, the levels of miR-190a-5p in HPAECs and HPMECs increased significantly on exposure to hypoxia, which might be partially explained by the fact that miR-190a-5p belongs to the hypoxamir family and is upregulated during hypoxia. The changes in the expression of KLF15, Arg2, and eNOS in HPAECs and HPMECs in response to hypoxia provide an insight into the endothelial dysfunction that occurs not only in the pulmonary artery but also in the pulmonary microcirculation. Furthermore, hypoxia-induced increases in the expression of miR-190a-5p were observed in HPAECs and HPMECs. This suggests that the ECs of the pulmonary artery and pulmonary microcirculation are both the main sources of miR-190a-5p under hypoxic conditions. Consistent with our in vitro findings, the levels of miR-190a-5p were also found to be significantly higher in the animal model of hypoxic PH than in the control. We observed that antagomir-190a-5p administration prevented the development of hypoxia-induced PH in a mouse model, supported by the absence of an elevation in RVSP and RV/(LV+S) in animals treated with antagomir-190a-5p. We believe that the main reason for this phenomenon is that the inhibition of miR-190a-5p modulates the expression of Arg2 and eNOS by targeting KLF15, which relieves the pulmonary endothelial dysfunction that is induced by hypoxia, reverses the hypoxia-induced endothelium-mediated vasoconstriction, and attenuates the RVSP and right ventricular hypertrophy.
In this study, the miR-190a-5p levels in patients with COPD-PH were significantly higher than those in healthy controls, while there were no obvious differences between the other PH groups and the healthy controls. Although the miR-190a-5p levels in the global cohort of patients with PH were higher than those in the controls, it appears that this difference can mostly be attributed to the patients with COPD-PH, who represent the largest subgroup. Similarly, ROC curve analysis of the diagnostic value of miR-190a-5p revealed that the AUC of circulating miR-190a-5p for the prediction of COPD-PH was significantly greater than that of total PH.
Multivariate Cox regression analyses revealed that PO2, FEV1, and miR-190a-5p expression were independent risk factors of all-cause mortality in patients with COPD-PH. In addition, lower levels of miR-190a-5p were predictive of event-free survival in the COPD-PH subgroup, but not in the global PH group; this indicates that miR-190a-5p has a unique prognostic value in COPD-PH. COPD-related PH has been classified as group 3 “pulmonary hypertension due to lung disease and/or hypoxia,”28 which suggests that exposure to hypoxic conditions plays a more important role in the pathophysiologic mechanism of COPD-PH than in other types of PH. This is an important reason why the circulating level of miR-190a-5p shows an independent prognostic value in patients with COPD-PH, but not in the global cohort of patients with PH. In addition, miR-190a-5p shows a negative correlation with KLF15 mRNA in patients with COPD-PH. Furthermore, higher levels of miR-190a-5p were associated with a lower PO2 and greater severity of COPD-PH. This implies that circulating miR-190a-5p not only serves as a marker for the diagnosis and prognosis of COPD-PH, but can also be used to evaluate the severity and progression of the disease.
Limitations
Some limitations of the present study should be taken into consideration. First, computational miRNA target prediction analysis indicates that the miR-190a-5p fragment pairs well with the fragment of the KLF15 3′-UTR in the human species, but not in the same manner as that in the mouse. However, this UTR is highly conserved in mammals,47 and a sequence (UGAUAUGUUUGAUAUAUUAGGU) within hsa-miR-190a-5p is the same as the seed sequence of the mmu-miR-190a-5p. Therefore, the differences between the miR-190a-5p in these two animal species are unlikely to reduce the validity of the data interpretation in this study. Second, although it would enable further confirmation of the current findings, we did not use microarray to examine the global expression of miRNA in this study. Third, the trial was carried out at a single center, and therefore, it is only representative of the local practices of managing COPD-PH. Therefore, the results of this study should not be directly extrapolated to other centers or countries. Finally, we could not collect the data on COPD patients with or without PH due to congenital heart disease; obtaining data on these patients could be helpful in gaining a comprehensive understanding of the role of miR-190a-5p in congenital heart disease.
Conclusion
Our findings indicate that hypoxia-induced PH is mediated by pulmonary endothelial dysfunction; activation of the KLF15, Arg2, and eNOS pathways; as well as increased expression of miR-190a-5p. Consistent with these findings, we observed that inhibition of miR-190a-5p attenuated the development of hypoxia-induced PH via the Arg2 and eNOS pathways by modulating the expression of KLF15. The circulating miR-190a-5p level not only has the potential to be a diagnostic and prognostic marker but can also serve as a parameter for the evaluation of the progression of COPD-PH.
Acknowledgments
The present study was supported by the National Natural Science Foundation of China (grant nos. 81560067 and 81860071) and “Medical Excellence Award” Funded by Creative Research Development Grant from the First Affiliated Hospital of Guangxi Medical University. This manuscript has been edited and proofread by Medjaden Bioscience Limited.
Disclosure
The authors report no conflicts of interest in this work.
References
Thenappan T, Ormiston ML, Ryan JJ, Archer SL. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ. 2018;360:j5492. | ||
Chen D, Gao W, Wang S, Ni B, Gao Y. Critical effects of epigenetic regulation in pulmonary arterial hypertension. Cell Mol Life Sci. 2017;74(20):3789–3808. | ||
Sakao S, Voelkel NF, Tatsumi K. The vascular bed in COPD: pulmonary hypertension and pulmonary vascular alterations. Eur Respir Rev. 2014;23(133):350–355. | ||
Wrobel JP, Thompson BR, Williams TJ. Mechanisms of pulmonary hypertension in chronic obstructive pulmonary disease: a pathophysiologic review. J Heart Lung Transplant. 2012;31(6):557–564. | ||
Nikitopoulou I, Orfanos SE, Kotanidou A, et al. Vascular endothelial-cadherin downregulation as a feature of endothelial transdifferentiation in monocrotaline-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2016;311(2):L352–L363. | ||
Ogoshi T, Tsutsui M, Kido T, et al. Protective Role of Myelocytic Nitric Oxide Synthases against Hypoxic Pulmonary Hypertension in Mice. Am J Respir Crit Care Med. 2018;198(2):232–244. | ||
Lim HK, Lim HK, Ryoo S, et al. Mitochondrial arginase II constrains endothelial NOS-3 activity. Am J Physiol Heart Circ Physiol. 2007;293(6):H3317–H3324. | ||
Krause BJ, Carrasco-Wong I, Caniuguir A, Carvajal J, Farías M, Casanello P. Endothelial eNOS/arginase imbalance contributes to vascular dysfunction in IUGR umbilical and placental vessels. Placenta. 2013;34(1):20–28. | ||
Swamynathan SK. Krüppel-like factors: three fingers in control. Hum Genomics. 2010;4(4):263–270. | ||
Morris VA, Cummings CL, Korb B, Boaglio S, Oehler VG. Deregulated KLF4 Expression in Myeloid Leukemias Alters Cell Proliferation and Differentiation through MicroRNA and Gene Targets. Mol Cell Biol. 2016;36(4):559–573. | ||
Lu Y, Zhang L, Liao X, et al. Kruppel-like factor 15 is critical for vascular inflammation. J Clin Invest. 2013;123(10):4232–4241. | ||
Patel SK, Ramchand J, Crocitti V, Burrell LM. Kruppel-Like Factor 15 Is Critical for the Development of Left Ventricular Hypertrophy. Int J Mol Sci. 2018;19(5):1303. | ||
Pandey D, Nomura Y, Rossberg MC, et al. Hypoxia Triggers SENP1 (Sentrin-Specific Protease 1) Modulation of KLF15 (Kruppel-Like Factor 15) and Transcriptional Regulation of Arg2 (Arginase 2) in Pulmonary Endothelium. Arterioscler Thromb Vasc Biol. 2018;38(4):913–926. | ||
Kaiser R, Frantz C, Bals R, Wilkens H. The role of circulating thrombospondin-1 in patients with precapillary pulmonary hypertension. Respir Res. 2016;17(1):96. | ||
Hamam R, Hamam D, Alsaleh KA, et al. Circulating microRNAs in breast cancer: novel diagnostic and prognostic biomarkers. Cell Death Dis. 2017;8(9):e3045. | ||
Jakob P, Kacprowski T, Briand-Schumacher S, et al. Profiling and validation of circulating microRNAs for cardiovascular events in patients presenting with ST-segment elevation myocardial infarction. Eur Heart J. 2017;38(7):511–515. | ||
Baptista R, Marques C, Catarino S, et al. MicroRNA-424(322) as a new marker of disease progression in pulmonary arterial hypertension and its role in right ventricular hypertrophy by targeting SMURF1. Cardiovasc Res. 2018;114(1):53–64. | ||
Chen W, Li S. Circulating microRNA as a novel biomarker for pulmonary arterial hypertension due to congenital heart disease. Pediatr Cardiol. 2017;38(1):86–94. | ||
Jin P, Gu W, Lai Y, Zheng W, Zhou Q, Wu X. The Circulating MicroRNA-206 level predicts the severity of pulmonary hypertension in patients with left heart diseases. Cell Physiol Biochem. 2017;41(6):2150–2160. | ||
Nallamshetty S, Chan SY, Loscalzo J. Hypoxia: a master regulator of microRNA biogenesis and activity. Free Radic Biol Med. 2013;64:20–30. | ||
Greco S, Martelli F. MicroRNAs in Hypoxia Response. Antioxid Redox Signal. 2014;21(8):1164–1166. | ||
Liu H, Yin T, Yan W, et al. Dysregulation of microRNA-214 and PTEN contributes to the pathogenesis of hypoxic pulmonary hypertension. Int J Chron Obstruct Pulmon Dis. 2017;12:1781–1791. | ||
Liu H, Tao Y, Chen M, et al. Upregulation of MicroRNA-214 Contributes to the Development of Vascular Remodeling in Hypoxia-induced Pulmonary Hypertension Via Targeting CCNL2. Sci Rep. 2016;6:24661. | ||
Hale AE, White K, Chan SY. Hypoxamirs in pulmonary hypertension: breathing new life into pulmonary vascular research. Cardiovasc Diagn Ther. 2012;2(3):200–212. | ||
Li SS, Ran YJ, Zhang DD, Li SZ, Zhu D. MicroRNA-190 regulates hypoxic pulmonary vasoconstriction by targeting a voltage-gated K+ channel in arterial smooth muscle cells. J Cell Biochem. 2014;115(6):1196–1205. | ||
Blissenbach B, Nakas CT, Krönke M, Geiser T, Merz TM, Pichler Hefti J. Hypoxia-induced changes in plasma micro-RNAs correlate with pulmonary artery pressure at high altitude. Am J Physiol Lung Cell Mol Physiol. 2018;314(1):L157–L164. | ||
Brown LM, Chen H, Halpern S, et al. Delay in recognition of pulmonary arterial hypertension: factors identified from the REVEAL Registry. Chest. 2011;140(1):19–26. | ||
Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J. 2015;46(4):903–975. | ||
Wang L, Jin YZ, Zhao QH, et al. Hemodynamic and gas exchange effects of inhaled iloprost in patients with COPD and pulmonary hypertension. Int J Chron Obstruct Pulmon Dis. 2017;12:3353–3360. | ||
Seeger W, Adir Y, Barberà JA, et al. Pulmonary hypertension in chronic lung diseases. J Am Coll Cardiol. 2013;62(25 Suppl):D109–D116. | ||
Lao BB, Grishagin I, Mesallati H, Brewer TF, Olenyuk BZ, Arora PS. In vivo modulation of hypoxia-inducible signaling by topographical helix mimetics. Proc Natl Acad Sci U S A. 2014;111(21):7531–7536. | ||
Kubli DA, Zhang X, Lee Y, et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. 2013;288(2):915–926. | ||
Pullamsetti SS, Doebele C, Fischer A, et al. Inhibition of microRNA-17 improves lung and heart function in experimental pulmonary hypertension. Am J Respir Crit Care Med. 2012;185(4):409–419. | ||
Brock M, Samillan VJ, Trenkmann M, et al. AntagomiR directed against miR-20a restores functional BMPR2 signalling and prevents vascular remodelling in hypoxia-induced pulmonary hypertension. Eur Heart J. 2014;35(45):3203–3211. | ||
Samareh Fekri M, Torabi M, Azizi Shoul S, Mirzaee M. Prevalence and predictors associated with severe pulmonary hypertension in COPD. Am J Emerg Med. 2018;36(2):277–280. | ||
Portillo K, Torralba Y, Blanco I, et al. Pulmonary hemodynamic profile in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2015;10:1313–1320. | ||
Takemoto M, Sun J, Hiroki J, Shimokawa H, Liao JK. Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation. 2002;106(1):57–62. | ||
Koudelka A, Ambrozova G, Klinke A, et al. Nitro-Oleic Acid Prevents Hypoxia- and Asymmetric Dimethylarginine-Induced Pulmonary Endothelial Dysfunction. Cardiovasc Drugs Ther. 2016;30(6):579–586. | ||
Duluc L, Ahmetaj-Shala B, Mitchell J, et al. Tipifarnib prevents development of hypoxia-induced pulmonary hypertension. Cardiovasc Res. 2017;113(3):276–287. | ||
Dubois M, Delannoy E, Duluc L, et al. Biopterin metabolism and eNOS expression during hypoxic pulmonary hypertension in mice. PLoS One. 2013;8(11):e82594. | ||
Kim SY, Lee JH, Huh JW, et al. Bortezomib alleviates experimental pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2012;47(5):698–708. | ||
Zhang R, Wang XJ, Zhang HD, et al. Profiling nitric oxide metabolites in patients with idiopathic pulmonary arterial hypertension. Eur Respir J. 2016;48(5):1386–1395. | ||
Hajian B, de Backer J, Vos W, et al. Pulmonary vascular effects of pulsed inhaled nitric oxide in COPD patients with pulmonary hypertension. Int J Chron Obstruct Pulmon Dis. 2016;11:1533–1541. | ||
Pandey D, Bhunia A, Oh YJ, et al. OxLDL triggers retrograde translocation of arginase2 in aortic endothelial cells via ROCK and mitochondrial processing peptidase. Circ Res. 2014;115(4):450–459. | ||
Sikka G, Pandey D, Bhuniya AK, et al. Contribution of arginase activation to vascular dysfunction in cigarette smoking. Atherosclerosis. 2013;231(1):91–94. | ||
Keymel S, Schueller B, Sansone R, et al. Oxygen dependence of endothelium-dependent vasodilation: importance in chronic obstructive pulmonary disease. Arch Med Sci. 2018;14(2):297–306. | ||
Shao NY, Hu HY, Yan Z, et al. Comprehensive survey of human brain microRNA by deep sequencing. BMC Genomics. 2010;11:409. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.