")
Back to Journals » OncoTargets and Therapy » Volume 12
MiR-145 inhibits EGF-induced epithelial-to-mesenchymal transition via targeting Smad2 in human glioblastoma
Authors Chen W, Huang B, Wang E, Wang X
Received 18 January 2019
Accepted for publication 7 March 2019
Published 23 April 2019 Volume 2019:12 Pages 3099—3107
DOI https://doi.org/10.2147/OTT.S202129
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Weijie Chen,1 Baochen Huang,1 Enqin Wang,2 Xingqiang Wang1
1Department of Neurosurgery, People’s Hospital of Rizhao, Jining Medical University, Rizhao 276826, People’s Republic of China; 2Clinical Skill Training Center, People’s Hospital of Rizhao, Jining Medical University, Rizhao 276826, People’s Republic of China
Background/Aims: MiR-145 and Smad2 have been widely reported in the development and progression of human malignancies. In the present study, we investigated the correlation between miR-145 and Smad2 in human glioblastoma multiforme (GBM).
Methods: The epithelial–mesenchymal transition (EMT) biomarkers and Smad2 were assessed by Western blot. The silencing of Smad2 was conducted by transfection of Smad2 siRNAs. The cell migration and invasion were evaluated using Transwell assays, respectively. The dual luciferase reporter assay was performed to identify whether Smad2 is a direct target of miR-145.
Results: The epidermal growth factor (EGF) activated the phosphorylation of Smad2 in U87 and U251 cells in a time- and dose-dependent manner. However, treatment with silencing of Smad2 or overexpression of miR-145 significantly inhibited the expressions of total Smad2, N-cadherin, Vimentin and matrix metallopeptidase 9, but induced the expression of E-cadherin. In addition, silencing of Smad2 or overexpression of miR-145 also inhibited the migration and invasion of U87 and U251 cells. Mechanistically, Smad2 was confirmed to be a target gene of miR-145 by bioinformatics analysis and luciferase reporter assay. Restored Smad2 expression also reversed miR-145-induced inhibition of EMT in U87 and U251 cells.
Conclusion: MiR-145 inhibits EGF-induced EMT via targeting Smad2 in human GBM. Therefore, miR-145 may be a promising biomarker and therapeutic target for GBM patients.
Keywords: MiR-145, Smad2, EGF, GBM, EMT
Introduction
Glioblastoma multiforme (GBM) is a kind of cerebral tumors, which is derived from astrocytes.1–3 The therapy option for GBM patients is based on surgery followed by radiation and chemotherapy with temozolomide. Despite these treatments, overall survival (about 15 months) and the 5-year survival rate (<5%) remain very low due to its histological heterogeneity, aggressive property and poor response to radio-/chemotherapy.4,5 Thus, it is essential to understand the molecular mechanisms of GBM to improve anti-cancer strategies in GBM patients.
Epithelial–mesenchymal transition (EMT) is a well-recognized biological process, which is associated with tumor metastasis.6,7 The underlying molecular mechanisms of EMT in various cancer cells are complicated, and Smad2 has been identified as a key factor in the induction of EMT.8–10 In addition, miRNAs, a class of small non-coding RNA, regulate gene expression by facilitating mRNA degradation or inhibiting translation of target mRNAs.11,12 MiRNAs have been demonstrated to be involved in various human diseases, and play important roles in cellular differentiation, proliferation, metastasis and EMT.12 Notably, aberrant miR-145 expression was also reported to be implicated in cancer progression by regulating target genes.13–15 Using bioinformatics analysis, we found that Smad2 was also a potential candidate target of miR-145. Therefore, we assumed that miR-145 might play a tumor suppressor role in EMT of GBM through targeting the Smad2 pathway.
In the present study, we explored the role of miR-145 in cell migration, invasion and EMT of U87 and U251 cells by transfecting miR-145 mimics. Then, we investigated the regulation of miR-145 on the expression of Smad2 using the dual luciferase reporter assay. This study will identify the role for miR-145 in regulation of tumorigenicity of GBM, and elucidate that miR-145 may be a vital therapeutic target for GBM.
Materials and methods
Cell culture
Human glioma cell lines U87 and U251 were obtained from the American Type Culture Collection. The U87 cells were grown in minimum essential medium (HyClone, Logan, UT, USA), and the U251 cells were maintained in Dulbecco’s high-glucose modified Eagle medium (HyClone). All cells were cultured with both 10% FBS (Gibco, GrandIsland, NY, USA) and 1% penicillin/streptavidin (Gibco) and cultured at 37°C in a humidified atmosphere at 5% CO2.
Cell transfection
For transfection, 2×105 cells per well were placed in a 6-well plate. After adhering for 24 hrs, miR-145 mimics and negative control miRNA (miR-NC) (RiboBio, Guangzhou, People’s Republic of China) were added to the transfection medium for 6 hrs at 37°C in a CO2 incubator. Loss of Smad2 expression was achieved using small interfering RNA (siRNA) of Smad2 (si-Smad2) (Santa Cruz Biotech, Santa Cruz, CA, USA). At the same time, control siRNAs (si-control) (Santa Cruz Biotech) were used as a negative control. The Smad2 cDNA was cloned into pcDNA 3.1 vector (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The constructs were verified by DNA sequencing. The construction and verification of the pcDNA 3.1-Smad2 plasmids was performed by Generay Biotech Co., Ltd (Shanghai, People’s Republic of China). Cell transfection was carried out using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The cells were supplemented with normal culture media and cultured at 37°C with 5% CO2 for up to 48 hrs before harvest.
Reverse transcription-quantitative PCR (qRT-PCR)
TRIzol Reagent (Thermo Fisher Scientific, Inc.) was used to extract total RNA from U251 cells, in accordance with the manufacturer’s instructions. A RevertAid First Strand cDNA Synthesis kit (Fermentas; Thermo Fisher Scientific, Inc., Pittsburgh, PA, USA) was used to reverse transcribe total RNA into cDNA, according to the manufacturer’s protocol. The miRNA expression was determined using a PrimeScript® miRNA RT-PCR kit (Takara Biotechnology Co., Ltd., Dalian, People’s Republic of China), in accordance with the manufacturer’s instructions. The PCR conditions were 95°C for 10 mins, and 40 cycles of denaturation at 95°C for 30 s and annealing/elongation at 60°C for 30 s. The primer sequences were purchased from Shanghai GenePharma Co., Ltd. (Shanghai, China). All miRNA data are expressed relative to a U6 small nuclear RNA from the same sample. Independent experiments were repeated three times. The relative expression levels of mRNA were analyzed by use of the 2−∆∆Cq method. The primer sequences were listed as below:
miR-145:
Forward primer 5ʹ-GGCGTCCAGTTTTCCCAG-3ʹ
Reverse primer 5ʹ-CAGTGCTGGGTCCGAGTGA-3ʹ
U6 SnRNA:
Forward primer 5ʹ-GCGCGTCGTGAAGCGTTC-3ʹ
Reverse primer 5ʹ-GTGCAGGGTCCGAGGT-3ʹ
Western blot
Cells were lysed by ice-cold lysis buffer (Cell Signaling Technology, Beverly, MA, USA) with added protease inhibitor cocktail (Sigma-Aldrich, St Louis, MO, USA). Extracts were centrifuged at 13,000 r.p.m. for 15 mins at 4°C and supernatant protein concentrations were determined using standard BSA method. Equal amounts of proteins were separated by Tris-glycine SDS-PAGE and electrotransferred to polyvinylidene fluoride membranes. Membranes were blocked with Tris-buffered saline containing 5% (wt/vol) non-fat dry milk for 1 hr and then immunoblotted overnight at 4°C with specific primary antibodies diluted in Tris-buffered saline with 5% (wt/vol) non-fat dried milk and 0.1% (vol/vol) Tween-20. After incubation with appropriate horseradish peroxidase-conjugated secondary antibody for 1 hr at room temperature, signals were detected with enhanced chemiluminescent and CL-XPosure film (Thermo Fisher Scientific, Inc.).
Migration and invasion assays
Firstly, cell migration assay was carried out using Tran-swell chambers without Matrigel®. However, the invasion assay was carried out using Transwell chambers (8 µm) containing Matrigel® (BD Biosciences, Franklin Lakes, NJ, USA). After 48 hrs incubation, 5×104 of cells in FBS-free DMEM were inoculated in the upper chambers. The bottom chambers were covered with 600 µL DMEM containing 20% FBS, which acted as the nutritional attractant. After 24 hrs of incubation at 37°C with 5% CO2, cells invading the filter membranes were fixed with 4% paraformaldehyde and stained with 0.05% crystal violet (Beyotime Institute of Biotechnology, Shanghai, China). Cells in at least five randomly selected visual fields were counted and expressed as the average number of cells per field of view using an inverted microscope (IX83; Olympus, Tokyo, Japan).
Bioinformatics
Analysis of potential miR-mRNA interactions was performed using the public databases TargetScan, PITA, miRIAD and picTAR. Potential direct interactions were considered probable when two or more algorithms returned a positive target prediction. In-silico analysis of miR expression levels was conducted using the intragenic microRNA database miRIAD (
Luciferase reporter assay
The 3′-UTR sequence of Smad2 containing the predicted miR-145 binding sites cloned into downstream of the luciferase gene in the luciferase reporter vector (Promega Corporation, Fitchburg, WI, USA) was named as Smad2 wild type (WT). The mutant type of Smad2 lacking complementarities with miR-145 binding sequence in the 3′-UTR region was also constructed and named as Smad2 mutant (MUT). Then, luciferase reporter vectors and miR-145 mimics were transfected into HEK-293T cells using Lipofectamine 2000 (Thermo Fisher Scientific). After 48 hrs, the luciferase activity was measured using a Dual-Luciferase Reporter detection System (Promega Corporation). The relative luciferase activity was expressed as the ratio of firefly luciferase to Renilla luciferase activity.
Statistical analysis
All experiments were conducted a minimum of three times independently, with similar results. Quantitative data is represented by means±standard error of the mean. Data were analyzed via GraphPad Prism 7.0. Statistical analysis was conducted by using one-way ANOVA followed by Dunnett’s multiple comparisons test. P-values for 95% CI were calculated. P<0.05 was considered statistically significant.
Results
EGF activates Smad2 in U87 and U251 cells
Previously, some studies demonstrated that EGF induced EMT in GBM cells. In view of the important role of Smad family in EMT, we firstly investigated the effect of EGF on the total expression and phosphorylation of Smad2. We found that EGF activated the phosphorylation of Smad2 in U87 and U251 cells in a time- and dose-dependent manner (P<0.001). Specifically, the level of phospho-Smad2 reached its maximum levels in U87 and U251 cells at a concentration of 30 ng/mL EGF after treatment with different dosage (Figure 1A). In addition, the level of phospho-Smad2 reached a peak at 8 hrs after EGF treatment in U87 and U251 cells (Figure 1B). However, EGF treatment did not affect the total expression level of Smad2 in U87 and U251 cells.
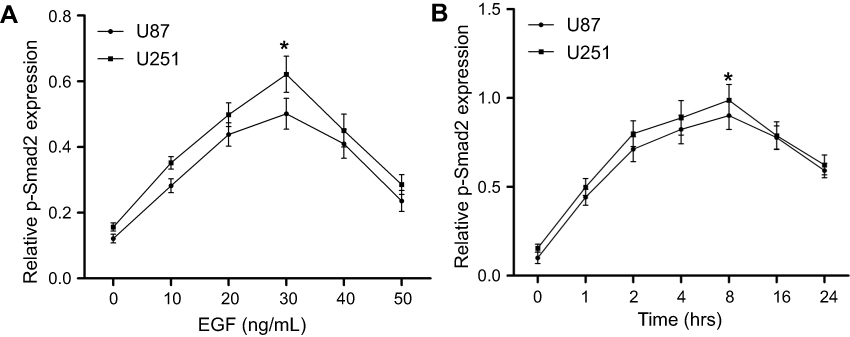 | Figure 1 EGF activates Smad2 in U87 and U251 cells. (A) U87 and U251 cells cultured in serum-free medium were treated with vehicle (0.1% DMSO) or increasing concentrations of EGF for 8 hrs. Western blot was incubated with anti-phospho-Smad2 and anti-Smad2 antibodies. (B) Cells were incubated with 30 ng/mL of EGF for the indicated time after serum starvation. The phosphorylation levels of Smad2 and total Smad2 were analyzed by Western blot at indicated time points. Phospho-Smad2 expression was normalized with total Smad2. All data represent the mean±SEM of three independent experiments with similar results. P-value was calculated compared with untreated control cells. *P<0.01 or a significant difference between the two test groups. Abbreviations: EGF, epidermal growth factor; SEM, standard error of the mean. |
Knockdown of Smad2 suppresses EGF-induced EMT
In this work, the role of Smad2 in EMT of GBM was investigated using a specific RNA interference. Here, U87 and U251 cells were transfected with si-control or si-Smad2. Firstly, we identified that si-Smad2 suppressed the expression of total Smad2 and the phosphorylation level of Smad2 in U87 and U251 cells (Figure 2A and B). In addition, EGF increased the expressions of N-cadherin, Vimentin and MMP9 in si-control-transfected U87 and U251 cells compared with those in control cells with the absence of EGF (Figure 2A and B). However, EGF-induced effects were partially repressed in si-Smad2-transfected U87 and U251 cells. In addition, Smad2 silencing increased the expression of E-cadherin in EGF-induced U87 and U251 cells. To further validate the impact of Smad2 on EMT, Transwell assay was carried out. Matrigel-coated (for invasion) or uncoated (for migration) Transwell assays showed that knockdown of Smad2 drastically decreased the migration (about 55%) and invasiveness (about 59%) of U87 and U251 cell lines (P<0.01, Figure 2C and D). These results confirmed that knockdown of Smad2 suppresses EGF-induced EMT.
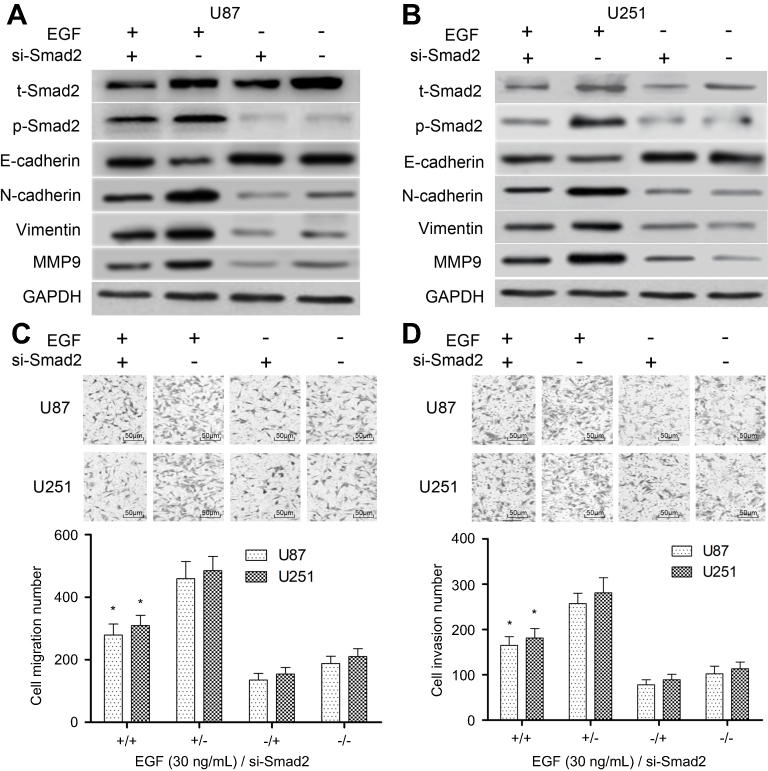 | Figure 2 Knockdown of Smad2 suppresses EGF-induced EMT. (A) U87 cells were transfected with the si-Smad2 or si-control, and subjected to vehicle (0.1% DMSO) or 30 ng/mL of EGF for 8 hrs, and the EMT markers (E-cadherin, N-cadherin, Vimentin and MMP9) were then detected by Western blotting. (B) U251 cells were transfected with the si-Smad2 or si-control, and subjected to vehicle (0.1% DMSO) or 30 ng/mL of EGF for 8 hrs, and the EMT markers were then detected by Western blotting. (C) Transwell assay was used to detect impaired migration ability of U87 and U251 cells under si-Smad2 or si-control treatment and their quantitative analysis. (D) Transwell assay was used to detect the invasion ability of U87 and U251 cells under si-Smad2 or si-control treatment and their quantitative analysis. Quantitative data are presented as the mean±SEM from three independent experiments. Experimental groups were compared to the negative control group. *P<0.01, compared with EGF alone or si-Smad2 alone group. “EGF +” means “30 ng/mL of EGF”; “EGF −” means “0.1% DMSO”; “si-Smad2 +” means “si-Smad2”; “si-Smad2 −” means “si-control”. Bar =50 μm. Abbreviations: EGF, epidermal growth factor; EMT, epithelial–mesenchymal transition; MMP9, matrix metallopeptidase 9; SEM, standard error of the mean. |
miR-145 suppresses the expression of total Smad2 to affect EMT
Previously, miR-145 mimics inhibited the EMT of U87 and U251 cells. To examine roles of miR-145 in regulating Smad2 and EMT, we transfected synthetic oligo mimics for miR-145 into U87 and U251 cells. Expression of miR-145 was increased around 10-fold after 48 hrs of transfection with its mimics (Figure 3A). To figure out whether miR-145 exerted a tumor-suppressor effect on GBM through targeting Smad2, we conducted gain of function and rescue experiments in U87 and U251 cells. We found that in the presence of EGF, overexpression of miR-145 significantly decreased endogenous Smad2 expression, and phosphorylation level as well as protein levels of EMT biomarkers in U87 (P<0.01, Figure 3B) and U251 cells (P<0.01, Figure 3C). Conversely, miR-145 mimics significantly induced E-cadherin expression, while the expressions of N-cadherin, Vimentin and MMP9 were significantly decreased in miR-145 mimics-induced U87 (P<0.01, Figure 3B) and U251 cells (P<0.01, Figure 3C). Intriguingly, using qRT-PCR, we found that Smad2 mRNA level did not change in miR-145 transfected cells compared with the control group, indicating that miR-145 regulates Smad2 only at a posttranslational level.
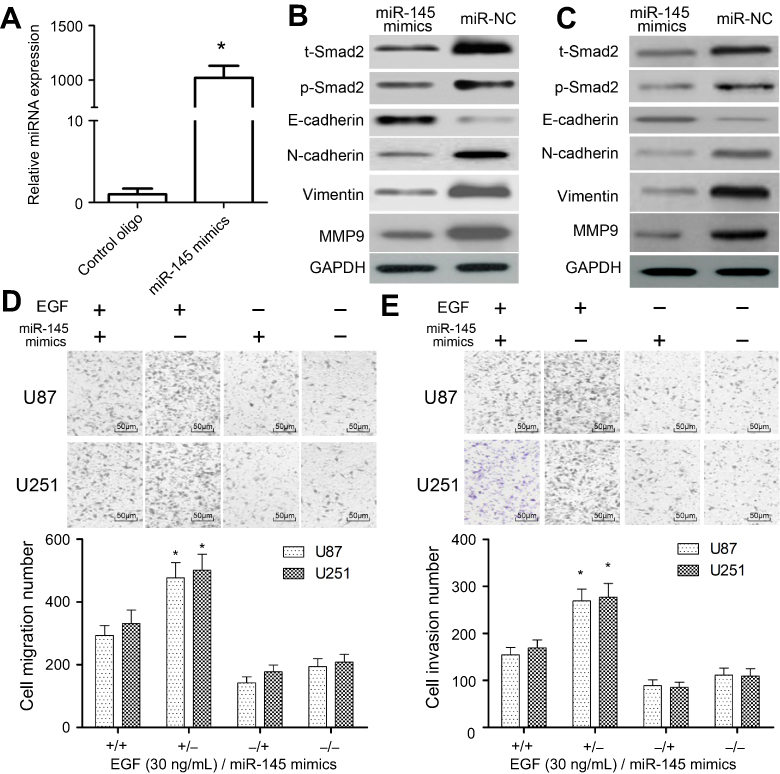 | Figure 3 Effects of miR-145 mimics on the expressions of Smad2 and EMT biomarkers. (A and B) U87 cells were co-transfected with miR-145 mimics or miR-NC. Cells were isolated and the expression of Smad2 and EMT biomarkers was analyzed by Western blotting after 48 hrs. (C) U251 cells were co-transfected with miR-145 mimics or miR-NC, and then the expressions of Smad2 and EMT biomarkers were analyzed by Western blotting after 48 hrs. (D) Transwell assay was used to detect impaired migration ability of U87 and U251 cells under si-Smad2 or si-control treatment and their quantitative analysis. (E) Transwell assay was used to detect the invasion ability of U87 and U251 cells under si-Smad2 or si-control treatment and their quantitative analysis. Quantitative data are presented as the mean±SEM from three independent experiments. Experimental groups were compared to the negative control group. *P<0.01, compared with EGF alone or miR-145 mimics alone group. “EGF +” means 30 ng/mL of EGF; “EGF −” means “0.1% DMSO”; “miR-145 mimics +” means “miR-145 mimics”; “miR-145 mimics −” means “miR-NC”. Bar =50 μm.Abbreviations: EMT, epithelial–mesenchymal transition; SEM, standard error of the mean. |
Further, we explored the impact of miR-145 on migration and invasion. Matrigel-coated (for invasion) or uncoated (for migration) Transwell assays showed that miR-145 overexpression drastically decreased the migration (about 55%) and invasiveness (about 56%) in U87 and U251 cell lines (P<0.01, Figure 3D and E). These findings confirmed miR-145 suppresses the expression level of total Smad2 to affect EMT.
miR-145 directly targets 3′-UTR of Smad2
Analysis of potential miR-mRNA interactions was performed using the public databases TargetScan, miRIAD and picTAR, and the results showed that Smad2 was a target gene of miR-145 (Figure 4A). To identify whether the predictive binding site of miR-145 on the 3′-UTR of Smad2 was required for this regulation, we cloned the WT or MUT Smad2 3′-UTR downstream of a luciferase reporter gene. Moreover, we co-transfected WT or MUT Smad2 vectors and miR-145 mimics into HEK-293T cells. We found that the co-transfection of WT Smad2 3′-UTR and miR-145 mimics into HEK-293T cells markedly reduced luciferase activity (P<0.01, Figure 4B). However, overexpression of miR-145 produced no significant change in luciferase activity in HEK-293T cells transfected with MUT Smad2 3′-UTR (P<0.01, Figure 4C).
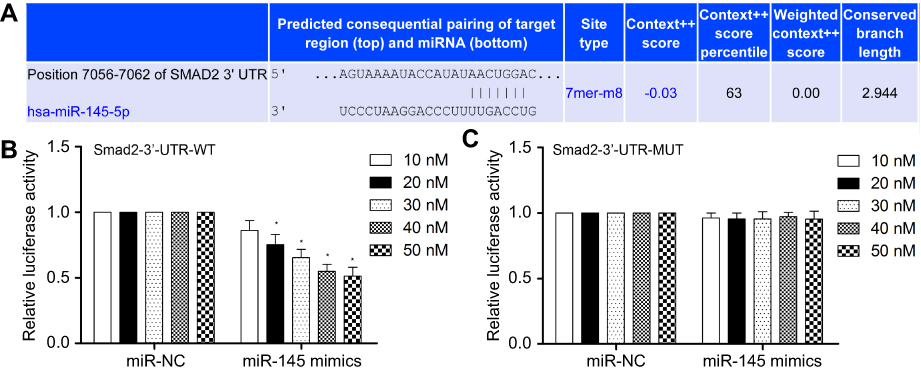 | Figure 4 MiR-145 directly targets the 3ʹ-UTR of Smad2 mRNA. (A) Base pairing between miR-145 and target sequences of Smad2. (B and C) HEK-293T cells were co-transfected with WT or MUT pMIR-Smad2 firefly luciferase reporter plasmids, pTK-Renilla luciferase plasmids, together with different concentrations of miR-145 mimics (10, 20, 30, 40 and 50 nM). Firefly luciferase activity was measured and normalized by Renilla luciferase activity after 36 hrs. Quantitative data are presented as the mean±SEM from three independent experiments. The data were subjected to Dunnett's Student’s t-test. *P<0.01 vs miR-NC. Abbreviations: WT, wild type; MUT, mutant; SEM, standard error of the mean. |
Restored Smad2 attenuates miR-145-inhibited EMT
Because Smad2 was a novel target of miR-145 in U87 and U251 cells, we further investigated whether Smad2 was involved in miR-145-inhibited EMT of U87 and U251 cells. As shown in Figure 5A and B, overexpression of Smad2 partially inhibited the miR-145-increased expression of E-cadherin and induced the miR-145-decreased expressions of N-cadherin, Vimentin and MMP9 in U87 and U251 cells. In addition, Transwell assay showed that the overexpression of Smad2 could reverse miR-145-inhibited migration and invasion compared with the vector group in U87 and U251 cells (P<0.01, Figure 5C and D).
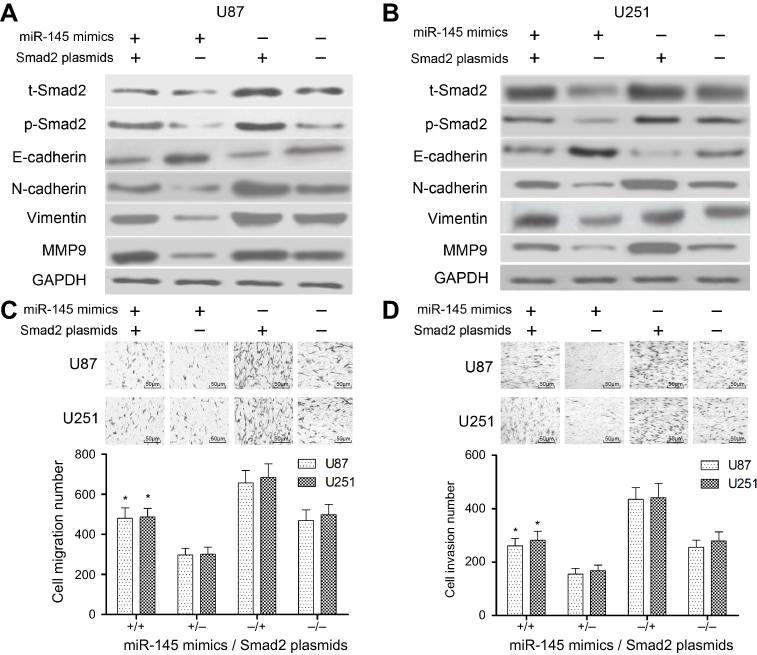 | Figure 5 The effect of miR-145 on EMT was mediated by Smad2. Cells in each group were treated with 30 ng/mL of EGF for 8 hrs. (A and B) The expression levels of EMT biomarkers after transfected with miR-145 mimics, Smad2 plasmids or control. (C) Uncoated Transwell assay was used to assess the migration effect of miR-145 mimics or Smad2 plasmids on U87 and U251cells. (D) Matrigel-coated Transwell assay was used to assess the invasiveness effect of miR-145 mimics or Smad2 plasmids on U87 and U251 cells. Each experiment was assayed in triplicate independently. Data were represented as mean±SEM. *P<0.01, compared with miR-145 mimics alone or Smad2 plasmids alone group. “miR-145 mimics +” means “miR-145 mimics”; “miR-145 mimics −” means “miR-NC”. “Smad2 plasmids +” means “Smad2 plasmids”; “Smad2 plasmids −” means “vector”. Bar =50 μm. Abbreviations: EMT, epithelial–mesenchymal transition; SEM, standard error of the mean. |
Discussion
To date, miRNAs have been reported to be implicated into some biological processes, including tumorigenesis and inflammatory response in numerous human diseases.15–17 As known to all, miR-145 has been reported to exert different effects on the development and progression of cancers. MiR-145 was additionally demonstrated to suppress TGF-β-induced EMT of non-small cell lung cancer by targeting MTDH and MAP3K1, or regulating the BAX/BCL-2 ratio and the caspase-3 cascade.16–18 Recently, it was demonstrated that miR-145 promoted esophageal cancer cells proliferation and metastasis by targeting SMAD5.19 Besides, Smad2/3 signals are translocated from the cytoplasm to the nucleus and to regulate matrix protein production, target gene transcription, and extracellular matrix synthesis and degradation.20 Smad2 pathway contributes to EMT in colorectal cancer and gastric cancer.21,22 EGF also activated the phosphorylation of Smad2 to participate in the EMT.23 Together with this analysis, miR-145 or Smad2 may play an anti-oncogenic role in GBM. However, the molecular mechanisms of miR-145 and Smad2 in GBM have been rarely reported till now.
The present study aimed to investigate the underlying mechanisms of miR-145 and Smad2 in the regulation of EMT of GBM. Firstly, we demonstrated that EGF effectively induced the phosphorylation of Smad2 in U87 and U251 cells in a time- and dose-dependent fashion. Based on the previous report, we further investigated the effect of miR-145 in EGF-induced EMT. The process of EMT in cancer cells confers properties that enhance their metastatic ability via increased motility and invasiveness, the ability to dismantle the extracellular matrix, and an increased potential to induce a stem cell-like state.9,10 Here, treatment with silencing of Smad2 or miR-145 mimics significantly inhibited the expressions of total Smad2 and E-cadherin, but induced N-cadherin, Vimentin and MMP9. In addition, either silencing of Smad2 or miR-145 mimics also inhibited the migration and invasion of U87 and U251 cells. These findings above indicated that miR-145 was indeed involved in cell migration and invasion through the regulation of EMT.
In most cancer cells, miRNAs regulate gene expression by switching off gene expression or degrading mRNA at the posttranscriptional level.6 Deregulated miRNAs function as either oncogenes or tumor suppressor genes in multiple cancer types.7 Among them, miR-145 has been reported to significantly affect oncogenicity in various neoplasms by binding to critical genes and signaling pathways, inhibiting the progression of cancers. Jiang et al reported that miR-145 targeted hepatitis B virus X-interacting protein to modulate breast cancer cell proliferation.15 Pan et al demonstrated that miR-145 suppressed the proliferation, invasion and migration of lung cancer cells by regulating the BAX/BCL-2 ratio and the caspase-3 cascade.17 Zhang et al suggested that miR-145 promotes esophageal cancer cells proliferation and metastasis by targeting SMAD5.19 In this work, miR-145 decreased Smad2 expression in U87 and U251 cells by directly targeting 3′-UTR of Smad2. The present study also confirmed Smad2 overexpression restored cell migration and invasion, and also effectively reversed the effect of miR-145 on GBM. Therefore, Smad2 is indeed a functional target gene of miR-145 in human GBM.
Conclusion
The findings above indicated that miR-145 repressed cell migration, invasion and EMT in human GBM cells by targeting Smad2 pathway. The present study confirmed the tumor-suppressive role of miR-145 in EMT of human GBM. Therefore, this study provided a novel insight into the underlying mechanisms of GBM progression, suggesting that miR-145 may be used as a novel candidate therapeutic target in GBM.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Taylor JW, Parikh M, Phillips JJ, et al. Phase-2 trial of palbociclib in adult patients with recurrent RB1-positive glioblastoma. J Neurooncol. 2018;140(2):477–483. doi:10.1007/s11060-018-2977-3
2. Lukas RV, Rodon J, Becker K, et al. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. J Neurooncol. 2018;140(2):317–328. doi:10.1007/s11060-018-2955-9
3. Marinelli A, Lamberti G, Cerbone L, et al. High-dose fotemustine in temozolomide-pretreated glioblastoma multiforme patients: a phase I/II trial. Medicine (Baltimore). 2018;97(27):e11254. doi:10.1097/MD.0000000000011254
4. Young JS, Bernal G, Polster SP, et al. Convection-enhanced delivery of polymeric nanoparticles encapsulating chemotherapy in canines with spontaneous supratentorial tumors. World Neurosurg. 2018;117:e698–e704. doi:10.1016/j.wneu.2018.06.114
5. Desjardins A, Gromeier M, Herndon JE
6. Stemmler MP, Eccles RL, Brabletz S, Brabletz T. Non-redundant functions of EMT transcription factors. Nat Cell Biol. 2019;21(1):102–112. doi:10.1038/s41556-018-0196-y
7. Conigliaro A, Cicchini C. Exosome-mediated signaling in epithelial to mesenchymal transition and tumor progression. J Clin Med. 2018;8(1):E26. doi:10.3390/jcm8010026
8. Guo S, Deng C-X. Effect of stromal cells in tumor microenvironment on metastasis initiation. Int J Biol Sci. 2018;14(14):2083–2093. doi:10.7150/ijbs.25720
9. Sisto M, Lisi S, Ribatti D. The role of the epithelial-to-mesenchymal transition (EMT) in diseases of the salivary glands. Histochem Cell Biol. 2018;150(2):133–147. doi:10.1007/s00418-018-1680-y
10. Yeh H-W, Hsu E-C, Lee S-S, et al. PSPC1 mediates TGF-β1 autocrine signalling and Smad2/3 target switching to promote EMT, stemness and metastasis. Nat Cell Biol. 2018;20(4):479–491. doi:10.1038/s41556-018-0062-y
11. Yachi K, Tsuda M, Kohsaka S, et al. miR-23a promotes invasion of glioblastoma via HOXD10-regulated glial-mesenchymal transition. Signal Transduct Target Ther. 2018;3:33. doi:10.1038/s41392-018-0033-6
12. Zeng W, Zhu J-F, Liu J-Y, et al. miR-133b inhibits cell proliferation, migration and invasion of esophageal squamous cell carcinoma by targeting EGFR. Biomed Pharmacother. 2019;111:476–484. doi:10.1016/j.biopha.2018.12.057
13. Sadeghiyeh N, Sehati N, Mansoori B, et al. MicroRNA-145 replacement effect on growth and migration inhibition in lung cancer cell line. Biomed Pharmacother. 2019;111:460–467. doi:10.1016/j.biopha.2018.12.094
14. Zhang H, Jiang M, Liu Q, Han Z, Zhao Y, Ji S. miR-145-5p inhibits the proliferation and migration of bladder cancer cells by targeting TAGLN2. Oncol Lett. 2018;16(5):6355–6360. doi:10.3892/ol.2018.9436
15. Jiang Y, Wang D, Ren H, Shi Y, Gao Y. MiR-145-targeted HBXIP modulates human breast cancer cell proliferation. Thorac Cancer. 2019;10(1):71–77. doi:10.1111/1759-7714.12903
16. Yin Q, Han Y, Zhu D, et al. miR-145 and miR-497 suppress TGF-β-induced epithelial-mesenchymal transition of non-small cell lung cancer by targeting MTDH. Cancer Cell Int. 2018;18:105. doi:10.1186/s12935-018-0601-4
17. Pan Y, Ye C, Tian Q, et al. miR-145 suppresses the proliferation, invasion and migration of NSCLC cells by regulating the BAX/BCL-2 ratio and the caspase-3 cascade. Oncol Lett. 2018;15(4):4337–4343. doi:10.3892/ol.2018.7863
18. Chang Y, Yan W, Sun C, Liu Q, Wang J, Wang M. miR-145-5p inhibits epithelial-mesenchymal transition via the JNK signaling pathway by targeting MAP3K1 in non-small cell lung cancer cells. Oncol Lett. 2017;14(6):6923–6928. doi:10.3892/ol.2017.7092
19. Zhang Q, Gan H, Song W, Chai D, Wu S. MicroRNA-145 promotes esophageal cancer cells proliferation and metastasis by targeting SMAD5. Scand J Gastroenterol. 2018;53(7):769–776. doi:10.1080/00365521.2018.1476913
20. Thien A, Prentzell MT, Holzwarth B, et al. TSC1 activates TGF-β-Smad2/3 signaling in growth arrest and epithelial-to-mesenchymal transition. Dev Cell. 2015;32(5):617–630. doi:10.1016/j.devcel.2015.01.026
21. Ren Y, Yang M, Chen M, et al. Microcystin-LR promotes epithelial-mesenchymal transition in colorectal cancer cells through PI3-K/AKT and SMAD2. Toxicol Lett. 2017;265:53–60. doi:10.1016/j.toxlet.2016.11.004
22. Zhou Q, Zheng X, Chen L, et al. Smad2/3/4 pathway contributes to TGF-β-induced MiRNA-181b expression to promote gastric cancer metastasis by targeting Timp3. Cell Physiol Biochem. 2016;39(2):453–466. doi:10.1159/000445638
23. Kim J, Kong J, Chang H, Kim H, Kim A. EGF induces epithelial-mesenchymal transition through phospho-Smad2/3-Snail signaling pathway in breast cancer cells. Oncotarget. 2016;7(51):85021–85032. doi:10.18632/oncotarget.13116
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.