")
Back to Journals » OncoTargets and Therapy » Volume 13
MiR-1299 Impedes the Progression of Non-Small-Cell Lung Cancer Through EGFR/PI3K/AKT Signaling Pathway
Authors Cao S, Li L, Li J, Zhao H
Received 18 February 2020
Accepted for publication 8 July 2020
Published 30 July 2020 Volume 2020:13 Pages 7493—7502
DOI https://doi.org/10.2147/OTT.S250396
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Shengya Cao,1,* Longfei Li,2,* Jia Li,3 Hongying Zhao4
1Department of Clinical Laboratory, Xuzhou Cancer Hospital, Xuzhou, Jiangsu, People’s Republic of China; 2Department of Thoracic Surgery, Xuzhou Cancer Hospital, Xuzhou, Jiangsu, People’s Republic of China; 3Department of Central Laboratory, Xuzhou Cancer Hospital, Xuzhou, Jiangsu, People’s Republic of China; 4Department of Oncology, Xuzhou Cancer Hospital, Xuzhou, Jiangsu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hongying Zhao
Department of Oncology, Xuzhou Cancer Hospital, Xuzhou, Jiangsu, People’s Republic of China
Email [email protected]
Background: Non-small-cell lung cancer (NSCLC) is one of the most malignant tumors. In which, numerous miRNAs had been reported to participate in the pathogenesis. However, the expression and function of miR-1299 in NSCLC are not clear.
Methods: To explore the roles of miR-1299 in NSCLC, we detected the levels of miR-1299 in clinical samples of NSCLC and investigated the role of miR-1299 in the regulation of the NSCLC cells proliferation, metastasis, and EMT. Luciferase reporter assay was employed to verify the target of miR-1299. Additionally, the proliferation, metastasis, and EMT of A549 and H1299 cells were analyzed after the overexpression and knockdown of miR-1299.
Results: We found that the miR-1299 expression negatively corresponded with the clinical stage and overall survival in NSCLC patients. Overexpression of miR-1299 inhibited the migration, invasion, and EMT of A549 and H1975 cells. Meanwhile, we proved that miR-1299 is the sponge of EGFR. Besides, our results suggested that miR-1299 inhibits the progression of NSCLC cells through the PI3K/Akt signal pathway.
Conclusion: We demonstrated that miR-1299 inhibits the progression of NSCLC through the EGFR/PI3K/Akt signal pathway. Therapeutic intervention targeting the miR-1299 may provide a potential strategy for the treatment of NSCLC.
Keywords: miR-1299, NSCLC, EGFR, PI3K/Akt
Introduction
Lung cancer is currently the malignant tumor with the highest morbidity and mortality. Among them, non-small-cell lung cancer (NSCLC) accounts for about 85%, which was traditionally treated with surgery and chemoradiotherapy.1 Surgery is often chosen for early NSCLC, but most NSCLC patients are diagnosed in the middle or late stage of this disease, missing the best opportunity for operation. Moreover, due to the high postoperative recurrence rate after surgical treatment, the 5-year survival rate of NSCLC patients is only about 65%.2 Although radiotherapy and chemotherapy based on cisplatin were recommended for patients with advanced NSCLC. However, these patients suffered from the side-effects of chemoradiotherapy, accompanied with a slight improvement of the survival rate.3
Consistent with the tumorigenesis of most cancers, the initiation of NSCLC is often driven by mutations activated oncogenes. Epidermal growth factor receptor (EGFR) is the dominant oncogene in NSCLC, which accounts for about 70% to 85% of total cases.4 Therapeutic drugs targeting EGFR can effectively interfere with the signal transduction pathway and inhibit the progression of tumors.5,6 Tyrosine kinase inhibitor (TKI) is the most representative one, which has been demonstrated that can significantly prolong the progression-free survival (PFS) and objective remission rate (ORR) in clinical trials.6,7 Whereas, it has been found lots of patients (57.3%) who were initially treated with EGFR targeted drugs will develop secondary drug resistance after about 14 months of PFS.8,9 Drug resistance of EGFR-TKIs has become a significant obstacle to the treatment of NSCLC.10,11
In recent years, as a new class of drug development targets, miRNAs have exhibited high potentials to serve as therapeutic targets in cancers. In lung cancer, numerous miRNAs, such as miRNA-21, miRNA-223, and miR-1253, have been found to participate in the pathogenesis of NSCLC. Yang etal12 reported that inhibition of miRNA-21 expression up-regulates the expression of PDCD4, thus inhibiting the proliferation, migration, and invasion of A549 cells. MiRNA-21 also induces acquired drug resistance of EGFR-TKI in NSCLC by activating PI3K/AKT signal pathway.13 Ma etal14 found that overexpression of miRNA-223 inhibits the protein expression of EGFR and PI3K, enhancing apoptosis of NSCLC cells. Besides, miR-1253 inhibits the proliferation, migration, and invasion of NSCLC cells by targeting Wnt5A.15 All these findings suggested that miRNAs may become therapeutic targets in NSCLC treatment. On the other hand, miR-1299 participates in the regulation of colon cancer,16 hepatocellular carcinoma,17 and prostate cancer.18 However, the expression and function of miR-1299 in NSCLC have not been studied.
In this study, we found that the expression level of miR-1299 in tumor tissues was lower than that in paracancerous tissues. Moreover, the overexpression of miR-1299 inhibited the migration, invasion, and EMT of A549 and H1975 cells. Mechanistically, EGFR was the target of miR-1299, and the expression level of EGFR and STAT3 was down-regulated by miR-1299. Besides, we demonstrated that miR-1299 inhibited the progression of NSCLC cells through the PI3K/Akt signal pathway. Based on these results, therapeutic interventions targeting the miR-1299 may provide a potential strategy for the treatment of NSCLC.
Materials and Methods
Cell Treatment
BEAS-2B (human bronchial epithelium cell lines) and four NSCLC cells, H1299, A549, H358, and H1975, were obtained from ATCC and cultured according to their recommendations. MiR-1299 mimic was purchased from GenePharm Co. Ltd. (Shanghai, China). The miR-1299 mimic sequences were: 5ʹ-UUC UGG AAU UCU GUG UGA GGGA-3ʹ (forward) and 5ʹ-CCU CAC ACA GAA UUC CAG AAUU-3ʹ (reverse). Under the specified plural number of infection and treatment time, this miRNA has no off-targeted effect, nor does it affect the adherence, shape or activity of lung cells mentioned above.
Clinical Specimens
Fresh NSCLC tissues were acquired from 56 patients at Xuzhou Cancer Hospital from June 2018 to March 2020. Moreover, the healthy lung tissues at the edge of the lung tumor were obtained as the paracancerous tissue (control). Subsequently, the specimens were quickly snap-frozen in liquid nitrogen and transferred to −80°C for preservation. All the patients signed informed consent, and the hospital ethical committee approved this study (Permit number:2018-03-14).
Quantitative Real-Time PCR
Cells were seeded in a 6-well plate and treated according to the experimental group. When the density reached 90%, the RNA extraction was performed by TRIzol reagent as described previously.17 According to the instructions, the extracted total RNA was reverse transcribed to cDNA by PrimeScript 1st Strand cDNA synthesis kit (Takara, Otsu, Japan). After the implementation of real-time reverse-transcription polymerase chain reaction, the CT value of the target gene expression was obtained compared with the control group. U6 was used as the internal control gene. The relative quantitative analysis was carried out by the 2–ΔΔCT method. The probes were synthesized as followed: miR-1299, 5ʹ-ACA CTC CAG CTG GGT TCT GGA AUU CTC-3ʹ (forward) and 5ʹ-CTC AAC TGG TGT CGT GGA GTC GGC AAT TCA GTT GAG TCC CTC AC-3ʹ (reverse); U6: 5ʹ-CCG CCC GCC GCC AGG CCCC-3ʹ (forward) and 5ʹ-ATA TGG AAC GCT TCA CGA ATT-3ʹ (reverse); EGFR, 5ʹ-TTG CCG CAA AGT GTG TAA CG-3ʹ (forward) and 5ʹ-GTC ACC CCT AAA TGC CAC CG-3ʹ (reverse).
Cell Viability Analysis
Cells were seeded in 96-well plates and cultured in 10% FBS DMEM medium. When the density reached 70%, cells were infected with miR-1299 mimic. After 48-h transfection, 10 μL CCK-8 solution (Beyotime Biotechnology, Nantong, China) was added to each well. After incubating 60 minutes, the absorbance of each well was detected at450nm by a microplate reader (Thermo Fisher, Massachusetts, USA).
Western Blot
To extract the total proteins, cells were lysed with RIPA Lysis Buffer (Beyotime Biotechnology, Nantong, China) as described previously.19 The lysate was centrifuged by 12,000 rpm at 4 °C for 10 minutes. The supernatant was transferred to a new tube to quantify the protein amount by BCA assay. Subsequently, electrophoresis was conducted with 12% SDS-PAGE, followed by transfer onto polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA, USA). After blocking with 5% (w/v) non-fat dry milk, the membranes were incubated with primary antibodies against GAPDH (1:2000 dilution), E-cadherin (1:1000 dilution), Vimentin (1:1000 dilution), Slug (1:1000 dilution), Twist1 (1:1000 dilution), EGFR (1:1000 dilution), PI3K (1:1000 dilution), p-PI3K (1:1000 dilution), AKT (1:1000 dilution) and p-AKT (1:1000 dilution). All antibodies were purchased from Abcam (Shanghai, China). Then, the appropriate HRP-conjugated secondary antibodies (1:5000 dilution, Proteintech, Wuhan, China) were applied. The protein bands were detected with chemiluminescence procedure (Pierce, Rockford, IL, USA) on Tanon 5200 Imaging system (Shanghai, China).
EdU Staining
Cells were seeded in a 6-well plate and treated according to the experimental group. After treatment, cells were fixed with 4% paraformaldehyde and permeated with 0.5% Triton X-100. After washed with 3% BSA PBS three times, the EdU staining solution (US Everbright, Suzhou, China) was added to cover the cells at room temperature for 30 minutes evenly. Subsequently, the nucleus was counterstained with DAPI, and the cells were observed with a fluorescence microscope (IX73, Olympus, Japan).
Cell Apoptosis Analysis
Cells (1 × 105) were seeded in 6-well plates and cultured in 5% CO2 and 37 °C incubators for 24 hours. After the transfection of miR-1299 mimic, the cells were collected by centrifugation at 1000 rpm for 5 minutes and washed twice with PBS buffer. Then re-suspended cells were added with 5 μL Annexin V-FITC and 5 μL PI and incubated at room temperature for 15 minutes. Flow cytometry analysis was carried out by a FACSCalibur flow cytometer (BD Bioscience, New Jersey, USA).
Wound Healing Migration Assay
Cells (3 × 106 cells), which had been infected with miR-1299 mimic, were seeded in a 6-well plate and grown to nearly 90% confluence. Subsequently, a linear scratch was made with a 200 μL pipette tip. After being washed with PBS three times, the cells were incubated with serum-free DMEM medium. All images were photographed at 0 and 24 h at 100 × magnification, and the size of the wound gap was measured. Each experiment was performed at least three times independently.
Transwell Chamber Assay
For the transwell chamber assay, a transwell chamber (8 μm, 24-well plate) that the insert membranes were coated with diluted Matrigel was used. Cells (1 × 105 cells), which had been infected with miR-1299 mimic, were added to the upper chamber and cultured for 24 hours. Subsequently, the insert membranes were cut and stained with crystal violet (Solarbio Science and Technology Ltd., Beijing, China). The images were taken by an inverted microscope, and the number of invading cells was counted in three wells per group.
Luciferase Reporter Gene Assay
The 3ʹ-UTRs (wild type) and the corresponding mutant 3ʹ-UTRs were amplified by PCR. Obtained target fragments were connected it to the pLVX report vector. HEK293T cells were inoculated into 24-well plate at the concentration of 1 × 105 cells/cm2. When the cell density reached 70–80%, reporter gene plasmids and miR-1299 were co-transfected into HEK293T cells by Lipofectamine 2000 (ThermoFisher, Shanghai, China). The cells were collected 48 h after transfection, and luciferase activity was detected.
Statistical Analysis
All experiments were repeated at least three times. Data were presented with mean ± SD. Statistical analysis was performed by GraphPad Prism 6 (GraphPad Software, Inc.), and differences between groups were analyzed by one-way ANOVA followed by Tukey’s test. P < 0.05 was considered to be statistically significant.
Results
Low Expression of MiR-1299 is Associated with the Pathogenesis of NSCLC
In order to explore the physiological expression of miR-1299, the levels of miR-1299 in 56 matched NSCLS tissues and corresponding paracancerous tissues were measured by qRT-PCR. As a result, the level of miR-1299 in tumor tissues was lower than that in paracancerous tissues (P < 0.01, Figure 1A). Additionally, the expression levels of miR-1299 in advanced cases (stage III and IV) was lower than those in early-stage cases (stage I and II) (Figure 1B). At the cellular levels, compared with BEAS-2B, the expression levels of miR-1299 in H1299, A549, H358, and H1975 cells were significantly down-regulated (Figure 1C), indicating that down-regulation of miR-1299 is positively correlated with the lung cancer progression. Kaplan-Meier analysis revealed that overall survival rates of NSCLC patients with the low miR-1299 level were decreased, compared to the high miR-1299 levels group (Figure 1D). These data suggest that the low expression of miR-1299 may have positive effects on the procession of NSCLC.
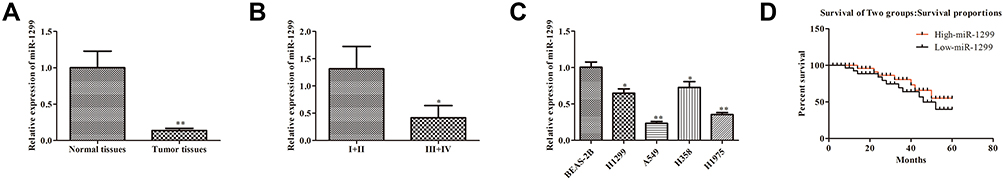 |
Figure 1 Expression and clinical signifificance of miR-1299 in NSCLC. (A) Expression of miR-1299 in cancer tissue and paracancerous tissue. (B) Expression of miR-1299 in different TNM stages. (C) Expression of miR-1299 in different lung cell lines, including BEAS-2B, H1299, A549, H358, and H1975 cells. (D) Overall survival rates of NSCLC patients. Data were represented as mean value ± SD. *P<0.05, **P<0.01 vs control group. |
MiR-1299 Inhibits the Proliferation and Promotes the Apoptosis of NSCLC Cells
Because of the decrease of miR-1299 in NSCLC, we further explore the effects of miR-1299 on the proliferation and apoptosis. As shown in Figure 2A, the expression of miR-1299 in A549 and H1975 cells was measured after the transfection of miR-1299 mimic and inhibitor. Meanwhile, CCK-8 assay showed that cell viability of A549 and H1975 cells were decreased after the transfection of miR-1299 mimic, while cell viability of them was increased after the transfection of miR-1299 inhibitor (Figure 2B). Furthermore, we further evaluated the effects of miR-1299 on the proliferation and apoptosis of these two cells by EdU assay and Annexin V/PI staining, respectively. After the transfection of miR-1299 mimics, we observed that red fluorescence intensity (EdU-positive) was decreased in these cells (Figure 2C). On the contrary, red fluorescence intensity was decreased in A549 and H1975 cells after the transfection of miR-1299 inhibitor, which indicated that the proliferation was negatively regulated by miR-1299. Moreover, the results of flow cytometry showed that the apoptosis of miR-1299 mimic-transfected cells was increased, while the opposite trend was observed in the cells after the transfection of miR-1299 inhibitor (Figure 2D). Above all, these results suggest that miR-1299 inhibited the proliferation, while promoted the apoptosis of NSCLC cells.
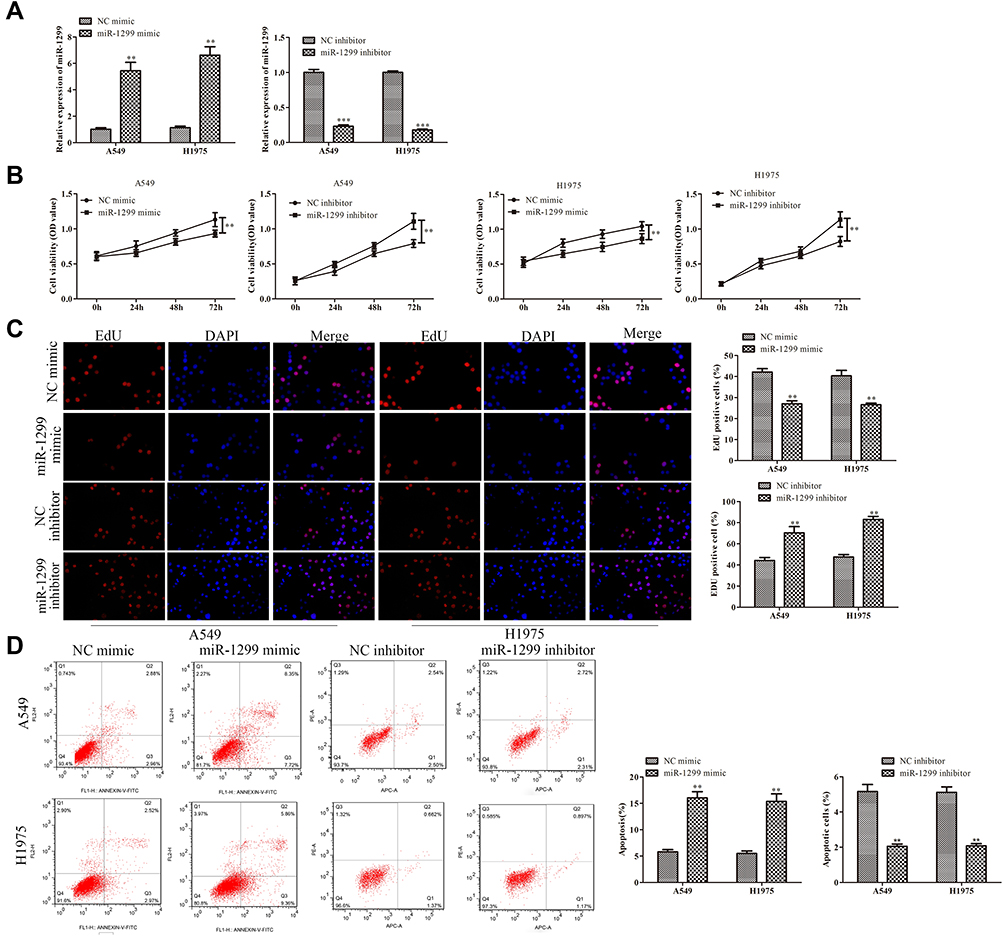 |
Figure 2 Effects of miR-1299 on proliferation and apoptosis of NSCLC cells. (A) Transfection efficiency of miR-1299 mimic and inhibitor in A549 and H1975 cells. (B) Cell viability, (C) proliferation, and (D) apoptosis of NSCLC cells were measured by CCK-8 assay, EdU staining, and Annexin V/PI staining, respectively. Statistical data are presented as means ± SD. n = 3 ***P<0.001, **P < 0.01 vs control group. The experiments were independently repeated three times. |
Overexpression of MiR-1299 Inhibits the Migration, Invasion, and EMT of NSCLC Cells
Tumor migration/invasion and EMT processes are critical factors for tumor progression. Then, we further explore the effects on the migration, invasion, and EMT of A549 and H1975 cells after the overexpression of miR-1299. As shown in Figure 3A, the wound closure percentages of the miR-1299 overexpression group were decreased, compared with these of NC mimic group (P < 0.01). On the contrary, the wound closure percentages were increased after the transfection of miR-1299 inhibitor (P < 0.01). Meanwhile, the transwell chamber assay showed the invasions of miR-1299-overexpressed A549 and H1975 cells were significantly inhibited, while the invasions were promoted after transfection of miR-1299 inhibitor (P < 0.01, Figure 3B). Furthermore, we evaluated the EMT of these two NSCLC cells by detecting the expression levels of its hallmarks, including E-cadherin, Vimentin, Slug, and Twist1. As shown in Figure 3C, the protein expression levels of E-cadherin were significantly increased, while the protein expression levels of Vimentin, Slug, and Twist1 were all decreased in A549 and H1975 cells after transfection of miR-1299 mimic (P < 0.01). As expected, the opposite trend was observed after transfection of miR-1299 inhibitor. Overall, these findings suggest that miR-1299 inhibited the migration, invasion, and EMT of NSCLC cells.
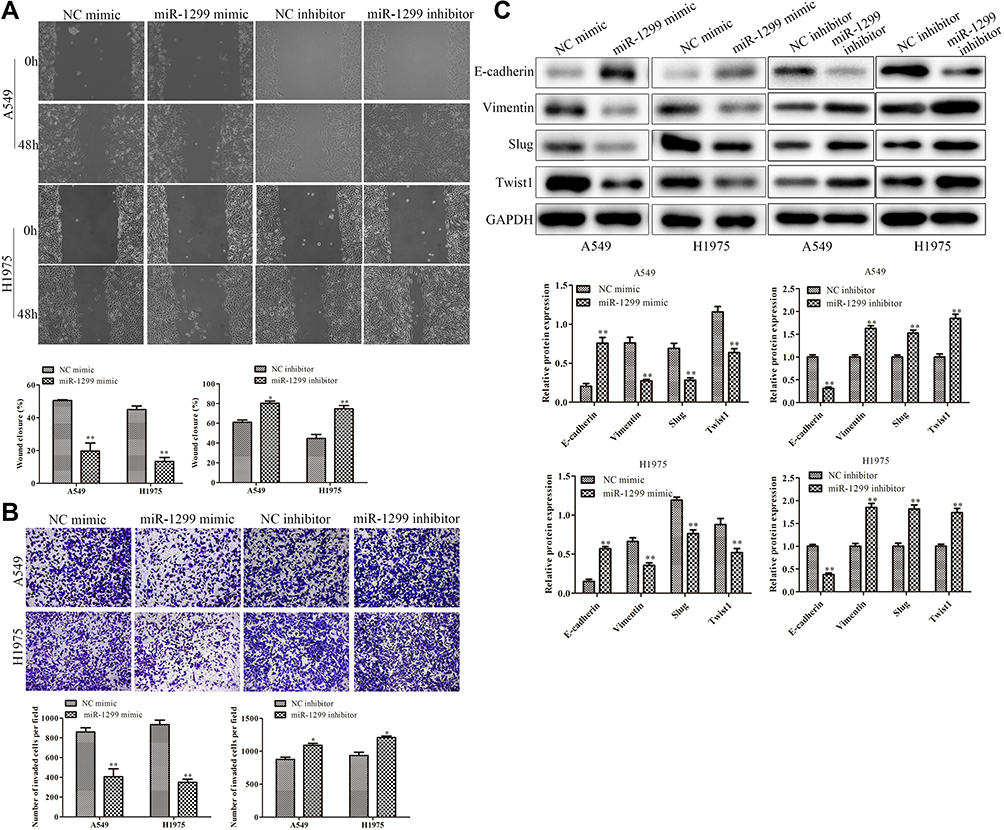 |
Figure 3 Effects of miR-1299 on migration, invasion, and EMT of NSCLC cells. (A) The migration of NSCLC cells was detected by wound healing migration assay. (B) The invasive ability of NSCLC cells was detected through the transwell membrane with Matrigel glue. (C) EMT of NSCLC cells was measured by Western Blot, including E-cadherin, Vimentin, Slug, and Twist1. Statistical data are presented as means ± SD. n = 3,*P<0.05, **P < 0.01 vs control group. The experiments were independently repeated three times. |
EGFR is the Target of MiR-1299
To further explore the molecular mechanism of miR-1299 involved in the migration and proliferation of NSCLC cells, we found that miR-1299 could target the EGFR under the prediction of online software TargetScan (https://www.targetscan.org) miRcode (https://www.mircode.org), and RegRNA 2.0 (http://regrna2.mbc.nctu.edu.tw/detection.html) (Figure 4A). Besides, we detected the expression of EGFR in different lung cells. The results showed that the expression of EGFR in four kinds of NSCLC cancer cells was higher than that in BEAS-2B cells (Figure 4B). Meanwhile, considering the critical roles of EGFR in NSCLC,11 we selected EGFR as our target. Subsequently, luciferase reporter assay showed that miR-1299 mimic significantly decreased the luciferase activity of EGFR 3ʹUTR, while this inhibition was blocked by mutation of the potential binding domains (Figure 4C). Furthermore, as shown in Figure 4D, the mRNA levels of EGFR were significantly down-regulated in A549 and H1975 cells overexpressed with miR-1299 (P < 0.01), while the mRNA level of EGFR was up-regulated after transfection of miR-1299 inhibitor (P < 0.01). Meanwhile, the trends of protein levels were consistent with the mRNA levels (Figure 4E). Overall, these results suggest that miR-1299 targeted EGFR and inhibited the expression of EGFR, which might lead to inhibiting the progression of NSCLC cells.
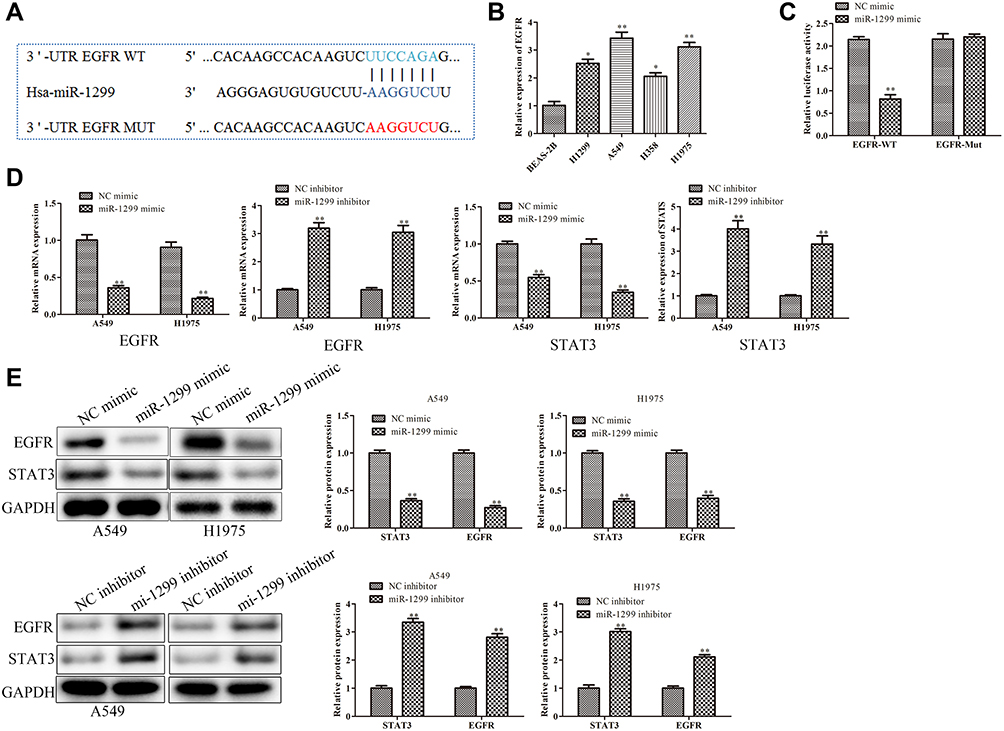 |
Figure 4 Target of miR-1299 in NSCLC cells. (A) Binding sites prediction between miR-1299 and EGFR by TargetScan. (B) Protein expression of EGFR in various lung cell lines, including BEAS-2B, H1299, A549, H358, and H1975 cells. (C) Binding capacity between miR-1299 and EGFR was detected by luciferase reporter assay. (D) mRNA expression and (E) protein expression of EGFR in NSCLC cells. Statistical data are presented as means ± SD. n = 3, *P < 0.05, **P < 0.01 vs control group. The experiments were independently repeated three times. |
MiR-1299 Negatively Regulates the Progression of NSCLC Through PI3K/AKT Pathway
In order to explore the downstream of miR-1299, we detected the phosphorylation of PI3K and AKT by Western blot. As shown in Figure 4A, the phosphorylation of PI3K and AKT were down-regulated after the transfection of miR-1299 mimic, while the phosphorylation of PI3K and AKT were up-regulated after transfection of miR-1299 inhibitor. Not surprisingly, the phosphorylation level of PI3K and AKT in H1975 cells was consistent with that in A549 cells (Figure 5A). To confirm the mediating role of PI3K/Akt signals in miR-1299 regulation, the 740Y-P (PI3K agonist) was employed to activate the PI3K activity. As shown in Figure 5B, miR-1299 mimic decreased the cell viability in A549 cells, whereas the inhibitory effect was blocked by 740Y-P. Besides, the EdU staining assay showed that the red fluorescence intensity of the miR-1299-overexpressed A549 cells treated with 740Y-P was much higher than that in the miR-1299 mimic alone group (Figure 5C). These results indicated that miR-1299 overexpression-induced inhibition on the NSCLC proliferation was blocked by 740Y-P. Furthermore, results of flow cytometry showed that apoptosis promotion of miR-1299-overexpressed A549 cells was also blocked by 740Y-P (Figure 5D).
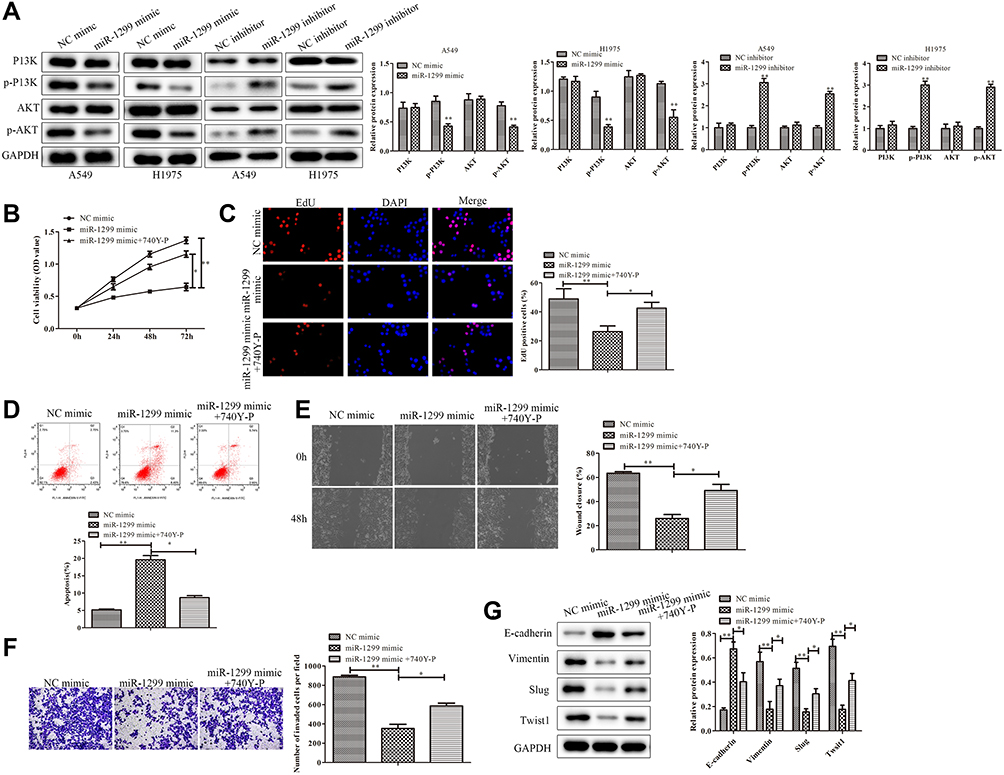 |
Figure 5 Mechanism of miR-1299 on the progression of NSCLC cells. (A) Regulation of PI3K/AKT pathway in A549 and H1975 cells. (B–D) Cell viability, proliferation, and apoptosis of A549 cells treated or not treated with 740Y-P were measured by CCK-8 assay, EdU staining and Annexin V/PI staining, respectively. (E) Migration of A549 cells treated or not treated with 740Y-P was detected by wound healing migration assay. (F) Invasive ability of A549 cells treated or not treated with 740Y-P was detected through the transwell membrane with Matrigel glue. (G) EMT of A549 cells treated or not treated with 740Y-P was evaluated by Western Blot, including E-cadherin, Vimentin, Slug, and Twist1. Statistical data are presented as means ± SD. n = 3, *P < 0.05, **P < 0.01 vs control group. The experiments were independently repeated three times. |
We further evaluated the migration, invasion, and EMT of miR-1299-regulated A549 cells. As shown in Figure 5E, the wound closure percentage of miR-1299-overexpressed A549 cells treated with 740Y-P was higher than that in the miR-1299 overexpression alone group (P< 0.05), which indicated that the miR-1299-inhibited cell migration was blocked by 740Y-P. Moreover, the transwell chamber assay showed that the invasion of miR-1299-overexpressed A549 cells was significantly inhibited by 740Y-P (P < 0.05, Figure 5F). Additionally, the EMT examination showed that the up-regulation of E-cadherin expression in miR-1299 mimic-treated A549 cells was significantly inhibited by 740Y-P (P < 0.05, Figure 5G). In contrast, the reduced expression levels of Vimentin, Slug, and Twist1 in miR-1299-overexpressed A549 cells were significantly increased by 740Y-P (P < 0.05, Figure 5G), indicating the PI3K pathway was involved in the miR-1299-inhibited EMT progression in A549 cells. Above all, these results suggest that miR-1299 negatively regulated the progression of NSCLC cells through PI3K/Akt signals.
Discussion
Tumors are new organisms formed by abnormal proliferation due to the loss of control at the gene level, which is primarily genetic diseases. Oncogenes are genes that have the potential to transform healthy cells into cancer cells. When oncogenes exist in an inactivated form in healthy cells, it was called proto-oncogenes. Most of them are cell growth factors and growth factor receptors that are very important for healthy cell growth, such as platelet growth factor (PGF), fibroblast growth factor (FGF), Epidermal Growth Factor (EGF), tyrosine kinase, nuclear regulatory proteins, etc. There are mainly two ways of proto-oncogenes activation: structural changes (mutations) of proto-oncogenes to produce oncoproteins with abnormal functions; overexpression of genes to produce excessive growth-promoting proteins with the correct structure.
In recent years, numerous studies had proved that miRNAs also involved in the mechanism of tumorigenesis. For instance, miR-1299 has been reported to regulate the progression of several cancers. Wang etal16 found that the level of miR-1299 in colon cancer cells is significantly decreased. Notably, overexpression of miR-1299 not only down-regulates the STAT3 pathway but also inhibits the growth of colon cancer cells. Another study reported that miR-1299 targets CDK6 and inhibits its expression, subsequently inhibiting the proliferation of hepatocellular carcinoma cells.17 However, the expression and function of miR-1299 in NSCLC remain elusive. In this study, we detected the expression level of miR-1299 in tumor tissues was lower than that in paracancerous tissues. Besides, the miR-1299 expression negatively corresponded with the clinical stage and overall survival in NSCLC patients. Further results showed that the miR-1299 expression was significantly decreased in the NSCLC cells, indicating that miR-1299 may participate in the progression of NSCLC.
The progression of lung cancer usually initiates with abnormal biological signaling pathways, including the overexpression and mutation of crucial signal proteins in the pathways. EGFR, as a transmembrane glycoprotein receptor with tyrosine kinase activity, is the expression product of proto-oncogene C-erb-1 (HER-1).20 Studies have shown that EGFR gene mutation and subsequent abnormal signal transduction lead to active oncogene expression and promote the progression of tumor cells.21 EGFR plays a regulatory role in the processes of tumor cells, including proliferation, differentiation, and anti-apoptosis. Its mutation or abnormal expression can promote tumor cell proliferation, adhesion, invasion, and metastasis.22 Signal transducer and activator of transcription 3 (STAT3) is one of the members of the family of signal transduction and transcription activation. EGFR indirectly activates c-Src by binding to Ral-GTPase and then activating the downstream transcriptional activator STAT3.23 Studies had shown that EGFR could promote the phosphorylation and expression of STAT3, which contributes to tumor development.24,25 For instance, miRNA-7 inhibits the expression of EGFR and thus leads to abnormal activation of STAT3 and expression.26 In this study, we found that EGFR was overexpressed in various kinds of NSCLC cells, compared to healthy lung tissue cells. Moreover, we predicted the EGFR is a target of miR-1299 by online software. The subsequent luciferase reporter assay confirmed this prediction. Furthermore, the results showed that the protein expression of EGFR in the miR-1299 overexpression group was significantly inhibited compared to the control group. Meanwhile, the expression of STAT3 was also inhibited in A549 and H1975 cells after the transfection of miR-1299 mimic. These results suggested that miR-1299 acts as a sponge of EGFR, thus participating in the progression of NSCLC.
As a downstream pathway of EGFR, hyperactivation of PI3K/AKT is a prominent feature of various human cancers.27,28 EGFR stimulates Ras protein after dimerization, leading to phosphorylation cascade reaction and activating PI3K/Akt signal pathway, which leads to the occurrence and development of tumors.29,30 Studies had shown that the PI3K/AKT pathway induces tumorigenesis through multiple mechanisms. Sos etal31 found that PTEN dephosphorylates PIP3 and converts it into PIP2, which inhibits the activation of Akt and blocks the PI3K/Akt/mTOR pathway. The deletion or mutation of the PTEN gene would lead to the continuous activation of the PI3K/Akt/mTOR pathway, thus promoting the proliferation of tumor cells. In this study, phosphorylation of PI3K and Akt were both down-regulated after the overexpression of miR-1299. Then, we employed an agonist of PI3K (740Y-P) toinvestigate whether PI3K is the downstream pathway of miR-1299. The results showed that 740Y-P blocked the down-regulation of PI3K and Akt protein phosphorylation in miR-1299-overexpressed A549 cells. Moreover, the inhibition of migration, invasion, and EMT processes in miR-1299-overexpressed A549 cells was also significantly blocked by 740Y-P. These results suggested that miR-1299 inhibited the progression of NSCLC cells through the PI3K/Akt signal pathway.
In conclusion, we found that miR-1299 down-regulation and EGFR up-regulation simultaneously existed in NSCLC cells. Subsequently, we demonstrated that EGFR is the targets of miR-1299, and its expression is decreased by miR-1299 overexpression. Moreover, our results suggested miR-1299 negatively regulated the PI3K/Akt pathway, which further affects the proliferation, apoptosis, migration, invasion, and EMT processes of NSCLC cells. Hence, this study demonstrated that miR-1299 impeded the progression of NSCLC cells through the EGFR/PI3K/Akt signal pathway.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel R, Ma J, Zou Z, etal. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi:10.3322/caac.21208
2. Kubo H, Suzuki T, Matsushima T, etal. Cyclin-dependent kinase-specific activity predicts the prognosis of stage I and stage II non-small cell lung cancer. BMC Cancer. 2014;14(1):755. doi:10.1186/1471-2407-14-755
3. Pisters KMW, Evans WK, Azzoli CG, etal. Cancer care ontario and american society of clinical oncology adjuvant chemotherapy and adjuvant radiation therapy for stages I-IIIA resectable non–small-cell lung cancer guideline. J Clin Oncol. 2007;25:5506–5518. doi:10.1200/JCO.2007.14.1226
4. Hirsch FR, Bunn PA. EGFR testing in lung cancer is ready for prime time. Lancet Oncol. 2009;10(5):432–433. doi:10.1016/S1470-2045(09)70110-X
5. El-Rayes BF, LoRusso PM. Targeting the epidermal growth factor receptor. Brit J Cancer. 2004;91:418–424. doi:10.1038/sj.bjc.6601921
6. Mok TSK, Lee K, Leung L. Targeting epidermal growth factor receptor in the management of lung cancer. Semin Oncol. 2014;41:101–109. doi:10.1053/j.seminoncol.2013.12.010
7. Yi HG, Kim HJ, Kim YJ, etal. Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) are effective for leptomeningeal metastasis from non-small cell lung cancer patients with sensitive EGFR mutation or other predictive factors of good response for EGFR TKI. Lung Cancer. 2009;65:80–84. doi:10.1016/j.lungcan.2008.10.016
8. Yang J, Chen H, Yan H, etal. Clinical modes of EGFR tyrosine kinase inhibitor failure and subsequent management in advanced non-small cell lung cancer. Lung Cancer. 2013;79:33–39. doi:10.1016/j.lungcan.2012.09.016
9. Paz-Ares L, Soulières D, Melezínek I, etal. Clinical outcomes in non-small-cell lung cancer patients with EGFR mutations: pooled analysis. J Cell Mol Med. 2010;14(1–2):51–69. doi:10.1111/j.1582-4934.2009.00991.x
10. Westover D, Zugazagoitia J, Cho BC, etal. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol. 2018;29:i10–i19. doi:10.1093/annonc/mdx703
11. Lee DH. Treatments for EGFR-mutant non-small cell lung cancer (NSCLC): the road to a success, paved with failures. Pharmacol Therapeut. 2017;174:1–21. doi:10.1016/j.pharmthera.2017.02.001
12. Yang Y, Meng H, Peng Q, etal. Downregulation of microRNA-21 expression restrains non-small cell lung cancer cell proliferation and migration through upregulation of programmed cell death 4. Cancer Gene Ther. 2015;22:23–29. doi:10.1038/cgt.2014.66
13. Li B, Ren S, Li X, etal. MiR-21 overexpression is associated with acquired resistance of EGFR-TKI in non-small cell lung cancer. Lung Cancer. 2014;83(2):146–153. doi:10.1016/j.lungcan.2013.11.003
14. Ma H, Kong W, Li X, etal. miRNA-223 is an anticancer gene in human non-small cell lung cancer through the PI3K/AKT pathway by targeting EGFR. Oncol Rep. 2019;41(3):1549–1559. doi:10.3892/or.2019.6983
15. Liu M, Zhang Y, Zhang J, etal. MicroRNA-1253 suppresses cell proliferation and invasion of non-small-cell lung carcinoma by targeting WNT5A. Cell Death Dis. 2018;9(2):189. doi:10.1038/s41419-017-0218-x
16. Wang Y, Lu Z, Wang N, etal. MicroRNA-1299 is a negative regulator of STAT3 in colon cancer. Oncol Rep. 2017;37(6):3227–3234. doi:10.3892/or.2017.5605
17. Zhu H, Wang G, Zhou X, etal. miR-1299 suppresses cell proliferation of hepatocellular carcinoma (HCC) by targeting CDK6. Biomed Pharmacother. 2016;83:792–797. doi:10.1016/j.biopha.2016.07.037
18. Zhang F, Du Y, Tian Y, etal. MiR-1299 functions as a tumor suppressor to inhibit the proliferation and metastasis of prostate cancer by targeting NEK2. Eur Rev Med Pharmacol. 2019;23:530. doi:10.26355/eurrev_201901_16865
19. Zhu Y, Gui W, Lin X, etal. Knock-down of circular RNA H19 induces human adipose-derived stem cells adipogenic differentiation via a mechanism involving the polypyrimidine tract-binding protein 1. Exp Cell Res. 2020;387(2):111753. doi:10.1016/j.yexcr.2019.111753
20. Baselga J, Arteaga CL. Critical update and emerging trends in epidermal growth factor receptor targeting in cancer. J Clin Oncol. 2005;23:2445–2459. doi:10.1200/JCO.2005.11.890
21. Voldborg BR, Damstrup L, Spang-Thomsen M, etal. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann Oncol. 1997;8(12):1197–1206. doi:10.1023/a:1008209720526
22. Lopez-Chavez A, Carter CA, Giaccone G. The role of KRAS mutations in resistance to EGFR inhibition in the treatment of cancer. Curr Opin Investig Drugs. 2009;10(12):1305–1314. doi:10.1016/j.clinthera.2009.12.012
23. Goi T, Shipitsin M, Lu Z, etal. An EGF receptor/Ral-GTPase signaling cascade regulates c-Src activity and substrate specificity. EMBO J. 2000;19:623–630. doi:10.1093/emboj/19.4.623
24. Fan Q, Cheng CK, Gustafson WC, etal. EGFR phosphorylates tumor-derived EGFRvIII Driving STAT3/5 and progression in glioblastoma. Cancer Cell. 2013;24(4):438–449. doi:10.1016/j.ccr.2013.09.004
25. Srivatsa S, Paul MC, Cardone C, etal. EGFR in tumor-associated myeloid cells promotes development of colorectal cancer in mice and associates with outcomes of patients. Gastroenterology. 2017;153(1):178–190. doi:10.1053/j.gastro.2017.03.053
26. Liu L, Liu F, Huang M, etal. Circular RNA ciRS-7 promotes the proliferation and metastasis of pancreatic cancer by regulating miR-7-mediated EGFR/STAT3 signaling pathway. Hepatob Pancreat Dis. 2019;18(6):580–586. doi:10.1016/j.hbpd.2019.03.003
27. He L, Liu X, Yang J, etal. Imbalance of the reciprocally inhibitory loop between the ubiquitin-specific protease USP43 and EGFR/PI3K/AKT drives breast carcinogenesis. Cell Res. 2018;28(9):934–951. doi:10.1038/s41422-018-0079-6
28. Wu DW, Wu TC, Chen CY, etal. PAK1 is a novel therapeutic target in tyrosine kinase inhibitor-resistant lung adenocarcinoma activated by the PI3K/AKT signaling regardless of EGFR mutation. Clin Cancer Res. 2016;22(21):5370–5382. doi:10.1158/1078-0432.CCR-15-2724
29. Ji M, Guan H, Gao C, etal. Highly frequent promoter methylation and PIK3CA amplification in non-small cell lung cancer (NSCLC). BMC Cancer. 2011;11:147. doi:10.1186/1471-2407-11-147
30. Kikuchi J, Kinoshita I, Shimizu Y, etal. Simultaneous blockade of AP-1 and phosphatidylinositol 3-kinase pathway in non-small cell lung cancer cells. Brit J Cancer. 2008;99:2013–2019. doi:10.1038/sj.bjc.6604782
31. Sos ML, Koker M, Weir BA, etal. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009;69:3256–3261. doi:10.1158/0008-5472.CAN-08-4055
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.