")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 14
miR-129-3p Targeting of MCU Protects Against Glucose Fluctuation-Mediated Neuronal Damage via a Mitochondrial-Dependent Intrinsic Apoptotic Pathway
Received 4 October 2020
Accepted for publication 12 December 2020
Published 15 January 2021 Volume 2021:14 Pages 153—163
DOI https://doi.org/10.2147/DMSO.S285179
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Antonio Brunetti
Bo Wang,1,2 Yang Li,3 Chao You1
1Department of Neurosurgery, West China School of Medicine/West China Hospital of Sichuan University, Chengdu, Sichuan 610041, People’s Republic of China; 2Department of Neurosurgery, Kunming Medical University First Affiliated Hospital, Kunming, Yunnan 650032, People’s Republic of China; 3Intensive Care Unit, West China School of Medicine/West China Hospital of Sichuan University, Chengdu, Sichuan 610041, People’s Republic of China
Correspondence: Chao You
Department of Neurosurgery, West China School of Medicine/West China Hospital of Sichuan University, Chengdu, Sichuan 610041, People’s Republic of China
Tel +86 28-85422026
Email [email protected]
Introduction: Glucose fluctuations have an adverse effect on several diabetes-related complications, especially for the nervous system, but the underlying mechanisms are not clear. MicroRNAs are critical regulators of posttranscription in many physiological processes, such as apoptosis. Our study clarified the neuroprotective effects of miR-129-3p targeting mitochondrial calcium uniporter (MCU) in glucose fluctuation-mediated neuronal damage and the specific mechanisms involved.
Methods: The expression of MCU and miR-129-3p was examined by real-time PCR and Western blot in the glucose fluctuation cell model. Dual-luciferase reporter assay was performed to confirm the transcriptional regulation of miR-129-3p by MCU. Fluorescent probe and assay kit assay was used to determine oxidative stress condition. Mitochondrial-dependent intrinsic apoptotic factors were examined by flow cytometry assay, enzyme-linked immunosorbent assay (ELISA), and gene and protein expression assays.
Results: We found an upregulation of MCU and downregulation of miR-129-3p in glucose fluctuation-treated primary hippocampal neuronal cells, and miR-129-3p directly targeted MCU. miR-129-3p overexpression produced a dramatic reduction in calcium overload, reactive oxygen species (ROS) generation, GSH-to-GSSG ratio, MMP-2 expression in the mitochondrial-dependent intrinsic apoptosis pathway and an increase in MnSOD activity. Increasing MCU expression rescued the effects of miR-129-3p overexpression. miR-129-3p downregulation produced a significant increase in calcium overload, reactive oxygen species (ROS) generation, MMP-2 expression, cytochrome c release and cell apoptosis, and antioxidant N-acetyl cysteine (NAC) rescued the effects of miR-129-3p downregulation.
Conclusion: Therefore, miR-129-3p suppressed glucose fluctuation-mediated neuronal damage by targeting MCU via a mitochondrial-dependent intrinsic apoptotic pathway. The miR-129-3p/MCU axis may be a promising therapeutic target for glucose fluctuation-mediated neuronal damage.
Keywords: miR-129-3p, MCU, calcium, ROS, mitochondrion, apoptosis, oxidative stress
Introduction
Diabetes is a common metabolic disease that severely affects human health worldwide.1 During the last several years, robust evidence showed that glucose fluctuations, independent of mean glucose levels, were implicated in the risk of diabetic complication, such as endothelial dysfunction and vascular injury.2,3 Notably, recent studies identified that glucose fluctuations were more harmful than stable hyperglycemia or hypoglycemia in neuronal and astroglial cells.4,5 Moreover, clinical and fundamental studies have demonstrated cognition and memory dysfunction in T1DM and T2DM, but its pathogenesis remains poorly understood.6 Previous studies have shown that glucose fluctuations in T2DM induced neuronal cell apoptosis in hippocampal tissues.7 Because glucose is the primary energy source for the brain, glucose fluctuations may play a critical role in central nervous system injuries induced by diabetic complications.5 Glucose fluctuation-mediated damage is primarily caused by ROS accumulation and an imbalance of the antioxidant defense system, leading to cellular functional impairment.8
Mitochondrial integrity and functionality are essential for neuronal health. Therefore, the development of new techniques for detecting mitochondrial dysfunction, especially the increased ROS production, is of great significance and has attracted vast research interest.9 MCU is located in the inner mitochondrial membrane (IMM) and promotes mitochondrial entry of Ca2+ from the cytosol. Therefore, it plays outstanding roles in mitochondrial homeostasis, and it is central to the brain’s physiological and pathological processes.10–12 MCU maintains mitochondrial Ca2+ homeostasis under physiological conditions, which is essential for the survival and energy supply of cells.13 Under pathological conditions, mitochondrial Ca2+ overload through MCU causes an accumulation of ROS, mitochondria dysfunction, a decrease in ATP, and cell apoptosis.14 MCU-mediated neuronal damage was described in brain ischemic stroke, traumatic brain injury, and conditions of high glucose levels.15–17 However, the mechanism of MCU in mediating neuronal cells damage after glucose fluctuation exposures is not well known.
miRNAs play a central role in developing, progressing, and treating human diseases.18 miR-129-3p was decreased in palmitic acid (PA) -induced cardiomyocytes, and overexpression of miR-129-3p inhibited inflammation and apoptosis in cardiomyocytes.19 miR-129-3p promoted cell proliferation and restrained apoptosis in breast cancer cells.20 However, the role of miR-129-3p in regulating neuronal cell damage after glucose fluctuation exposure and its underlying mechanism is not known.
Therefore, the present study examined the effect of miR-129-3p on apoptosis of hippocampal neuronal cells via the targeting MCU during glucose fluctuation and elucidated the underlying molecular mechanisms involved.
Materials and Methods
Cell Culture and Incubation
Rat primary hippocampal neuronal cells (iCell, China) were cultured in a primary neuronal cell culture system (iCell, China) at 37 °C in 5% CO2. According to a previous study, The cells were treated with different concentrations of glucose.4 Briefly, treatments with 10.0 mM, 2.0 mM, 25 mM glucose for 10 h were considered the normal glucose (control, CT), low glucose (LG), high glucose (HG) conditions, respectively. To simulate t the abrupt short-term glucose fluctuations (GV) that can occur in a 24-h period in diabetic patients, we treated cells with LG for 1 h followed by 4 h in HG, and this protocol was repeated twice for a total of 10 h. The cells were transfected with miR-129-3p mimic, miR-129-3p inhibitor, pcDNA 3.1-MCU (lacks the 3′ UTR) or their respective NCs (NC mimic and pcDNA3.1 vector) (GenePharma, China) were used. All cells were transfected using Lipofectamine 2000 (Invitrogen, USA) according to standard manufacturer’s protocols.
Western Blotting
Western blotting procedures were performed as described previously.21 Briefly, the rat primary hippocampal neuronal cell’s total protein was extracted from RIPA lysis buffer, and the total protein concentration was detected by the BCA method. Samples were heated in boiling water to denature and then subjected to electrophoresis and transferred to PVDF membranes (Millipore, USA). After the membranes were incubated in 5% BSA at room temperature for 2 h, and then incubated with primary antibodies against MCU (1:1000, Cell Signaling Technology, USA), caspase-9 (1:1000, Cell Signaling Technology, USA), caspase-3 (1:1000, Cell Signaling Technology, USA), MMP-2 (1:1000, Cell Signaling Technology, USA), and GAPDH (1:1000, Abcam, USA) were used. The secondary antibodies were HRP-conjugated goat anti-rabbit IgG (1: 2000, BOSTER, China) and HRP-conjugated goat anti-mouse IgG (1: 2000, BOSTER, China).
Quantitative Real-Time PCR
Total RNA extraction, cDNA synthesis, and quantitative RT‐PCR were performed as described previously.21 Briefly, total RNA was isolated from each group of cells using TRIzol reagent (Invitrogen, USA), and the RNA concentration and purity of RNA were obtained using a spectrophotometer. First-strand cDNA synthesis was performed using a cDNA synthesis kit (Fermentas, USA), and qPCR was performed using SYBR FAST qPCR Master Mix (Roche, Switzerland): the reaction system (0.5 ul forward primer, 0.5 ul reverse primer, 5ul Master mix, 1ul cDNA and 4 ul nuclease-free water); the reaction conditions (95 °C for 30 s, 95 °C for 5 s and 60 °C for 31 s for 40 cycles); melt-Curve analysis (1 cycle of 95°C for 15 s, 60°C for 30 s, and 95°C for 15 s). Ct values were calculated to analyze the relative expression levels using the 2–ΔΔCt method with β-actin as an endogenous control. The following primer sequences were used: β-actin forward 5ʹ-CATTGCT-GACAGGATGCAGA-3ʹ, and reverse, 5ʹ-CTGCTGGAAGGTGGACAGTGA-3ʹ; MCU forward 5ʹ-CAAGGATGCAATTGCTCAGG-3ʹ, and reverse, 5ʹ- TTCCTTTTCTCCGATCTGTCG-3ʹ; U6 forward 5ʹ-CTCGCTTCGGCAGCACA-3ʹ, and reverse, 5ʹ-AACGCTTCACGAATTTGCGT-3ʹ; miR-129-3p RT Stem-loop: 5ʹ-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGATAC-TTTT-3ʹ, forward 5ʹ-ACACTCCAGCTGGGAAGCCCTTACCCCAAA-3ʹ, and reverse, 5ʹ- TGGTGTCGT-GGAGTCG-3ʹ; MMP-2 forward 5ʹ-CTATTCTGTCAGCACTTTGG-3ʹ, and reverse, 5ʹ- CAGACTT-TGGTTCTCCAACTT-3ʹ; caspase-9 forward 5ʹ-TCCTGGTACATCGAGACCTTG-3ʹ, and reverse, 5ʹ-AAGTCCCTTTCGCAGAAACAG-3ʹ; caspase-3 forward 5ʹ- GGTTCATCCAGTCCCT-TTGC-3ʹ, and reverse, 5ʹ-ACGGGATCTGTTTCTTTGCG-3ʹ.
Dual-Luciferase Assay
The 3′‐UTR of MCU was synthesized and inserted into the psiCHECK‐2 vector (Ambion). The possible miR-129-3p binding sites in the 3′-UTR (MCU: AAGGGCT) were replaced with TCACTGA and validated via sequencing. The two reporter plasmids named MCU-WT and MCU-MUT were co-transfected into rat primary hippocampal neuronal cells with miR-129-3p mimic or NC mimic, respectively. The Dual Luciferase Reporter Assay kit (Promega, USA) with renilla luciferase as an internal control was used to perform luciferase assays, and The dual-luciferase activity was detected by Promega’s Digital-Luciferase Reporter Assay System (Promega, USA).
Isolation of Mitochondria and Cytosol
Mitochondrial and cytosolic fractions were isolated from hippocampal neuronal cells using the Cell Mitochondria Isolation Kit (Beyotime, China), according to the manufacturer’s protocol. Briefly, after proper treatment, cells were incubated in cell lysis buffer (250mM of sucrose; 1mM of DTT; 10mM of KCl; 1mM of EDTA; 1mM of ethylene glycol bis(aminoethylether)-tetraacetic acid (EGTA); 1.5mM of MgCl2; phenylmethylsulfonyl fluoride; 20mM of HEPES, pH 7.4) at 4°C and disrupted with a glass tissue grinder. The cell lysate was centrifuged at 800g for 10 min, and the supernatant was further centrifuged at 10,000 for 20 min-the resulting final supernatants and pellets, containing the cytosolic and mitochondrial fractions, respectively.
Measurement of Mitochondrial Ca2+ and O2− Levels
Mitochondrial Ca2+ and O2− were examined using the fluorescent probe Rhod-2/AM (Abcam, USA) and MitoSOX (Invitrogen, USA), respectively, according to the manufacturer’s guidance. Mitochondrial fractions of hippocampal neuronal cells were washed with 0.1% PBS-polyvinyl alcohol (PVA) and incubated in the darkroom for 15 min in neuronal cell medium mixed with 100 μM Rhod-2 dye. Mitochondrial fractions of hippocampal neuronal cells were washed three times in 0.1% PVA-PBS. Washed mitochondrial fractions were cultured in 50 μL of neuronal cell medium supplemented with 5 μM Mito-SOX for 10 min, protected from light. After three washing three times of all samples in 0.1% PVA-PBS. All images were captured using a fluorescence microscope (Leica, Germany), and ImageJ software to determine fluorescence intensities.
Detection of GSH-to-GSSG Ratio and MnSOD Activities
According to the manufacturer’s instructions, the GSH-to-GSSG ratio was examined using a GSH and GSSG assay kit (Beyotime, China). Briefly, after proper treatment, samples were frozen and thawed five times, then centrifuged at 10000g for 5 min. The supernatant was collected for use. Absorbance at 412 nm was measured using a microplate reader (Thermo, USA). Reduced GSH was calculated as a difference between total glutathione and GSSG, and GSH/GSSG was determined. The enzyme activities of MnSOD were detected using a Cu/Zn-SOD and Mn-SOD Assay Kit with WST-8 (Beyotime, China), according to the manufacturer’s specifications. Briefly, SOD inhibitor A and B were successively added to the samples, followed by incubation with the WST-8/enzyme working solution at 37°C for 30 min. The protein concentrations were determined using the BCA assay. The absorption at 450 nm was recorded using a microplate reader (Thermo, USA).
Flow Cytometry
Quantitation of apoptotic cells was obtained using the Annexin V-FITC Apoptosis Detection Kit (Beyotime, China), according to the manufacturer’s protocol. Briefly, the samples treated with Tm or β-ME were harvested and were stained with Annexin-V-FITC at room temperature for 10 min. Then, the cells were centrifuged at 1000g for 5 min. After washed, cells were stained with propidium iodide (PI). The percentage of apoptotic cells was analyzed by flow cytometer (BD bioscience, USA). The data were analyzed using WinMDI 2.9 software.
ELISA
According to the manufacturer’s instructions, cytochrome c levels were determined using the Mouse/Rat Cytochrome C ELISA Kit (Abcam, USA). Briefly, after proper treatment, samples were treated with antibody cocktail and incubate with shaking for 1 h. Then, 100 µL of TMB substrate was added to samples and incubated with shaking for 10 min in the dark. Finally,100 µL of Stop Solution was added to the samples. The absorption at 450 nm was recorded using a microplate reader (Thermo, USA).
Statistical Analysis
All data are expressed as the means ± SD. Multiple group comparisons were analyzed using independent sample t-test or one-way ANOVA with Tukey’s post hoc test. P < 0.05 was considered significant. P < 0.01 was considered strongly significant. All statistical analyses and graphs were performed using GraphPad Prism 8.0.1 software (GraphPad Software Inc., USA).
Results
miR-129-3p Directly Regulates MCU Expression in Glucose Fluctuation-Exposed Hippocampal Neuronal Neuronal Cells
To investigate the alterations of key molecules underlying hippocampal neuronal cells exposed to GV, we determined the expression of MCU and miR-129-3. We first analyzed MCU protein levels in hippocampal neuronal cells treated with different glucose conditions. As shown in Figure 1A and B, the exposure of hippocampal neuronal cells to GV promoted a significant increase in MCU protein levels compared to the other groups. The mRNA expression of miR-129-3p in neuronal cells exposed to GV was lower than the other group (Figure 1C). These findings collectively indicate that MCU upregulation and miR-129-3p downregulation occur in hippocampal neuronal cells exposed to GV.To further identify the mechanism of miR-129-3p in hippocampal neuronal cells, we used TargetScan to predict the binding sites between miR-129-3p and MCU-3′ UTR (Figure 1D). Dual luciferase assays were performed to functionally verify whether miR-129-3p directly targeted MCU in hippocampal neuronal cells. The dual luciferase assay showed that miR-129-3p overexpression (miR-129-3p mimics) significantly inhibited the relative luciferase activity of MCU with the WT 3′ UTR (WT), but the mutant 3′ UTR (MUT) had no effect on the activity (Figure 1E). miR-129-3p mimics and miR-129-3p inhibitor downregulated and upregulated MCU expression compared to the miR-NC group, respectively (Figure 2F). Therefore, miR-129-3p directly targeted MCU and inhibited its expression.
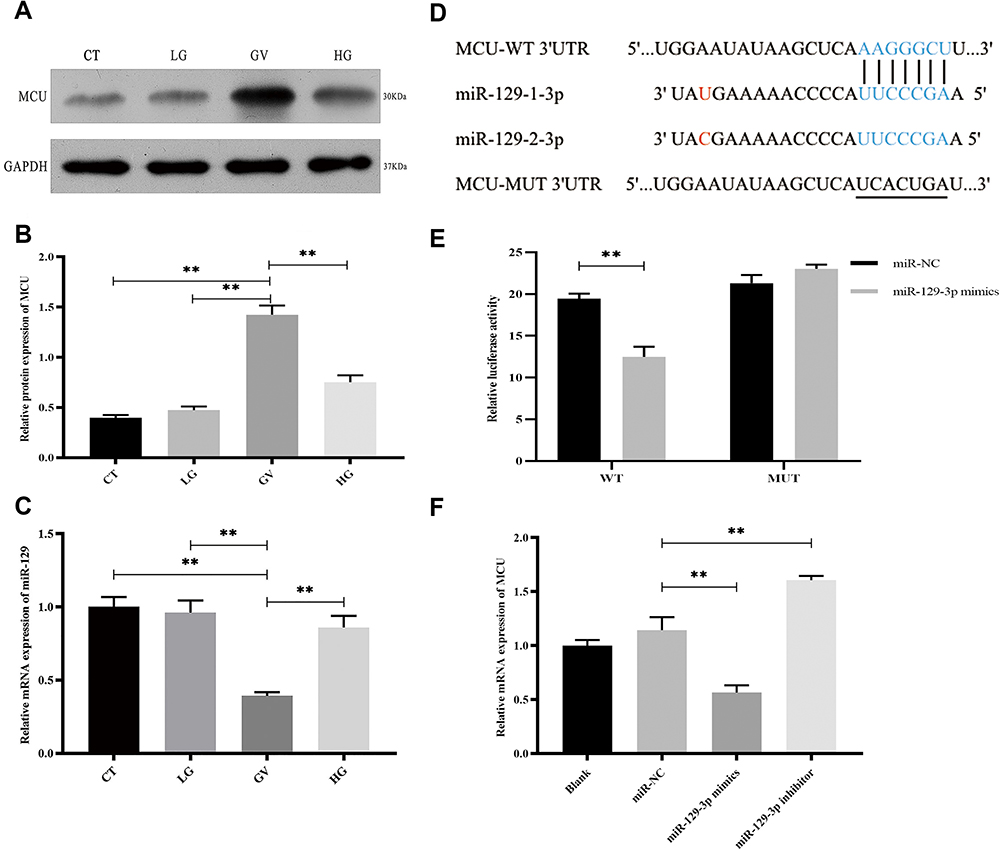 |
Figure 1 miR-129-3p directly regulates MCU expression in glucose fluctuation-exposed hippocampal neuronal neuronal cells. Rat primary hippocampal neuronal cells were exposed to normal glucose (control, CT), low glucose (LG), high glucose (HG) and glucose fluctuation (GV) conditions. (A and B) Protein levels of MCU. (C) mRNA levels of miR-129-3p. (D) Predicted mi R-129-3p target sequence in the MCU-3 ‘ UTR. Target sequences of MCU-3 ‘UTR were mutated. (E) Luciferase assay of cells transfected with MCU-WT or MCU-MUT reporter together with miR-129-3p mimic or miR-NC. (F) mRNA levels of MCU in cells transfected with miR-129-3p inhibitor or mimics and miR-NC. Data are presented as the means ± SD; ** p< 0.01; (n = 5). |
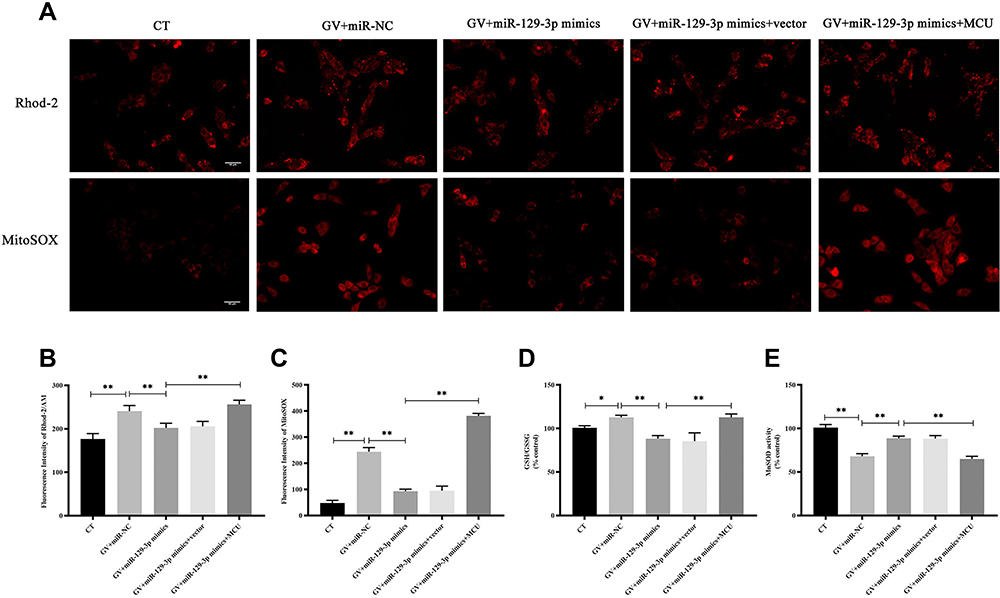 |
Figure 2 miR-129-3p overexpression alleviates oxidative stress in glucose fluctuation-exposed hippocampal neuronal cells by targeting MCU. Rat primary hippocampal neuronal cells were transfected with miR-NC, miR-129-3p mimics, miR-129-3p mimics+vector, or miR-129-3p mimics+MCU followed by glucose fluctuation treatment. (A–C) The concentration of mitochondrial Ca2+ and O2− was detected using immunofluorescence. Scale bars = 50 μm. (D) GSH/GSSG ratio. (E) MnSOD activity. Data are presented as the means ± SD; *p< 0.05, **p< 0.01; (n = 5). CT, normal glucose; GV, glucose fluctuation; vector, MCU negative control (pcDNA3.1 vector). |
miR-129-3p Overexpression Alleviates Oxidative Stress in Glucose Fluctuation-Exposed Hippocampal Neuronal Cells by Targeting MCU
To study the influence of miR-129-3p on mitochondrial calcium and ROS levels in glucose fluctuation-exposed hippocampal neuronal cells, we used two well-known fluorescent probes, Rhod-2/AM and MitoSOX. Mitochondrial calcium and ROS levels were significantly elevated in hippocampal neuronal cells exposed to GV, and miR-129-3p upregulation (miR-129-3p mimics) inhibited calcium overload and oxidative stress (Figure 2A–C and Supplementary Figure S1). Forced MCU expression (miR-129-3p mimics+MCU) partially abolished the miR-129-5p overexpression (miR-129-3p mimics)-induced inhibition of calcium overload and oxidative stress (Figure 2A–C and Supplementary Figure S1). These results suggest that miR-129-3p inhibited mitochondrial calcium overload and oxidative stress by targeting MCU. We then investigated the effects of miR-129-5p overexpression on the antioxidant system in glucose fluctuation-exposed hippocampal neuronal cells. The results showed that GV treatment significantly increased the mitochondrial GSH-to-GSSG ratio (an indicator of intracellular redox status), and miR-129-3p overexpression (miR-129-3p mimics) produced the opposite effect, which was a significant reversal via MCU overexpression (miR-129-3p mimics+MCU) (Figure 2D). GV treatment also reduced MnSOD activity (one of the primary defenses against ROS), and miR-129 overexpression (miR-129-3p mimics) significantly reversed these effects in GV-treated hippocampal neuronal cells (Figure 2E). MCU overexpression (miR-129-3p mimics+MCU) partially inhibited miR-129-3p-enhanced MnSOD activity (Figure 2E). Collectively, these findings suggest that miR-129-3p alleviates mitochondrial oxidative stress by inhibiting mitochondrial calcium overload and ROS production and restoring antioxidant system homeostasis by targeting MCU.
miR-129-3p Overexpression Inhibits Apoptosis in Glucose Fluctuation-Exposed Hippocampal Neuronal Cells by Targeting MCU
Flow cytometry analysis showed that the enhanced expression of miR-129-3p (miR-129-3p mimics) significantly reduced the apoptotic rate in GV-mediated hippocampal neuronal cells, but cotransfection with pcDNA 3.1-MCU (miR-129-3p mimics+MCU) partially reversed this effect (Figure 3). miR-129-3p overexpression (miR-129-3p mimics) in GV-treated hippocampal neuronal cells markedly reduced cytochrome c release (Figure 4A and B). The upregulation of miR-129-3p significantly (miR-129-3p mimics) decreased the expression levels of MMP-2, MCU, caspase-9 and caspase-3, and cotransfection with pcDNA 3.1-MCU (miR-129-3p mimics+MCU) reversed these effects (Figure 4C–H and Supplementary Figure S2). Therefore, miR-129-3p suppresses mitochondria-dependent intrinsic apoptosis by targeting MCU.
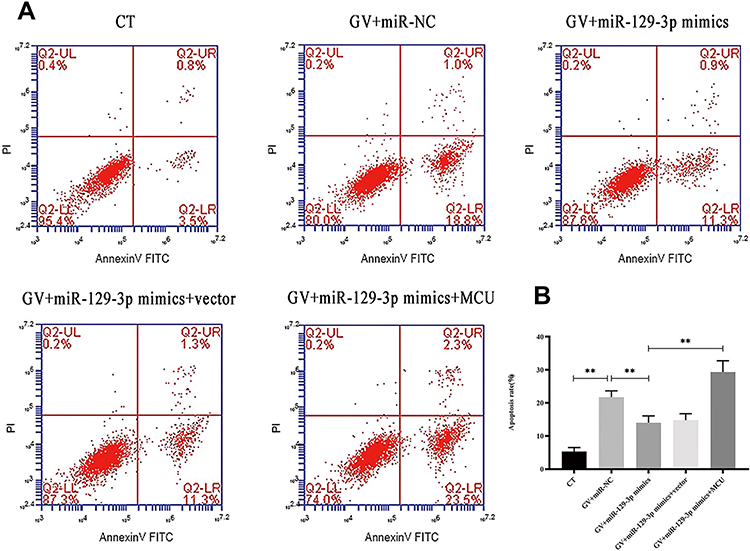 |
Figure 3 miR-129-3p overexpression inhibits apoptosis in glucose fluctuation-exposed hippocampal neuronal cells apoptosis by targeting MCU. Rat primary hippocampal neuronal cells were transfected with miR-NC, miR-129-3p mimics, miR-129-3p mimics+vector, or miR-129-3p mimics+MCU followed by glucose fluctuation treatment. (A) Flow cytometry was performed to detect apoptosis of hippocampal neuronal cells. (B) Apoptosis rate (%). Data are presented as the means ± SD; **p< 0.01 (n = 5). CT, normal glucose; GV, glucose fluctuation; vector, MCU negative control (pcDNA3.1 vector). |
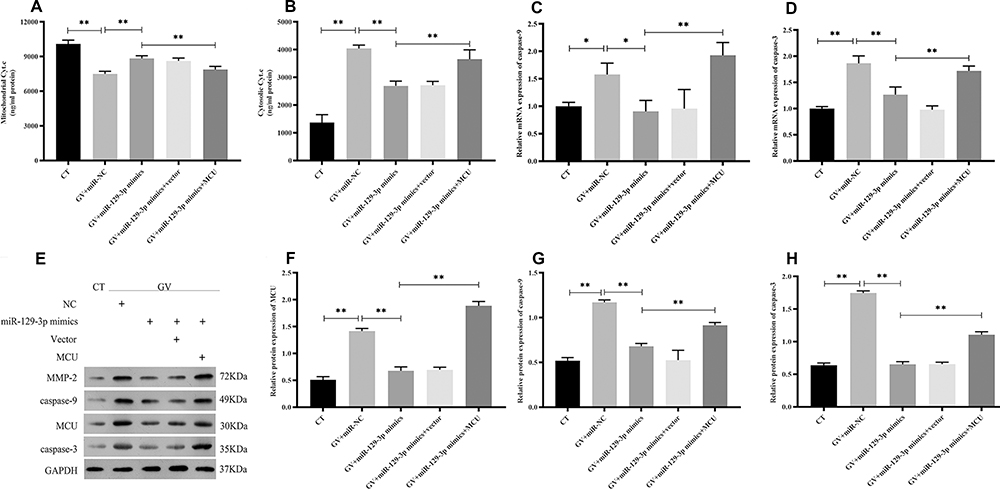 |
Figure 4 miR-129-3p overexpression inhibits the mitochondrial-dependent intrinsic apoptosis pathway in glucose fluctuation-treated hippocampal neuronal cells by targeting MCU. Rat primary hippocampal neuronal cells were transfected with miR-NC, miR-129-3p mimics, miR-129-3p mimics+vector, or miR-129-3p mimics+MCU followed by glucose fluctuation treatment. (A) Mitochondrial cytochrome c, and (B) cytosolic cytochrome c. mRNA levels of (C) caspase-9 and (D) caspase-3. (E–H) Protein levels of MCU, caspase-9, and caspase-3. Data are presented as the means ± SD; *p< 0.05, **p< 0.01; (n = 5). CT, normal glucose; GV, glucose fluctuation; vector, MCU negative control (pcDNA3.1 vector). |
miR-129-3p Inhibits ROS-Mediated Apoptosis in Glucose Fluctuation-Exposed Hippocampal Neuronal Cells
ROS is a critical regulator of glucose fluctuation-mediated damage. Therefore, we determined whether ROS played an essential role in miR-129-3p-mediated anti-apoptosis in GV-treated hippocampal neuronal cells. Notably, we demonstrated that NAC significantly reduced calcium overload (Figure 5A, B and Supplementary Figure S3a). Our data also demonstrated that antioxidant NAC (miR-129-3p inhibitor+antio) remarkably blocked the promoting oxidation effect of miR-129-3p downregulation (miR-129-3p inhibitor) in GV-treated hippocampal neuronal cells (Figure 5A, C and Supplementary Figure S3b). NAC treatment (miR-129-3p inhibitor+antio) decreased cytochrome c release and inhibited mRNA and protein levels of MMP-2, caspase-9, and caspase-3 (Figure 6 and Supplementary Figure S4). Therefore, miR-129-3p inhibited ROS-mediated apoptosis.
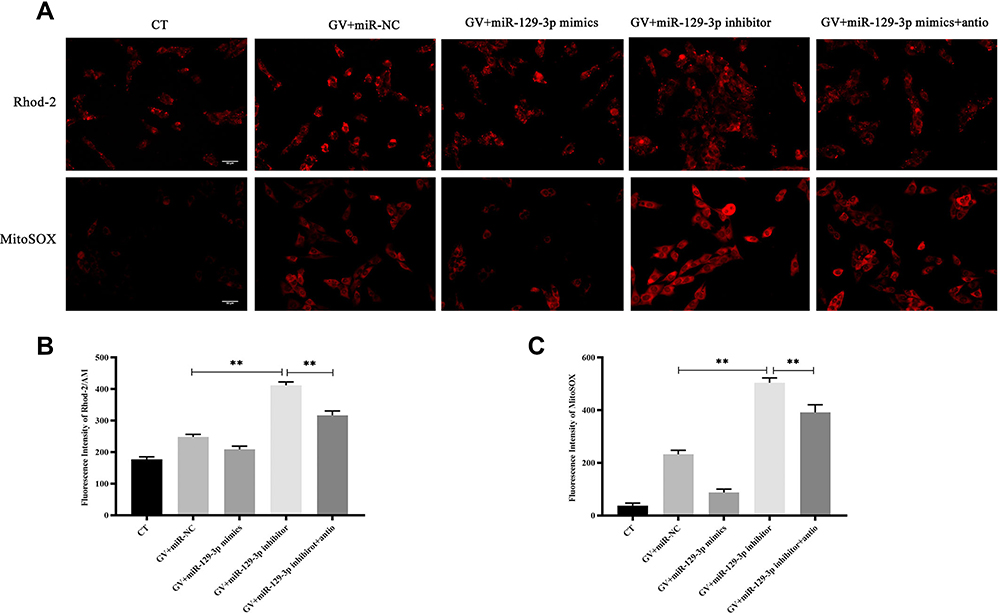 |
Figure 5 Silencing of miR-129-3p inhibits calcium overload and oxidative stress in glucose fluctuation-exposed hippocampal neuronal cells. Rat primary hippocampal neuronal cells were transfected with miR-NC, miR-129-3p mimics, miR-129-3p inhibitor with or without NAC followed by glucose fluctuation treatment. (A–C) The concentration of mitochondrial Ca2+ and O2− was detected using immunofluorescence. Scale bars = 50 μm. Data are presented as the means ± SD; **p< 0.01; (n = 5). CT, normal glucose; GV, glucose fluctuation; antio, NAC. |
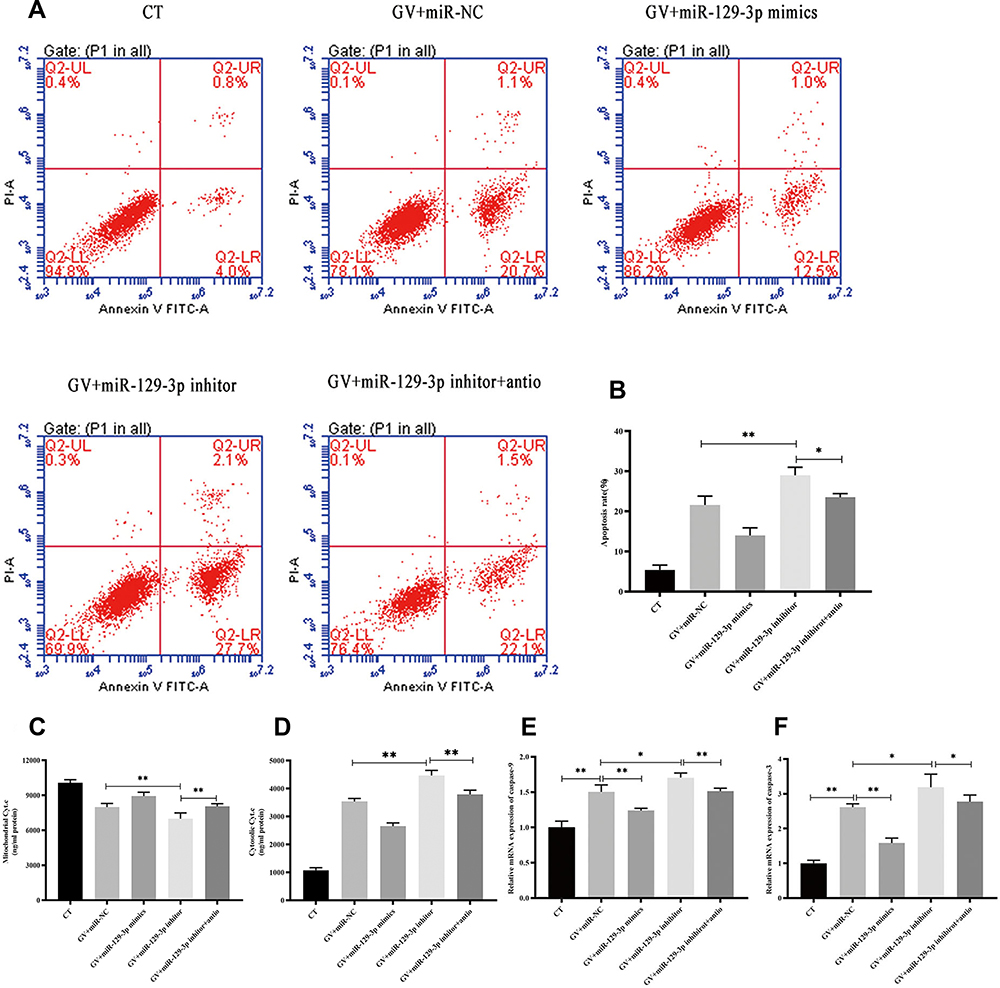 |
Figure 6 miR-129-3p inhibits ROS-mediated apoptosis in glucose fluctuation-exposed hippocampal neuronal cells. Rat primary hippocampal neuronal cells were transfected with miR-NC, miR-129-3p mimics, miR-129-3p inhibitor with or without NAC, followed by glucose fluctuation treatment. (A) Flow cytometry was performed to detect the apoptosis of hippocampal neuronal cells. (B) Apoptosis rate (%). (C) Mitochondrial cytochrome c and (D) cytosolic cytochrome c. mRNA levels of (E) caspase-9 and (F) caspase-3. Data are presented as the means ± SD; *p< 0.05, **p< 0.01; (n = 5). CT, normal glucose; GV, glucose fluctuation; antio, NAC. |
Discussion
Our study provides evidence for an anti-apoptotic effect of miR-129-3p by targeting MCU expression, relieving mitochondrial Ca2+ overload and oxidative stress, and inactivating the mitochondrial-dependent intrinsic apoptosis pathway in hippocampal neuronal cells exposed to GV. First, MCU was upregulated in GV-treated hippocampal neuronal cells, and miR-129-3p was downregulated. Second, the dual-luciferase assay showed that MCU was a direct target of miR-129-3p. Third, miR-129-3p alleviated mitochondrial Ca2+ and ROS elevation by targeting MCU, contributing to the restoration of antioxidant system homeostasis. Fourth, miR-129-3p conferred significant inhibition of the mitochondrial-dependent intrinsic apoptosis pathway. Last, ROS mediated the neuroprotective effect of miR-129-3p, and there is crosstalk between calcium and ROS.
miR-129-3p is a mature miRNA that is formed from the miR-129 precursor. Previous studies recognized miR-129-3p as a tumor suppressor, which is associated with increased apoptosis-related cleaved caspase-9 and caspase-3 proteins in colorectal cancer.22,23 miR-129-3p dramatically arrested the lung cancer cell cycle in S phrase.22 However, the functional roles of miR-129-3p in pathological conditions, including glucose fluctuation, are mostly unknown. The current study demonstrated that downregulated miR-129-3p in GV-treated hippocampal neuronal cells contributed to neuronal apoptosis. Consistent with our results, Wu et al showed that miR-129 was downregulated at the mRNA and protein levels in hippocampal neurons, and its deficiency sensitized neurons to cells.24
MCU is a crucial protein located in the IMM that mediates Ca2+ uptake into the mitochondrial matrix to regulate Ca2+ signaling and control aerobic metabolism.13 Under pathological conditions, excessive mitochondrial Ca2+ uptake through MCU causes ROS production, reduced ATP and loss of MMP.25,26 Oxidative stress induces oxidation of cellular macromolecules, such as cardiolipin anchoring cytochrome c, promoting the release of cytochrome c from mitochondria.27 Released cytochrome c is a pro-apoptotic factor that binds to Apaf-1, pro-caspase-9, and dATP in the cytoplasm to activate downstream caspase 9 or 3/7.28,29 Our results are consistent with a previous study that showed that GV-mediated enhancement of MCU induced the mitochondrial-dependent intrinsic apoptosis pathway by elevating mitochondrial Ca2+ uptake, which potentiated ROS formation. In contrast, the overexpression of miR-129-3p reversed this effect by targeting MCU.
Our study found that GV treatment significantly enhanced the ratio of GSH-to-GSSG, and miR-129-3p overexpression produced the opposite effect, which was a significant reversal via MCU overexpression. That is different from the traditional point that oxidative damage is related to lower antioxidant system activity.30,31 One of the critical roles of Ca2+ is the regulation of oxidative stress via misbalancing ROS generation and ROS detoxification. Ca2+ modulates the antioxidant defense systems via the promotion of nicotinamide adenine dinucleotide phosphate (NADPH) regeneration, which donates electrons to regenerate GSH from GSSG.32,33 Therefore, It is reasonable to draw a hypothesis that miR-129-3p/MCU-mediated Ca2+ uptake enhances the antioxidant system, which may be a metabolic alteration associated with the adaptive response in pathological conditions.34
A recent study showed that glucose fluctuation was related to brain dysfunction and cognitive impairment.35 Our study found that miR-129-3p reduced MMP-2 expression in GV-treated hippocampal neuronal cells. Matrix metalloproteinases (MMP) are zinc- and calcium-dependent endopeptidases that degrade the extracellular matrix (ECM).36 MMP-2 may play a role in Alzheimer’s disease (AD) pathology.37 An MMP-2 inhibition prevented beta-amyloid (Aβ)-induced neuronal cell death.38 Moreover, MMP-2 is a modulator of neuronal precursor activity and cognitive and motor behaviors, and it was increased in the hippocampus of the postoperative cognitive dysfunction (POCD) model.39,40 This evidence indicated the crucial role of MMP-2 in cognitive function. Notably, the expression of MMP-2 is ROS-dependent, and MMP-2, but not MMP-9, is related to MCU activity.41 Collectively, we draw a hypothesis that miR-129-3p improved glucose fluctuation-induced cognitive impairment via a miR-129 −3p/MCU/ROS/MMP-2 pathway. However, this hypothesis needs further clinical and/or animal model studies to confirm our findings.
It is widely accepted that ROS plays a critical role in GV-mediated damage.8 We demonstrated that the ROS scavenger NAC, which shows antioxidant properties by replenishing cellular GSH and participating in endogenous antioxidant systems,42 reduced GV-treated neuronal cells apoptosis and ROS levels in GV-treated hippocampal neuronal cells and alleviated the mitochondrial Ca2+ uptake. There is a feed-forward, self-amplified loop between the Ca2+-modulated generation of ROS and the ROS-induced increase of Ca2+.14 Dong et al showed that the crosstalk between ROS accumulation and MCU activity was a positive feedback mechanism, and MCU is a mitochondrial redox sensor through cysteine 97 (cys-97).43 These results strongly suggest that ROS and mitochondrial Ca2+ are closely linked and support the outstanding role of miR-129-3p/MCU in the treatment of GV-mediated neuronal damage.
Conclusion
Our study provides evidence that miR-129-3p protects GV-exposed hippocampal neurons by reducing MCU expression and subsequently easing mitochondrial Ca2+ overload, oxidative stress, and mitochondrial-dependent intrinsic apoptosis. We are the first to elucidate the role of miR-129-3p in GV-treated hippocampal neuronal cells. Our findings provide novel insights for the miRNAs’ role in coping with oxidative stress and may represent new targets for GV-mediated neuronal damage.
Data Sharing Statement
The data used to support the findings of this study are available from the corresponding author upon request.
Funding
This study is supported by Yunnan clinical medical center of nervous system diseases(ZX2019-03-05).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, Nogueira-Machado JA. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018;9(2):119. doi:10.1038/s41419-017-0135-z
2. Quagliaro L, Piconi L, Assaloni R, Martinelli L, Motz E, Ceriello A. Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells: the role of protein kinase C and NAD(P)H-oxidase activation. Diabetes. 2003;52(11):2795–2804. doi:10.2337/diabetes.52.11.2795
3. Liu TS, Pei YH, Peng YP, Chen J, Jiang SS, Gong JB. Oscillating high glucose enhances oxidative stress and apoptosis in human coronary artery endothelial cells. J Endocrinol Invest. 2014;37(7):645–651. doi:10.1007/s40618-014-0086-5
4. Russo VC, Higgins S, Werther GA, Cameron FJ. Effects of fluctuating glucose levels on neuronal cells in vitro. Neurochem Res. 2012;37(8):1768–1782. doi:10.1007/s11064-012-0789-y
5. Quincozes-Santos A, Bobermin LD, de Assis AM, Gonçalves CA, Souza DO. Fluctuations in glucose levels induce glial toxicity with glutamatergic, oxidative and inflammatory implications. Biochim Biophys Acta Mol Basis Dis. 2017;1863(1):1–14. doi:10.1016/j.bbadis.2016.09.013
6. Cox DJ, Kovatchev BP, Gonder-Frederick LA, et al. Relationships between hyperglycemia and cognitive performance among adults with type 1 and type 2 diabetes. Diabetes Care. 2005;28(1):71–77. doi:10.2337/diacare.28.1.71
7. Wang H, Deng J, Chen L, Ding K, Wang Y. Acute glucose fluctuation induces inflammation and neurons apoptosis in hippocampal tissues of diabetic rats. J Cell Biochem. 2019. doi:10.1002/jcb.29523
8. Ceriello A, Ihnat MA. ‘Glycaemic variability’: a new therapeutic challenge in diabetes and the critical care setting. Diabet Med. 2010;27(8):862–867. doi:10.1111/j.1464-5491.2010.02967.x
9. Cardoso S, Correia S, Carvalho C, et al. Perspectives on mitochondrial uncoupling proteins-mediated neuroprotection. J Bioenerg Biomembr. 2015;47(1–2):119–131. doi:10.1007/s10863-014-9580-x
10. De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476(7360):336–340. doi:10.1038/nature10230
11. Baughman JM, Perocchi F, Girgis HS, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476(7360):341–345. doi:10.1038/nature10234
12. Llorente-Folch I, Rueda CB, Pardo B, Szabadkai G, Duchen MR, Satrustegui J. The regulation of neuronal mitochondrial metabolism by calcium. J Physiol. 2015;593(16):3447–3462. doi:10.1113/JP270254
13. De Stefani D, Patron M, Rizzuto R. Structure and function of the mitochondrial calcium uniporter complex. Biochim Biophys Acta. 2015;1853(9):2006–2011. doi:10.1016/j.bbamcr.2015.04.008
14. Peng TI, Jou MJ. Oxidative stress caused by mitochondrial calcium overload. Ann N Y Acad Sci. 2010;1201:183–188. doi:10.1111/j.1749-6632.2010.05634.x
15. Zhang K, Yan J, Wang L, et al. The Pyk2/MCU pathway in the rat middle cerebral artery occlusion model of ischemic stroke. Neurosci Res. 2018;131:52–62. doi:10.1016/j.neures.2017.09.002
16. Zhang L, Wang H, Zhou X, Mao L, Ding K, Hu Z. Role of mitochondrial calcium uniporter-mediated Ca(2+) and iron accumulation in traumatic brain injury. J Cell Mol Med. 2019;23(4):2995–3009. doi:10.1111/jcmm.14206
17. Liu ZJ, Zhao W, Lei HY, et al. High glucose enhances bupivacaine-induced neurotoxicity via MCU-mediated oxidative stress in SH-SY5Y cells. Oxid Med Cell Longev. 2019;2019:7192798.
18. Vishnoi A, Rani S. MiRNA biogenesis and regulation of diseases: an overview. Methods Mol Biol. 2017;1509:1–10.
19. Zou Y, Kong M. Tetrahydroxy stilbene glucoside alleviates palmitic acid-induced inflammation and apoptosis in cardiomyocytes by regulating miR-129-3p/Smad3 signaling. Cell Mol Biol Lett. 2019;24:5. doi:10.1186/s11658-018-0125-x
20. Zhang Y, Wang Y, Wei Y, et al. MiR-129-3p promotes docetaxel resistance of breast cancer cells via CP110 inhibition. Sci Rep. 2015;5:15424. doi:10.1038/srep15424
21. Guo J, Sang Y, Yin T, et al. miR-1273g-3p participates in acute glucose fluctuation-induced autophagy, dysfunction, and proliferation attenuation in human umbilical vein endothelial cells. Am J Physiol Endocrinol Metab. 2016;310(9):E734–E743. doi:10.1152/ajpendo.00444.2015
22. Wu YF, Ou CC, Chien PJ, Chang HY, Ko JL, Wang BY. Chidamide-induced ROS accumulation and miR-129-3p-dependent cell cycle arrest in non-small lung cancer cells. Phytomedicine. 2019;56:94–102. doi:10.1016/j.phymed.2018.09.218
23. Karaayvaz M, Zhai H, Ju J. miR-129 promotes apoptosis and enhances chemosensitivity to 5-fluorouracil in colorectal cancer. Cell Death Dis. 2013;4(6):e659. doi:10.1038/cddis.2013.193
24. Wu DM, Zhang YT, Lu J, Zheng YL. Effects of microRNA-129 and its target gene c-Fos on proliferation and apoptosis of hippocampal neurons in rats with epilepsy via the MAPK signaling pathway. J Cell Physiol. 2018;233(9):6632–6643. doi:10.1002/jcp.26297
25. Vergun O, Votyakova TV, Reynolds IJ. Spontaneous changes in mitochondrial membrane potential in single isolated brain mitochondria. Biophys J. 2003;85(5):3358–3366. doi:10.1016/S0006-3495(03)74755-9
26. Paupe V, Prudent J. New insights into the role of mitochondrial calcium homeostasis in cell migration. Biochem Biophys Res Commun. 2018;500(1):75–86. doi:10.1016/j.bbrc.2017.05.039
27. Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci U S A. 2002;99(3):1259–1263. doi:10.1073/pnas.241655498
28. Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86(1):147–157. doi:10.1016/S0092-8674(00)80085-9
29. Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11(9):621–632. doi:10.1038/nrm2952
30. Prasai PK, Shrestha B, Orr AW, Pattillo CB. Decreases in GSH:GSSG activate vascular endothelial growth factor receptor 2 (VEGFR2) in human aortic endothelial cells. Redox Biol. 2018;19:22–27. doi:10.1016/j.redox.2018.07.015
31. Soni KK, Zhang LT, Choi BR, et al. Protective effect of MOTILIPERM in varicocele-induced oxidative injury in rat testis by activating phosphorylated inositol requiring kinase 1α (p-IRE1α) and phosphorylated c-Jun N-terminal kinase (p-JNK) pathways. Pharm Biol. 2018;56(1):94–103. doi:10.1080/13880209.2017.1421672
32. Kohlhaas M, Liu T, Knopp A, et al. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121(14):1606–1613. doi:10.1161/CIRCULATIONAHA.109.914911
33. Chen Y, Csordás G, Jowdy C, et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ Res. 2012;111(7):863–875. doi:10.1161/CIRCRESAHA.112.266585
34. Russo GL, Tedesco I, Russo M, Cioppa A, Andreassi MG, Picano E. Cellular adaptive response to chronic radiation exposure in interventional cardiologists. Eur Heart J. 2012;33(3):408–414. doi:10.1093/eurheartj/ehr263
35. Xia W, Luo Y, Chen YC, Chen H, Ma J, Yin X. Glucose fluctuations are linked to disrupted brain functional architecture and cognitive impairment. J Alzheimer’s Dis. 2020;74(2):603–613. doi:10.3233/JAD-191217
36. Nakaji K, Ihara M, Takahashi C, et al. Matrix metalloproteinase-2 plays a critical role in the pathogenesis of white matter lesions after chronic cerebral hypoperfusion in rodents. Stroke. 2006;37(11):2816–2823. doi:10.1161/01.STR.0000244808.17972.55
37. Brkic M, Balusu S, Libert C, Vandenbroucke RE. Friends or foes: matrix metalloproteinases and their multifaceted roles in neurodegenerative diseases. Mediators Inflamm. 2015;2015:620581. doi:10.1155/2015/620581
38. Haorah J, Ramirez SH, Schall K, Smith D, Pandya R, Persidsky Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J Neurochem. 2007;101(2):566–576. doi:10.1111/j.1471-4159.2006.04393.x
39. Li Q, Michaud M, Shankar R, Canosa S, Schwartz M, Madri JA. MMP-2: A modulator of neuronal precursor activity and cognitive and motor behaviors. Behav Brain Res. 2017;333:74–82. doi:10.1016/j.bbr.2017.06.041
40. Hu N, Guo D, Wang H, et al. Involvement of the blood-brain barrier opening in cognitive decline in aged rats following orthopedic surgery and high concentration of sevoflurane inhalation. Brain Res. 2014;1551:13–24. doi:10.1016/j.brainres.2014.01.015
41. Liu D, Zhang R, Wu J, et al. Interleukin-17A promotes esophageal adenocarcinoma cell invasiveness through ROS-dependent, NF-κB-mediated MMP-2/9 activation. Oncol Rep. 2017;37(3):1779–1785. doi:10.3892/or.2017.5426
42. Miranda-Díaz AG, García-Sánchez A, Cardona-Muñoz EG. Foods with potential prooxidant and antioxidant effects involved in Parkinson’s disease. Oxid Med Cell Longev. 2020;2020:6281454. doi:10.1155/2020/6281454
43. Dong Z, Shanmughapriya S, Tomar D, et al. Mitochondrial Ca(2+) uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol Cell. 2017;65(6):1014–1028.e1017. doi:10.1016/j.molcel.2017.01.032
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.