")
Back to Journals » OncoTargets and Therapy » Volume 11
miR-1 inhibits the proliferation of breast cancer stem cells by targeting EVI-1
Authors Wu L, Wang T, He D, Li X, Jiang Y
Received 26 September 2018
Accepted for publication 3 November 2018
Published 6 December 2018 Volume 2018:11 Pages 8773—8781
DOI https://doi.org/10.2147/OTT.S188836
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjeev K. Srivastava
Lei Wu,1 Tianyi Wang,2 Dongning He,2 Xiaoxi Li,1 Youhong Jiang1
1Molecular Oncology Laboratory of Cancer Research Institute, The First Hospital of China Medical University, Shenyang 110001, China; 2Department of Medical Oncology, The First Affiliated Hospital of Jinzhou Medical University, Jinzhou 121001, China
Purpose: Breast cancer stem cells (BCSCs) have been regarded as the key factor for treatment failure in breast cancer. The abnormal expression of miRNAs plays a significant role in different tumor types. However, the role of miR-1 in breast cancer remains poorly understood. The purpose of this study was to evaluate the effects of miR-1 on the proliferation and apoptosis of BCSCs.
Materials and methods: CD44+/CD24-/low/epithelial-specific antigen+ BCSCs were isolated by flow cytometry. Real-time PCR and Western blotting were used to determine the expression of miRNAs, mRNAs, and epithelial–mesenchymal transition (EMT)-related genes. Cell proliferation and apoptosis were measured using the Cell Counting Kit-8 assay and Annexin V-fluorescein isothiocyanate flow cytometry, respectively. Luciferase reporter assay was used to verify whether miR-1 targeted ecotropic virus integration-1 (EVI-1). The role of miR-1 in breast cancer in vivo was evaluated using BCSCs xenograft mouse models.
Results: In this study, we demonstrated that miR-1 was significantly downregulated in breast cancer tissues compared to the adjacent non-tumor tissues. The luciferase reporter assay verified that EVI-1 was a direct target of miR-1, and upregulation of miR-1 negatively correlated with the expression of EVI-1 in BCSCs at both the transcriptional and posttranslational levels. Furthermore, overexpression of miR-1 inhibited BCSCs proliferation and promoted apoptosis, which was reversed by the overexpression of EVI-1. In addition, we demonstrated that aberrant expression of miR-1 could regulate EMT-related genes in BCSCs. Finally, immunohistochemical staining demonstrated that EVI-1 expression was decreased in BCSCs tumors following intratumoral miR-1 agomir treatment compared to the control group.
Conclusion: miR-1 can negatively regulate the expression of EVI-1 and, thus, affect BCSCs proliferation, apoptosis, and EMT-related markers. Taken together, these findings demonstrate that miR-1 could be employed as a therapeutic strategy in the treatment of breast cancer.
Keywords: breast cancer, miR-1, proliferation, apoptosis, EVI-1
Introduction
Breast cancer is a leading cause of cancer-related death in women.1 Conventional treatment options for breast cancer include surgery, chemotherapy, radiotherapy, and endocrinotherapy. These therapeutic modalities have some disadvantages, including a high incidence of drug-related side effects, primary or secondary drug resistance, and postoperative complications.2 To overcome these side effects, novel therapies are needed. Recent studies have demonstrated that breast cancer stem cells (BCSCs) constitute a rare subpopulation of tumor cells. Due to their high tumorigenicity and high invasive and metastatic potential, BCSCs may play an important role in the initiation, progression, metastasis, and recurrence of breast cancer.3,4 Al-Hajj et al5 first discovered CD44+/CD24−/low/epithelial-specific antigen (ESA)+ breast cancer cells, which exhibited higher tumorigenicity compared to other cell types. These cells are considered BCSCs, and the proportion of BCSCs in the total tumor cell population is closely related to the prognosis of breast cancer patients. Therefore, the study of stem cells has become more important in the field of breast cancer.
miRNAs are a class of noncoding RNAs that are 19–25 nucleotides in length. They regulate cell proliferation, differentiation, and apoptosis via either perfect or imperfect base pairing with the 3′-untranslated region (3′UTR) of their target mRNAs.6–8 Certain miRNAs are important molecular markers for breast cancer.9,10 For instance, the expression of miR-206 is downregulated in estrogen receptor-α-positive breast cancer cells, and the upregulation of miR-206 dramatically inhibits the growth of MCF-7 breast cancer cells;11 miR-1 is also a tumor suppressor, and its expression is downregulated in several types of cancer, including breast cancer.12 However, the particular role of miR-1 in breast cancer requires further investigation.
Ecotropic virus integration-1 (EVI-1) is an oncogenic transcription factor.13 EVI-1 expression is associated with tumor progression and poor prognosis in breast cancer patients.14 Overexpression of EVI-1 has been frequently observed in hematological malignancies15–17 and several solid tumors.18,19 Interestingly, the relationship between EVI-1 and miRNAs is gradually being discovered. Previous studies have reported that EVI-1 is a target of miR-22 in ovarian cancer.20 miRNA-206/133b might facilitate cancerous growth by increasing the expression of EVI-1.21 However, very little is known about how EVI-1 and miR-1 regulate breast cancer oncogenesis.
In the present study, we demonstrated that miR-1 can negatively regulate EVI-1 expression and affect BCSCs proliferation, apoptosis, and epithelial–mesenchymal transition (EMT)-related markers. Our study provides potential new insights into the treatment of breast cancer.
Materials and methods
Patient samples
Twenty-one matched pairs of human breast cancer and adjacent non-tumor tissue were obtained from the First Affiliated Hospital of China Medical University. All patients had not received preoperative adjuvant therapy. Written informed consent was obtained from each patient. This study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of China Medical University.
Cell culture
Human MDA-MB-231 breast cancer cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cells were maintained in L-15 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (HyClone Laboratories Inc., Logan, UT, USA) at 37°C in a humidified atmosphere without CO2.
Identification of BCSCs
MDA-MB-231 cells were harvested by trypsinization at 80%–90% confluence. The cells were labeled with anti-human CD44-fluorescein isothiocyanate (FITC), anti-human CD24-phycoerythrin, and anti-human ESA-PerCP-Cy5.5-A (peridinin–chlorophyll–protein complex) antibodies (BioLegend Inc., San Diego, CA, USA) for 20 minutes in the dark according to the manufacturer’s instructions. The BCSCs were identified as CD44+/CD24−/low/ESA+ cells using flow cytometry (BD, Franklin Lakes, NJ, USA). The BCSCs were maintained in DMEM/F12 (1:1) medium (Thermo Fisher Scientific) supplemented with 20 μg/L basic fibroblast growth factor, 10 μg/L epidermal growth factor, and 2% B27, and incubated at 37°C under 5% CO2.
Cell transfection
The sequence of the miR-1 agomir was 5′-UGGAAUGUAAAGAAGUAUGUAU-3′, and the miR-1 antagomir sequence was 5′-AUACAUACUUCUUUACAUUCCA-3′. Both oligonucleotides were synthesized by GenePharma (Shanghai, China). The EVI-1 expressing vector pcDNA3.1-EVI-1 and EVI-1-shRNA (pSilencer 2.1-EVI-1) were designed and synthesized by Sangon Biotech Co., Ltd. (Shanghai, China). The primers used in this study were – pcDNA3.1-EVI-1 forward, 5′-CCGGAATTCATGATCTTAGACGAATTTTACA-3′; pcDNA3.1-EVI-1 reverse, 5′-CGCGGATCC TCATACGTGGGGATAGCACTGGA-3′; pSilencer2.1-EVI-1 forward, 5′-CCTACGATCAGTCCTACCA-3′; pSilencer2.1-EVI-1 reverse, 5′-TGGTAGGACTGATCGTAGG-3′. Transfection was performed using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Dual luciferase reporter assay
The wild-type (WT) or mutant (MUT) 3′UTR of EVI-1 was cloned downstream of the luciferase coding region of the pGL3-basic vector (Promega Corporation, Fitchburg, WI, USA) by Jrdun Biotechnology Co., Ltd. (Shanghai, China). The BCSCs were transfected with miR-1 or negative control (NC) agomir, and pGL3-EVI-1-3′UTR-WT or pGL3-EVI-1-3′UTR-MUT using Lipofectamine 2000 according to the manufacturer’s protocol. Forty-eight hours post-transfection, Firefly and Renilla luciferase activities were measured using the Dual-Luciferase® Reporter Assay System (Promega Corporation).
Cell proliferation assay
Cell viability was assessed using the Cell Counting Kit-8 assay (CCK-8; Beyotime Institute of Biotechnology, Jiangsu, China). Transfected BCSCs were seeded in 96-well plates at 5,000 cells/well. At designated time points following transfection (24, 48, and 72 hours), 10 μL CCK-8 assay solution was added to each well, and the plates were incubated for 2 hours. The absorbance was measured at 450 nm using a microplate reader (Thermo Fisher Scientific).
Flow cytometry analysis of cell apoptosis
Forty-eight hours after transfection, 2×105 BCSCs were collected, and apoptosis was measured using the Annexin V-FITC Apoptosis Detection Kit (Beyotime Institute of Biotechnology). The apoptosis rate was measured by flow cytometry (BD Biosciences, San Jose, CA, USA).
Quantitative real-time PCR
Total RNA was isolated from BCSCs or tissues with RNAiso Plus (Takara Biotechnology Co., Ltd., Dalian, China) according to the manufacturer’s instructions. To determine the expression of miR-1, the One Step PrimeScript 1 miRNA cDNA Synthesis Kit and SYBR® Premix Ex Taq™ II (Takara Biotechnology Co. Ltd.) were used to conduct real-time PCR. For mRNA detection, PrimeScript™ RT Reagent Kit with gDNA Eraser and SYBR® Premix Ex Taq™ II were used to perform real-time PCR according to the manufacturer’s instructions. The expression levels of U6 and GAPDH were used as an internal control for miRNA and mRNA, respectively. The sequences of the primers used for real-time PCR are shown in Table 1. Quantitative PCR data were analyzed using the 2−ΔΔCT method.
 | Table 1 Primers used in the real-time PCR |
Western blotting
Total protein was isolated from BCSCs using RIPA lysis buffer (Beyotime Institute of Biotechnology). Equal amounts of protein were loaded onto 8% SDS-PAGE gels and transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% nonfat milk for 2 hours at room temperature and then incubated with rabbit anti-EVI-1 (1:1,000), rabbit anti-E-cadherin (1:1,000), or rabbit anti-N-cadherin (1:1,000). GAPDH (1:2,000) was used as an internal control to ensure equal protein loading. All antibodies were purchased from Abcam (Cambridge, UK). The signal was detected by ECL (Thermo Fisher Scientific) after incubation with the corresponding goat anti-rabbit secondary antibody.
BCSCs xenografts in nude mice
Animal experiments were approved by the Committee on Animal Care of the First Affiliated Hospital of China Medical University and conducted according to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Female athymic BALB/c nude mice (4–5 weeks old) were bred under specific pathogen-free conditions in the Laboratory Animal Center of China Medical University. BCSCs (5×106) were resuspended in PBS and implanted subcutaneously in the right armpit of the mice. After 2 weeks, the mice were randomly divided into two groups (n=3). The mice were intratumorally injected with 10 nmol miR-1 agomir or NC-miR agomir in 20 μL PBS every 3 days for a total of six injections. The tumors were measured using calipers. The tumor volume was calculated using the formula: V=L×W2/2.
Immunohistochemistry
Xenograft tissues from nude mice were paraffin-embedded and sectioned. The tumor sections were incubated with primary antibody (polyclonal rabbit anti-EVI-1 antibody; 1:100) overnight at 4°C. The tumor sections were incubated with goat anti-rabbit secondary antibody (1:100) for 1 hour at room temperature. The sections were stained with diaminobenzidine, and the nucleus was counterstained with hematoxylin.
Statistical analyses
All experiments were repeated three times independently, and the data were presented as mean ± SD. The difference between any two groups was analyzed by the Student’s t-test. P<0.05 was considered statistically significant.
Results
miR-1 and EVI-1 were inversely expressed in human breast cancer tissues
The levels of miR-1 expression were significantly decreased in the 21 cases of human breast cancer compared to the matched adjacent normal tissue (Figure 1A). In contrast, EVI-1 mRNA expression was increased in these breast cancer tissues compared to the matched normal tissues (Figure 1B). In addition, we evaluated the expression levels of miR-1 and EVI-1 following the transfection of different vectors into BCSCs. The expression of miR-1 was markedly enhanced by transfection with the miR-1 agomir and significantly reduced by transfection with the miR-1 antagomir (Figure 1C and D). The expression of EVI-1 was noticeably increased or reduced in BCSCs transfected with pcDNA3.1-EVI-1 or pSilencer 2.1-EVI-1 (Figure 1E).
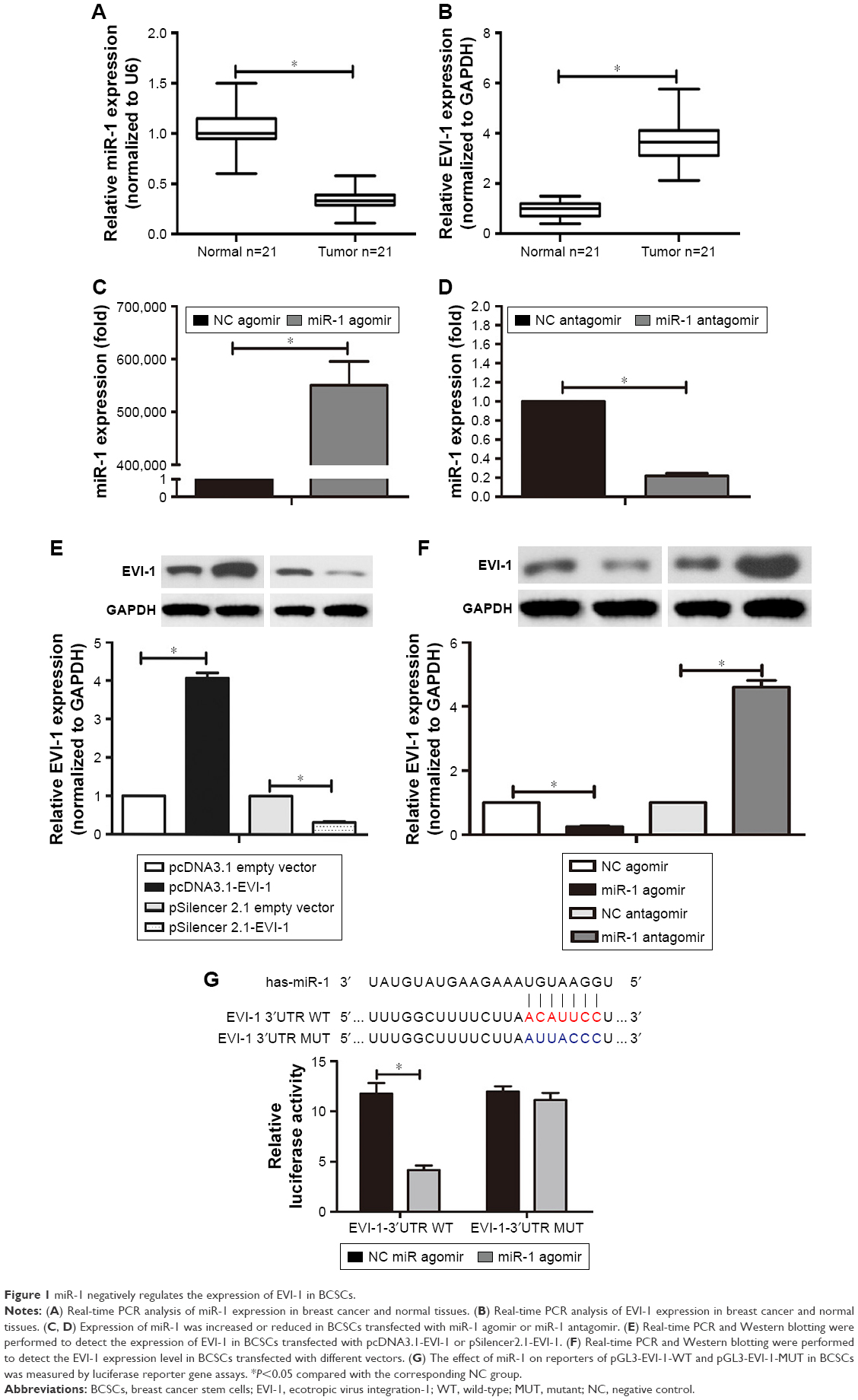 | Figure 1 miR-1 negatively regulates the expression of EVI-1 in BCSCs. |
miR-1 inhibits the expression of EVI-1 through direct targeting of its 3′UTR
To confirm the effect of miR-1 on EVI-1 expression, we evaluated the expression of EVI-1 in transfected BCSCs. EVI-1 expression was significantly decreased in miR-1 overexpressing BCSCs. However, EVI-1 expression was increased upon miR-1 silencing in BCSCs (Figure 1F). To further determine whether miR-1 directly targeted EVI-1, we performed bioinformatics prediction analysis using TargetScan (http://www.targetscan.org/) and identified potential binding sites for miR-1 in the 3′UTR of the EVI-1 mRNA. The sequences containing WT or MUT 3′UTR of EVI-1 were cloned downstream of the pGL3-basic dual luciferase reporter gene (pGL3-EVI-1-WT or pGL3-EVI-1-MUT, respectively). Luciferase activity was decreased by co-transfection of miR-1 agomir and WT EVI-1-3′UTR. However, the miR-1 agomir did not affect luciferase activity when co-transfected with EVI-1-3′UTR-MUT (Figure 1G). Overall, miR-1 negatively regulated EVI-1 expression at the mRNA and protein levels, suggesting that EVI-1 is a target gene of miR-1.
miR-1 inhibits proliferation and promotes apoptosis in BCSCs through downregulation of EVI-1 expression
The overexpression of miR-1 notably inhibited the proliferation of the BCSCs. Conversely, miR-1 silencing promoted cell proliferation. However, proliferation was nearly restored to the levels of the NC group by expression of EVI-1 in miR-1 overexpressing cells or silencing of EVI-1 in miR-1-silenced cells (Figure 2A). Flow cytometry was performed to determine the effect of miR-1 on apoptosis. The results showed that miR-1 overexpression increased and miR-1 silencing decreased the number of apoptotic cells compared to the NC groups. Furthermore, the number of apoptotic cells returned to levels similar to that of the NC group following ectopic EVI-1 expression in miR-1 overexpressing cells or silencing of EVI-1 expression in miR-1-silenced cells (Figure 2B).
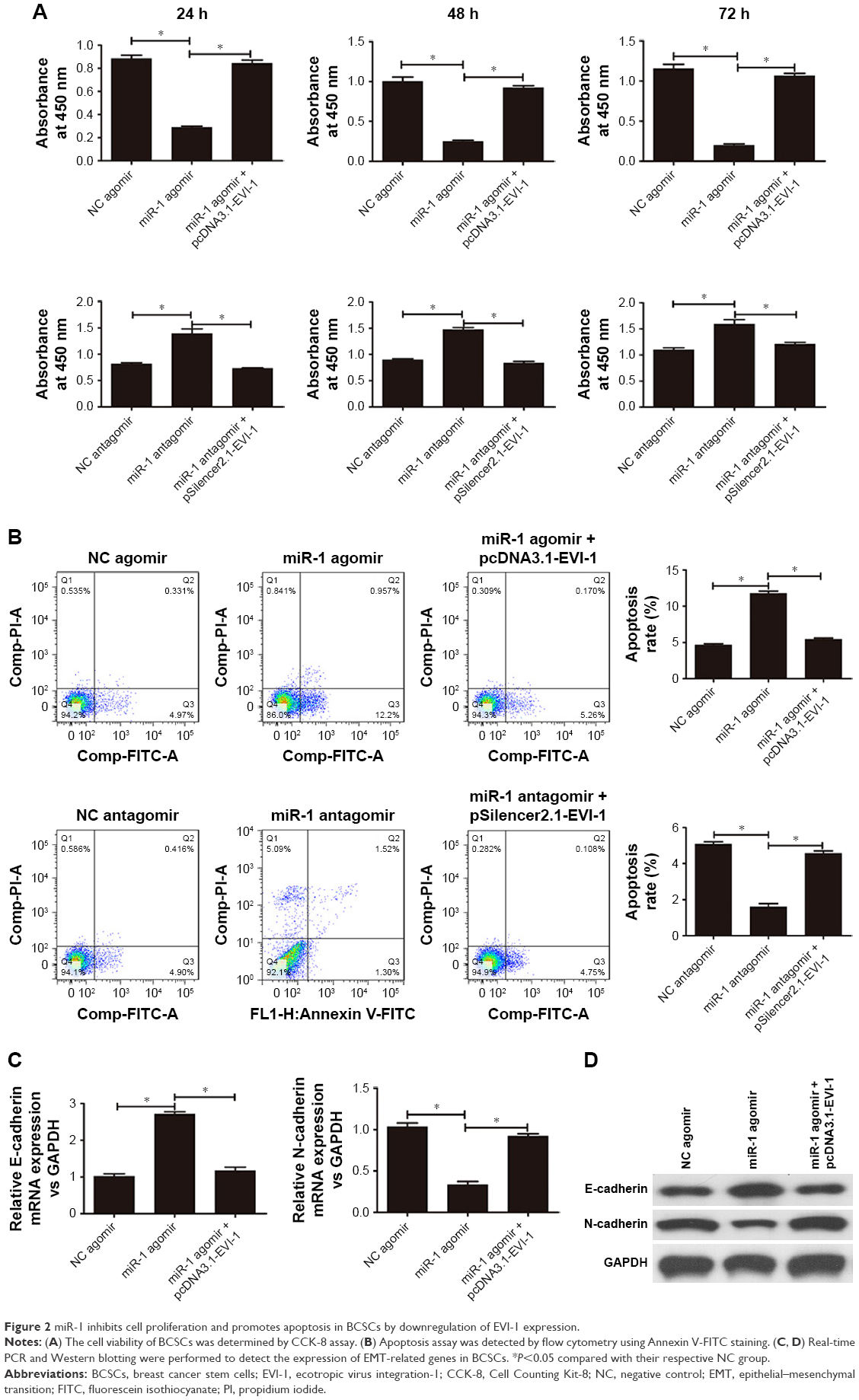 | Figure 2 miR-1 inhibits cell proliferation and promotes apoptosis in BCSCs by downregulation of EVI-1 expression. |
miR-1 regulates EMT-related marker expression in BCSCs via EVI-1
To evaluate the role of miR-1 in the EMT of BCSCs, we determined the expression of EMT markers (E-cadherin and N-cadherin) in BCSCs by real-time PCR and Western blotting following miR-1 overexpression or silencing. As shown in Figure 2C, there was a significant increase in E-cadherin mRNA expression and a significant decrease in N-cadherin mRNA expression in the BCSCs after the upregulation of miR-1 compared to the NC group. This effect was abrogated by the co-overexpression of miR-1 agomir and pcDNA3.1-EVI-1. Similar effects were observed at the protein level (Figure 2D).
miR-1 inhibits breast cancer growth in vivo
To further verify the suppressive effect of miR-1 in vivo, tumor xenografts were established from BCSCs isolated from MDA-MB-231 cells. Intratumor injection with miR-1 agomir significantly repressed the growth of the xenografts (Figure 3A and B). Immunohistochemistry demonstrated that EVI-1 levels were dramatically reduced in xenograft tissues injected with miR-1 agomir compared to NC agomir (Figure 3C).
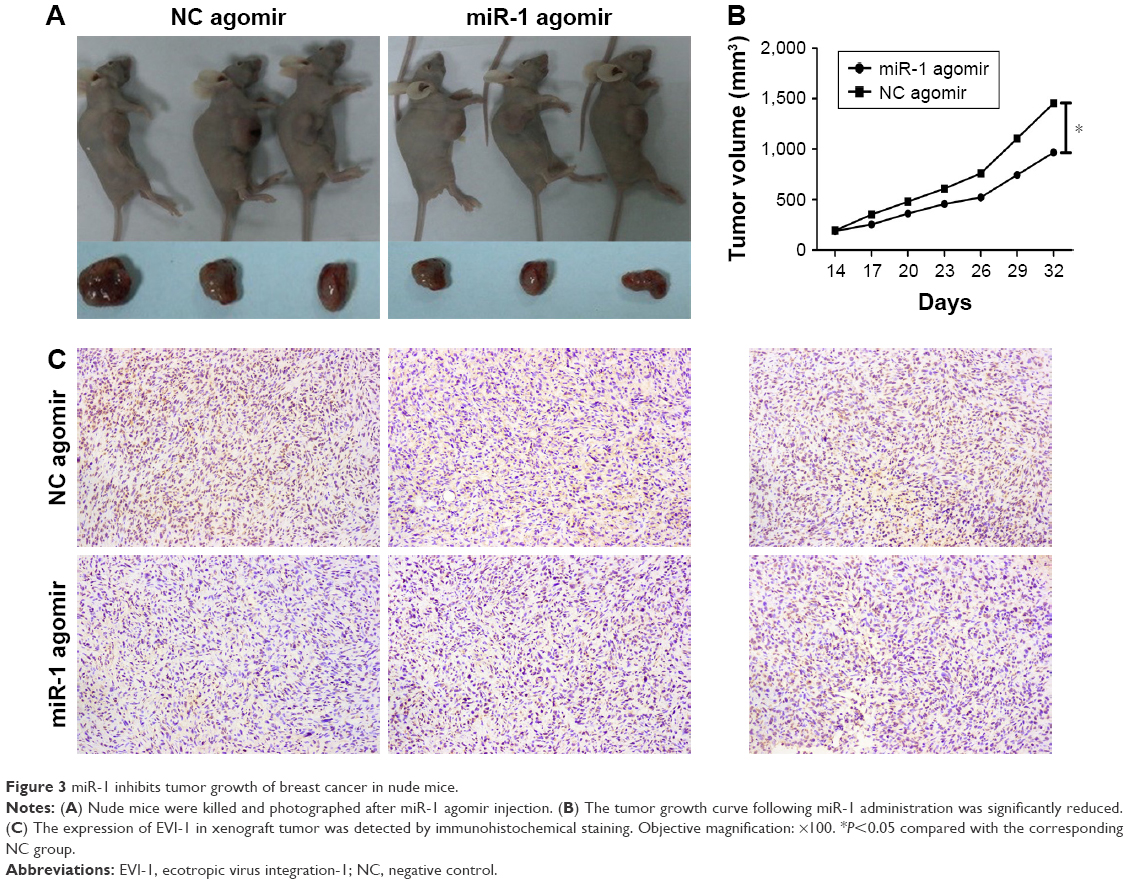 | Figure 3 miR-1 inhibits tumor growth of breast cancer in nude mice. |
Discussion
BCSCs are a special subpopulation of tumor cells that have a self-renewal capacity and are highly tumorigenic; both of these properties can contribute to treatment failure, tumor recurrence, and metastasis in breast cancer.22,23 miRNAs affect tumor initiation and progression, similar to the functions of oncogenes and tumor suppressors.24,25 Abnormal expression of miRNAs has been reported in breast cancer.26–28 Given the critical role of miRNAs in tumorigenesis, relationships between miRNAs and BCSCs have been investigated. Shimono et al29 investigated the low expression level of miR-200 family members in BCSCs and found that the proportion of BCSCs in breast cancer cells decreased in vitro upon induction of miR-200c expression. Therefore, studying the relationship between BCSCs and miRNAs should help unravel the mechanisms underlying miRNA-regulated BCSCs proliferation and metastasis, potentially leading to the discovery of novel therapeutic options for the treatment of breast cancer.
EVI-1 is a transcription factor that plays a critical role in oncogenesis.13 Overexpression of EVI-1 is closely correlated with high-risk tumor factor and worse prognosis.30,31 In this study, we demonstrated that miR-1 was expressed at low levels in human breast cancer tissues, and had a negative correlation with EVI-1 expression. In addition, we found that miR-1 inhibited the expression of EVI-1 by specifically binding to the 3′UTR of the EVI-1 mRNA.
Previous studies have indicated that some miRNAs can affect the proliferation of cancer cells.32–34 Using the CCK-8 assay, we demonstrated that miR-1 overexpression suppressed the proliferation of BCSCs. Conversely, the inhibition of miR-1 expression increased the proliferation of BCSCs. In vivo assays showed that miR-1 could function as a tumor suppressor for breast cancer in nude mice. The relationship between miRNAs and cancer cell apoptosis has been frequently reported in recent years.35–37 In our study, we found that overexpression of miR-1 increased apoptosis in BCSCs whereas its inhibition decreased apoptosis in BCSCs.
EMT plays an important role in tumor migration and metastasis to distant organs.38,39 Emerging evidence has shown that a number of miRNAs are involved in the regulation of EMT.40–43 Tumor cells frequently lose E-cadherin. Several studies have demonstrated that reduced expression of E-cadherin could promote EMT.44,45 Restoration of its expression can inhibit metastasis. In this study, we demonstrated that miR-1 overexpression can increase E-cadherin expression but decrease N-cadherin expression at both the mRNA and protein levels.
Conclusion
In summary, our study showed that there is a negative relationship between miR-1 and EVI-1, and miR-1 could affect the biological behavior of BCSCs by regulating the expression of EVI-1. miR-1 could potentially be used as a novel therapeutic target for breast cancer treatment.
Acknowledgments
The authors would like to thank Professor Ping Ma (Molecular Oncology Laboratory of Cancer Research Institute, The First Affiliated Hospital of China Medical University, Shenyang, China) for designing the study and excellent technical assistance. This work was kindly supported by a grant from the National Natural Science Foundation of China (no 81372812).
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Stebbing J, Ellis P. An overview of drug development for metastatic breast cancer. Br J Nurs. 2012;21(4):S18–S22. | ||
Velasco-Velázquez MA, Homsi N, De La Fuente M, Pestell RG. Breast cancer stem cells. Int J Biochem Cell Biol. 2012;44(4):573–577. | ||
Dandawate PR, Subramaniam D, Jensen RA, Anant S. Targeting cancer stem cells and signaling pathways by phytochemicals: novel approach for breast cancer therapy. Semin Cancer Biol. 2016;40–41:192–208. | ||
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. | ||
Sayed D, Abdellatif M. MicroRNAs in development and disease. Physiol Rev. 2011;91(3):827–887. | ||
Kwak PB, Iwasaki S, Tomari Y. The microRNA pathway and cancer. Cancer Sci. 2010;101(11):2309–2315. | ||
Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet. 2012;13(4):271–282. | ||
Wu X, Somlo G, Yu Y, et al. De novo sequencing of circulating miRNAs identifies novel markers predicting clinical outcome of locally advanced breast cancer. J Transl Med. 2012;10:42. | ||
Halvorsen AR, Helland Å, Gromov P, et al. Profiling of microRNAs in tumor interstitial fluid of breast tumors – a novel resource to identify biomarkers for prognostic classification and detection of cancer. Mol Oncol. 2017;11(2):220–234. | ||
Adams BD, Cowee DM, White BA. The role of miR-206 in the epidermal growth factor (EGF) induced repression of estrogen receptor-alpha (ERalpha) signaling and a luminal phenotype in MCF-7 breast cancer cells. Mol Endocrinol. 2009;23(8):1215–1230. | ||
Beltran AS, Russo A, Lara H, Fan C, Lizardi PM, Blancafort P. Suppression of breast tumor growth and metastasis by an engineered transcription factor. PLoS One. 2011;6(9):e24595. | ||
Wieser R. The oncogene and developmental regulator EVI1: expression, biochemical properties, and biological functions. Gene. 2007;396(2):346–357. | ||
Patel JB, Appaiah HN, Burnett RM, et al. Control of EVI-1 oncogene expression in metastatic breast cancer cells through microRNA miR-22. Oncogene. 2011;30(11):1290–1301. | ||
Daghistani M, Marin D, Khorashad JS, et al. EVI-1 oncogene expression predicts survival in chronic-phase CML patients resistant to imatinib treated with second-generation tyrosine kinase inhibitors. Blood. 2010;116(26):6014–6017. | ||
Gröschel S, Lugthart S, Schlenk RF, et al. High EVI1 expression predicts outcome in younger adult patients with acute myeloid leukemia and is associated with distinct cytogenetic abnormalities. J Clin Oncol. 2010;28(12):2101–2107. | ||
Vázquez I, Maicas M, Cervera J, et al. Down-regulation of EVI1 is associated with epigenetic alterations and good prognosis in patients with acute myeloid leukemia. Haematologica. 2011;96(10):1448–1456. | ||
Koos B, Bender S, Witt H, et al. The transcription factor evi-1 is overexpressed, promotes proliferation, and is prognostically unfavorable in infratentorial ependymomas. Clin Cancer Res. 2011;17(11):3631–3637. | ||
Deng X, Cao Y, Liu Y, et al. Overexpression of Evi-1 oncoprotein represses TGF-β signaling in colorectal cancer. Mol Carcinog. 2013;52(4):255–264. | ||
Nagaraja AK, Creighton CJ, Yu Z, et al. A link between mir-100 and FRAP1/mTOR in clear cell ovarian cancer. Mol Endocrinol. 2010;24(2):447–463. | ||
Wang L, Wang J. MicroRNA-mediated breast cancer metastasis: from primary site to distant organs. Oncogene. 2012;31(20):2499–2511. | ||
Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17(3):313–319. | ||
Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008;10(2):R25. | ||
Nikitina EG, Urazova LN, Stegny VN. MicroRNAs and human cancer. Exp Oncol. 2012;34(1):2–8. | ||
Farazi TA, Spitzer JI, Morozov P, Tuschl T. miRNAs in human cancer. J Pathol. 2011;223(2):102–115. | ||
Hu Q, Gong JP, Li J, et al. Down-regulation of miRNA-452 is associated with adriamycin-resistance in breast cancer cells. Asian Pac J Cancer Prev. 2014;15(13):5137–5142. | ||
Ell B, Qiu Q, Wei Y, et al. The microRNA-23b/27b/24 cluster promotes breast cancer lung metastasis by targeting metastasis-suppressive gene prosaposin. J Biol Chem. 2014;289(32):21888–21895. | ||
Zhu J, Zheng Z, Wang J, et al. Different miRNA expression profiles between human breast cancer tumors and serum. Front Genet. 2014;5:149. | ||
Shimono Y, Zabala M, Cho RW, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009;138(3):592–603. | ||
Lugthart S, van Drunen E, van Norden Y, et al. High EVI1 levels predict adverse outcome in acute myeloid leukemia: prevalence of EVI1 overexpression and chromosome 3q26 abnormalities underestimated. Blood. 2008;111(8):4329–4337. | ||
Barjesteh van Waalwijk van Doorn-Khosrovani S, Erpelinck C, van Putten WL, et al. High EVI1 expression predicts poor survival in acute myeloid leukemia: a study of 319 de novo AML patients. Blood. 2003;101(3):837–845. | ||
Zhang G, Liu Z, Xu H, Yang Q. miR-409-3p suppresses breast cancer cell growth and invasion by targeting Akt1. Biochem Biophys Res Commun. 2016;469(2):189–195. | ||
Zhao W, Sun Q, Yu Z, et al. MiR-320a-3p/ELF3 axis regulates cell metastasis and invasion in non-small cell lung cancer via PI3K/Akt pathway. Gene. 2018;670:31–37. | ||
Chou NH, Lo YH, Wang KC, Kang CH, Tsai CY, Tsai KW. MiR-193a-5p and -3p Play a Distinct Role in Gastric Cancer: miR-193a-3p suppresses gastric cancer cell growth by targeting ETS1 and CCND1. Anticancer Res. 2018;38(6):3309–3318. | ||
Kim JS, Choi DW, Kim CS, et al. MicroRNA-203 induces apoptosis by targeting Bmi-1 in YD-38 oral cancer cells. Anticancer Res. 2018;38(6):3477–3485. | ||
Nakagawa R, Leyland R, Meyer-Hermann M, et al. MicroRNA-155 controls affinity-based selection by protecting c-MYC+ B cells from apoptosis. J Clin Invest. 2016;126(1):377–388. | ||
Zhang J, Liang J, Huang J. Downregulated microRNA-26a modulates prostate cancer cell proliferation and apoptosis by targeting COX-2. Oncol Lett. 2016;12(5):3397–3402. | ||
Li X, Wang X, Tan Z, Chen S, Guan F. Role of glycans in cancer cells undergoing epithelial-mesenchymal transition. Front Oncol. 2016;6:33. | ||
Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells – an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005;5(9):744–749. | ||
Siemens H, Jackstadt R, Hünten S, et al. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle. 2011;10(24):4256–4271. | ||
Santisteban M, Reiman JM, Asiedu MK, et al. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res. 2009;69(7):2887–2895. | ||
Tang J, Li Y, Wang J, Wen Z, Lai M, Zhang H. Molecular mechanisms of microRNAs in regulating epithelial-mesenchymal transitions in human cancers. Cancer Lett. 2016;371(2):301–313. | ||
Zhao N, Sun H, Sun B, et al. miR-27a-3p suppresses tumor metastasis and VM by down-regulating VE-cadherin expression and inhibiting EMT: an essential role for Twist-1 in HCC. Sci Rep. 2016;6:23091. | ||
Kim SH, Kim JM, Shin MH, et al. Correlation of epithelial-mesenchymal transition markers with clinicopathologic parameters in adenocarcinomas and squamous cell carcinoma of the lung. Histol Histopathol. 2012;27(5):581–591. | ||
Matsubara D, Kishaba Y, Yoshimoto T, et al. Immunohistochemical analysis of the expression of E-cadherin and ZEB1 in non-small cell lung cancer. Pathol Int. 2014;64(11):560–568. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.