")
Back to Journals » Drug Design, Development and Therapy » Volume 13
miR- 26a Sensitizes Melanoma Cells To Dabrafenib Via Targeting HMGB1-Dependent Autophagy Pathways
Authors Yu Y, Xiang N, Lin M , Huang JW, Zhang J, Cheng B, Ji C
Received 1 August 2019
Accepted for publication 2 October 2019
Published 29 October 2019 Volume 2019:13 Pages 3717—3726
DOI https://doi.org/10.2147/DDDT.S225671
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Yan Yu,1,* Niu Xiang,2,* Min Lin,2 Jin-Wen Huang,2 Jing Zhang,2 Bo Cheng,2 Chao Ji2
1Department of Dermatology, First Hospital of Jilin University, Changchun, Jilin 130021, People’s Republic of China; 2Department of Dermatology, The First Affiliated Hospital of Fujian Medical University, Fuzhou, Fujian 350005, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Chao Ji
Department of Dermatology, The First Affiliated Hospital of Fujian Medical University, Fuzhou, Fujian 350005, People’s Republic of China
Email [email protected]
Bo Cheng
Department of Dermatology, The First Affiliated Hospital of Fujian Medical University, Fuzhou, Fujian 350005, People’s Republic of China
Email [email protected]
Background: Melanoma is known as the most aggressive and lethal type of cutaneous cancer due to its rapid development of drug resistance to chemotherapy drugs.
Methods: In our study, we conducted a variety of studies, including quantitative PCR, Western blot, and autophagy and apoptosis assays to investigate the involvement of miR-26a and HMGB1 in modulation of dabrafenib sensitivity in human melanoma cell lines.
Results: Our studies revealed that the expressions of miR-26a and HMGB1 were altered in two melanoma cell lines after dabrafenib treatment. Additionally, dabrafenib caused autophagy in melanoma and this autophagic process was regulated by miR-26a via modifying HMGB1 expression. Furthermore, silencing HMGB1-inhibited autophagy induced by dabrafenib in melanoma cells. Last, we verified that treatment with a miR-26a mimic and HMGB1 shRNA could increase the efficacy of dabrafenib in melanoma cells.
Conclusion: Taken together, we showed that miR-26a is involved in the regulation of dabrafenib efficacy via a HMGB1-dependent autophagy pathway in melanoma cells. These results shed light on a novel treatment for conventional dabrafenib-based chemotherapy for melanoma.
Keywords: melanoma, miR-26a, HMGB1, dabrafenib, autophagy, apoptosis
Introduction
Melanoma is a type of cutaneous cancer which arises from melanocytes of the skin or mucous membranes.1 Although it accounts for a limited percentage of all skin cancers, melanoma has already become an indisputable crisis to human health worldwide due to its poor prognosis and high mortality rate. In the last two decades, many efforts have been put into the discovery of new anti-tumor drugs against melanoma.2–5 Some small-molecule-targeted therapies have been developed, such as dabrafenib.6,7 Dabrafenib, a BRAF inhibitors (BRAFi), was clinically approved by the FDA for the treatment of late-stage melanoma. It selectively causes cell death of melanoma cells bearing the V600-mutation and improves the overall survival rates of BRAF‐mutation melanoma patients.8 However, the majority of patients will develop drug resistance within several months after dabrafenib treatment where resistance mechanisms are not yet fully understood.9,10 Therefore, there is an urgent need for new therapies and medications that can target this drug resistance.
MicroRNAs (miRNAs), a class of small noncoding RNAs of 19–24 nucleotides in length, have been verified as post-transcriptional regulators of gene expression via binding to complementary sequences in the 3ʹ-untranslated region (3ʹUTR) of target mRNAs, leading to their degradation.11,12 Recent studies revealed that miRNAs play important roles in various diseases and cellular processes.13 For example, in many types of cancers, thousands of miRNAs have been verified as oncosuppressors and oncogenes, which are largely involved in cancer cell proliferation, apoptosis, invasion, and metastasis.14,15 One such miRNA is miR-26a. A large number of studies support that miR-26a is a pivotal regulator in tumor development and contributes to chemosensitivity via many target transcripts including PTEN, ULK1, NRAS, EZH2, GSK3β, SMAD1, and high mobility group box 1 (HMGB1).16–20 Among these target transcripts, HMGB1 became a focus of our interest. It is a highly conserved and ubiquitously expressed nuclear protein that functions as a regulator in DNA repair, inflammation, cell differentiation, cell migration, and invasion.21 Additionally, it has been reported that HMGB1 is a key regulator of autophagy and plays a critical role in chemotherapy resistance in many types of cancer cells.22 However, how it regulates the sensitivity of cells to dabrafenib in melanoma has yet to be established.
In this study, we sought to investigate the potential role of miR-26a in sensitizing melanoma cells to dabrafenib chemotherapy. We first tested the expression of miR-26a and HMGB1 in two melanoma cell lines after treatment with dabrafenib. Second, we explored whether dabrafenib could cause autophagy in melanoma and whether this autophagic process was regulated by miR-26a via modifying HMGB1 expression. Furthermore, we sought to test whether silencing HMGB1 could inhibit autophagy induced by dabrafenib in melanoma cells. Last, we verified that miR-26a and HMGB1 could increase the efficacy of dabrafenib in treating melanoma cells. Taken together, our study suggests that miR-26a is involved in the regulation of melanoma dabrafenib efficacy via a HMGB1-dependent autophagy pathway. These results shed light on a novel, dabrafenib-based chemotherapy for melanoma.
Materials And Methods
Cell Lines And Culture
The melanoma cell lines A375 and MEL624 were purchased from American Type Culture Collection (ATCC, Manassas, VA) and cultured in Dulbecco’s modified Eagle's medium (DMEM) (Invitrogen Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) (Biomeda, Foster City, CA, USA) and 1% penicillin/streptomycin/glutamine (Gibco/Invitrogen, Carlsbad, CA, USA). All cell lines were maintained in a humidified incubator with 5% CO2 at 37°C.
Antibodies And Reagents
Dabrafenib, 3-methyladenosine (3-MA) (#M9218), and chloroquine (CQ) (#C6628) were purchased from Sigma (St. Louis, MO, USA). The primary antibodies used in this study were rabbit anti-cleaved PARP (#5625, 1:1000, Cell Signaling Technology, Danvers, MA, USA), mouse anti-β-actin (#3700, 1:3000, Cell Signaling Technology, Danvers, MA, USA), mouse anti-LC3I/II (NB600-1384, 1:500, Novus, Littleton, CO, USA), mouse anti-p62 (SC-48389, 1:500, Santa Cruz, CA, USA), and rabbit anti-HMGB1 (ab79823, 1:200, Abcam, Cambridge, MA, USA). Species-specific secondary antibodies were obtained from LI-COR Biosciences.
Quantitative PCR
Total RNA was isolated by using Trizol (#15596026, Invitrogen Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions and cDNA was synthesized by using a High-Capacity cDNA Reverse Transcription Kit (Life Technologies/Applied Biosystem).
Ten ng cDNA was then conducted in qPCR using SYBR green kits (Thermo Fisher) on a 480 real-time PCR machine (Roche Applied Science, Penzberg, Germany). qPCR results were calculated using the ∆-Ct method and normalized to the GAPDH as housekeeping gene. The sequences of the forward and reverse primers used in qPCR were as follows: primers for HMGB1 (forward: 5ʹ-TATGGCAAAAGCGGACAAGG-3ʹ, reverse: 5ʹ-CTTCGCAACATCACCAATGGA-3ʹ) and GAPDH (forward: 5ʹ-ATCAGCAATGCCTCCTGCAC-3ʹ, reverse: 5ʹ-CGTCAAAGGTGGAGGAGTGG-3ʹ).
Autophagy Assays And Apoptosis Assay
GFP-tagged microtubule-associated protein 1 light chain 3 (GFP-MAP1LC3) was used to determine the degree of autophagy. Briefly, the melanoma cells were seeded into 6-well plates (2×105 cells/well) and transiently transfected with GFP-MAP1LC3 plasmid using Lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol. These cells were cultured for 24 hrs before different treatments and then fixed in 4% paraformaldehyde, and analyzed under Nikon PCM 2000 confocal microscope. Melanoma cells that undergo autophagic process were observed to have significant numbers of GFP-positive puncta. The degree of apoptosis in melanoma cells was measured by Western blot analysis of cleaved PARP.
Cell Transfection
Short hairpin RNA (shRNA) against HMGB1 (Sigma), non-target control (NTC) shRNA (Sigma), negative control miRNA (miR-NC) (Dharmacon), and miR-26a mimic (miR-26a) (Dharmacon) were transfected into melanoma cells using Lipofectamine 2000 Transfection Reagent in accordance with the manufacturer’s instructions. In all studies, cells were transfected 48 hrs before all treatments.
Western Blot
The methods have been described previously.23,24 In brief, after each treatment, the whole-cell lysates were prepared with ice-cold cell lysis buffer and cleared by centrifugation. The total protein concentration was measured with the bicinchoninic acid assay Kit (Bio-Rad Laboratories). Samples consisting of 40 μg of protein were resolved on a denaturing 8–12% SDS-PAGE gel (Bio-Rad) and then transferred to polyvinylidene fluoride membranes by electroblotting. The membrane was then blocked in PBST containing 5% dried milk at room temperature for 1 hr, incubated with primary antibodies at 4°C overnight. Blots were then incubated with appropriate secondary antibodies at room temperature for 1 hr the next day. The signals were detected by ECL reagents. β-Actin protein was used as an equivalent loading control.
Cell Viability Assay
This method has been described previously.23,24 In brief, cells were plated at a density of 5×104 cells/well in 96-well plates in 100 μL medium. After each treatment, cell viability was measured by the Cell Counting Kit-8 (Dojindo, Japan) test in accordance with the manufacturer’s instructions.
Statistical Analysis
All data represented in this study are the mean values ± SEM of at least three separate experiments. P values were calculated with the appropriate statistical tests using GraphPad Prism software 7.0. A significant difference was considered to be present at p<0.05.
Results
Dabrafenib Induces Autophagy Which Protects Cells From The Cytotoxicity Of Dabrafenib In Melanoma Cells
A growing interest exists in the protective role of autophagy during the chemotherapies in cancers. In our study, we first examined whether dabrafenib would trigger autophagy in melanoma cells. A375 and MEL624 cells transfected with a GFP-MAP1LC3 reporter plasmid were treated with dabrafenib (100nM) for 24 hrs. The GFP-LC3 puncta formation was then observed and assessed under confocal microscopy. Dense LC3-positive puncta were found when treated with dabrafenib (Figures 1A and B). In consistence with the results above, Western blot analysis revealed that autophagic signaling was significantly activated by dabrafenib in a dose-dependent manner (Figure 1C–E). To further prove the role of autophagy in sensitivity of melanoma cells to dabrafenib, we compared treatments with dabrafenib alone, co-treatment with dabrafenib and inhibitors of autophagy, 3-MA or CQ. We found inhibition of autophagy will enhance the cytotoxicity of dabrafenib in melanoma cells (Figure 1F and G).
miR-26a Is Downregulated While HMGB1 Is Upregulated When Treated With Dabrafenib In Melanoma Cells
miR-26a is known to be downregulated in some types of cancers including colorectal cancer, cutaneous squamous cell carcinoma, and retinoblastoma.25–29 We sought to determine the expression of miR-26a and HMGB1 in melanoma cells when treated with dabrafenib. A375 and MEL624 cells were treated with dabrafenib and the levels of miR-26a and HMGB1were assessed by qPCR. Consistent with the data above, HMGB1 expression was also found significantly increased in response to dabrafenib treatment in the protein level (Figure 4A and B). The results showed that miR-26a was significantly downregulated (Figure 2A and B) in melanoma cells while HMGB1 was upregulated (Figure 2C and D) when treated with dabrafenib.
miR-26a Attenuates Autophagy And Promotes Apoptosis Induced By Dabrafenib Via Targeting HMGB1
To confirm the inhibitory effect of miR-26a on autophagy in melanoma, cells were first transfected with control miRNA or a miR-26a mimic followed by the treatment with dabrafenib. Overexpression of miR-26a significantly diminished the GFP-LC3 puncta formation induced by dabrafenib treatment (Figure 3A–C). Meanwhile, we assessed the apoptosis levels in melanoma cells by measuring cleaved PARP levels via Western blot (Figure 3D and E). Cleaved PARP levels were robustly elevated in miR-26a-overexpressing cells. Moreover, protein levels of HMGB1 were found decreased in miR-26a-overexpressing cells correspondingly (Figure 3D). Taken together, these results demonstrate that miR-26a is a negative regulator of autophagy in dabrafenib-based chemotherapy in melanoma and this effect is possibly modulated by HMGB1.
HMGB1 Deficiency Impairs Autophagy And Augments Apoptosis Signaling In Melanoma Cells
To gain further insight into autophagy regulated by miR-26a in dabrafenib-based chemotherapy in melanoma, we performed a series of studies using shRNA-mediated HMGB1-deficient melanoma cells. We observed reduced levels of LC3II in response to dabrafenib in HMGB1shRNA-expressing cells relative to controls expressing NTC shRNA (Figure 5B–D). Correspondingly, GFP-LC3 puncta formation induced by dabrafenib treatment was also found attenuated by HMGB1 deficiency (Figure 5E–G). As anticipated, dabrafenib treatment elicited significantly higher levels of cleaved PARP in HMGB1-deficient cells relative to controls (Figure 4C and D). Therefore, we determined that HMGB1 is a key regulator of the miR-26a-mediated drug efficacy via autophagy pathways.
miR-26a Sensitizes Melanoma Cells To Dabrafenib While HMGB1 Attenuates Dabrafenib Cytotoxicity In Melanoma
Based on the results above, we would expect that miR-26a would augment dabrafenib cytotoxicity while HMGB1 has the opposite effect. Cells transfected with miR-NC, miR-26a mimic, NTC shRNA, or HMGB1 shRNA were treated with dabrafenib at 100nM for 24 hrs. We then assessed cell viability by CCK8 test and found that melanoma cells transfected with the miR-26a mimic and HMGB1 shRNA were more sensitive to the dabrafenib chemotherapy in comparison to control groups (Figure 6). These results indicate that miR26a possibly sensitizes dabrafenib-induced anti-tumor activity via HMGB1.
Discussion
Melanoma is known as the most aggressive and lethal type of cutaneous cancer. In the last two decades, a large number of chemotherapeutic drugs have been developed including BRAF inhibitors such as dabrafenib. However, melanoma of advanced stages still has poor prognosis and high mortality rates due to adaptive or acquired drug resistance. Our study demonstrated the underlying mechanisms of miR-26a-mediated chemo-resistance regulation to dabrafenib via a HMGB1-dependent autophagy pathway for the first time.
Recent interest has focused on understanding the molecular basis of drug resistance to BRAFi in melanoma cells. It is a complex process that involves a large amount of genes and the pathways. Our group has recently focused on the roles of miRNAs in melanoma drug resistance as they are major post-transcriptional regulators and their dysregulation is observed in a wide range of human diseases, cancers in particular. miR-26a is one such example. miR-26a was found downregulated in a variety of cancers including hepatocellular carcinoma (HCC), breast cancer, anaplastic thyroid cancer, nasopharyngeal cancer, colon cancer, and melanoma.30–35 In addition, miR-26a was also implicated in chemotherapy resistance in HCC, breast cancer, lung cancer, and gastric cancer through different target transcripts.16,36–38 In the present study, we searched and tried several putative targets of miR-26a that are previously documented in other studies including EZH2, PTEN (data not shown), and HMGB1.18,39,40 miR-26a was found to inhibit HMGB1 expression and protect cardiomyocytes against ischemia-reperfusion injury and inflammatory injury.41,42 In our current study, we found that miR-26a was downregulated while HMGB1 was upregulated post-dabrafenib treatment in two melanoma cell lines.
HMGB1 is well established as a key regulator of autophagy and apoptosis. Autophagy is a cellular process of self-digestion of cytoplasmic components through which cells can maintain cellular homeostasis.43 Recent studies have revealed that autophagy is also an adaptive response to certain stresses including chemotherapies and radiation therapies.44 However, the exact roles of miR-26a/HMGB1 and autophagy in determining the sensitivity of melanoma cells to dabrafenib remain enigmatic. Our results demonstrated that dabrafenib caused autophagy in melanoma and this autophagic process was regulated by miR-26a via modifying HMGB1 expression. We further determined that silencing HMGB1 inhibited dabrafenib-induced autophagy in melanoma cells. We also verified that treatment with a miR-26a mimic or HMGB1 shRNA could increase the efficacy of dabrafenib treatment in melanoma cells.
In sum, our studies identified that miR-26a is involved in the regulation of dabrafenib sensitivity in melanoma via a HMGB1-dependent autophagy pathway. These findings may provide new insight into how autophagy regulates the BRAFi resistance in melanoma cells. Therefore, miR-26a/HMGB1 might be a promising therapeutic target for dabrafenib-based melanoma therapy.
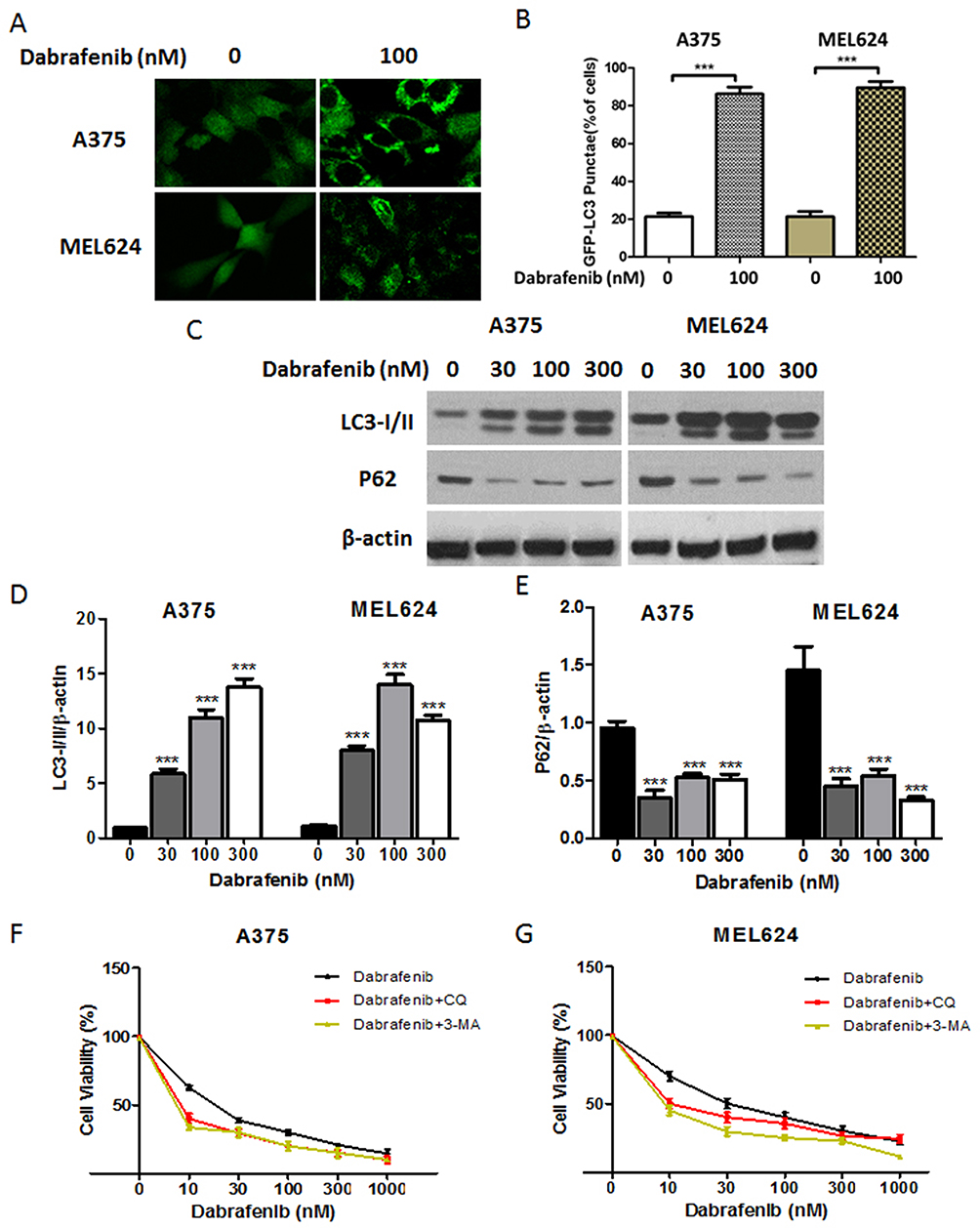 |
Figure 1 Dabrafenib triggers autophagy in melanoma cells. (A) A375 and MEL624 cells transfected with a GFP-MAP1LC3 reporter plasmid were treated with dabrafenib (100 nM) for 24 hrs. Confocal images showing GFP-LC3 puncta formation induced by dabrafenib treatment. (B) The percentage of cells showing accumulation of GFP-LC3 puncta. (C) Western blot analysis of LC3-I/II, p62, and β-actin proteins after dabrafenib treatment at the indicated concentrations in A375 (left panel) and MEL624 (right panel) cells. (D) LC3-I/II and (E) p62 levels were quantified. (F) A375 and (G) MEL624 cells were treated with dabrafenib at the indicated concentrations for 24 hrs with or without 3-MA (2 mM) or CQ (2.5 µM). Inhibition of autophagy sensitizes melanoma cells to dabrafenib. ***p < 0.001 compared with control group. |
 |
Figure 2 Expression of miR-26a and HMGB1 in melanoma cells. A375 (left panel) and MEL624 (right panel) cells were treated with dabrafenib (100 nM) for 24 hrs. miR-26a levels were then assayed by reverse transcription-quantitative polymerase chain reaction (qRT-PCR) in (A) A375 and (B) MEL624 cells. HMGB1 mRNA levels were also detected in (C) A375 and (D) MEL624 cells by qRT-PCR. miR-26a was downregulated in melanoma cell lines while HMGB1 expression was upregulated when treated with dabrafenib. ***p < 0.001 compared with control group. |
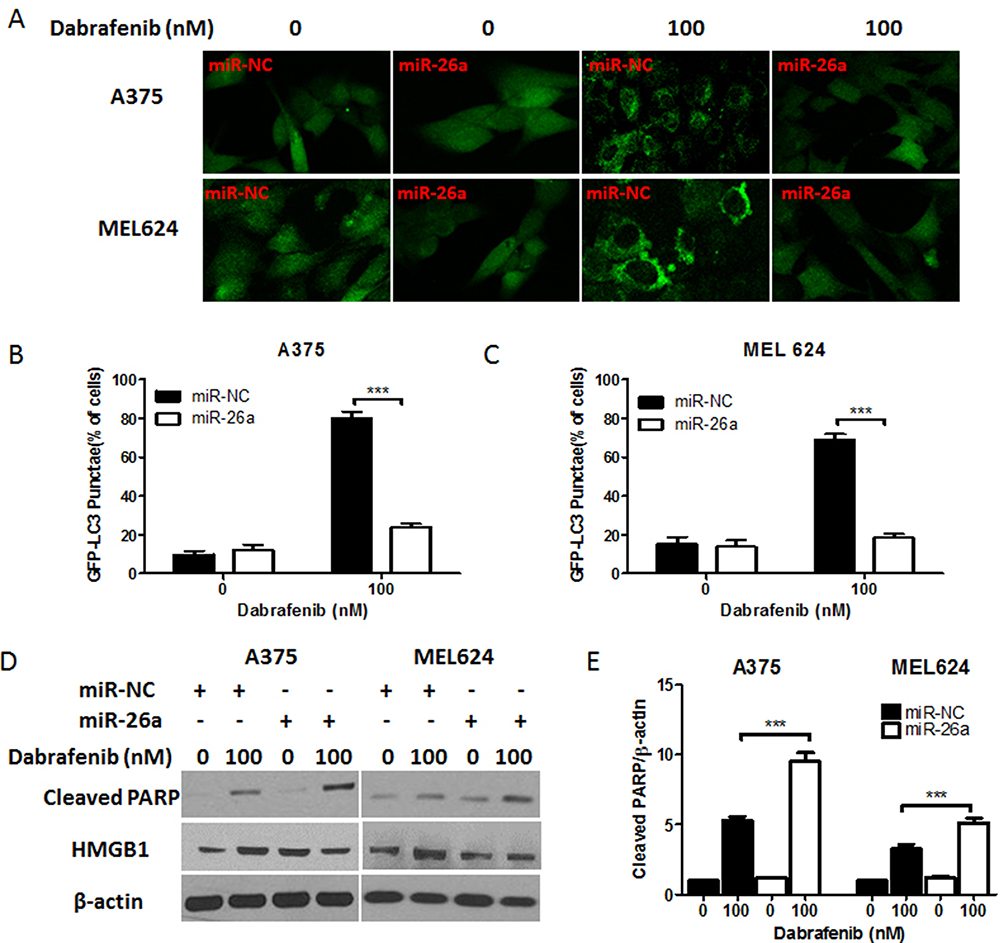 |
Figure 3 miR-26a inhibits autophagy and induces apoptosis in melanoma cells in response to dabrafenib via a HMGB1-dependent pathway. Melanoma cells were first transfected with a GFP-MAP1LC3 reporter plasmid and control miRNA or a miR-26a mimic. These cells were then treated with dabrafenib (100 nM) for 24 hrs. (A) Confocal images showing GFP-LC3 puncta formation induced by dabrafenib treatment. The percentage of cells showing accumulation of GFP-LC3 puncta in (B) A375 and (C) MEL624 cells. (D) Western blot analysis of cleaved PARP, HMGB1, and β-actin proteins after dabrafenib treatment in A375 (left panel) and MEL624 (right panel) cells. Apoptosis levels in melanoma cells were analyzed by measuring cleaved PARP levels via Western blot. Cleaved PARP levels (E) were elevated while HMGB1 expression was decreased in miR-26a-overexpressing cells. ***p < 0.001 compared with control group. |
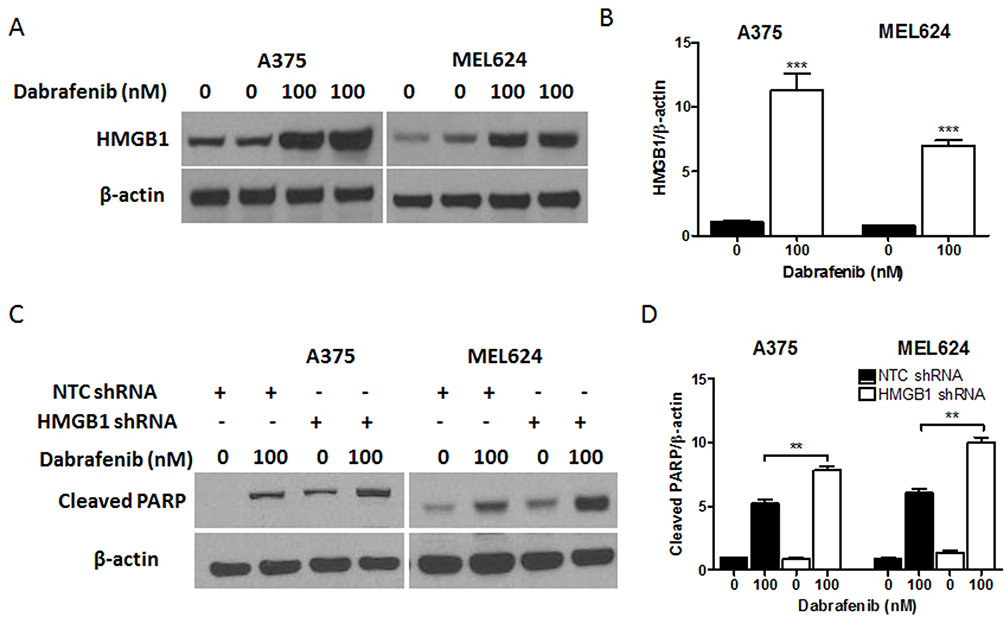 |
Figure 4 HMGB1 deficiency promotes apoptosis signaling in melanoma cells in response to dabrafenib. Melanoma cells were first transfected with NTC shRNA or HMGB1 shRNA. These cells were then treated with dabrafenib (100 nM) for 24 hrs. (A) HMGB1 levels were detected by Western blot and (C) apoptosis was assayed by measuring cleaved PARP. (B) HMGB1 and (D) cleaved PARP levels were quantified. **p < 0.01, ***p < 0.001 compared to control group. |
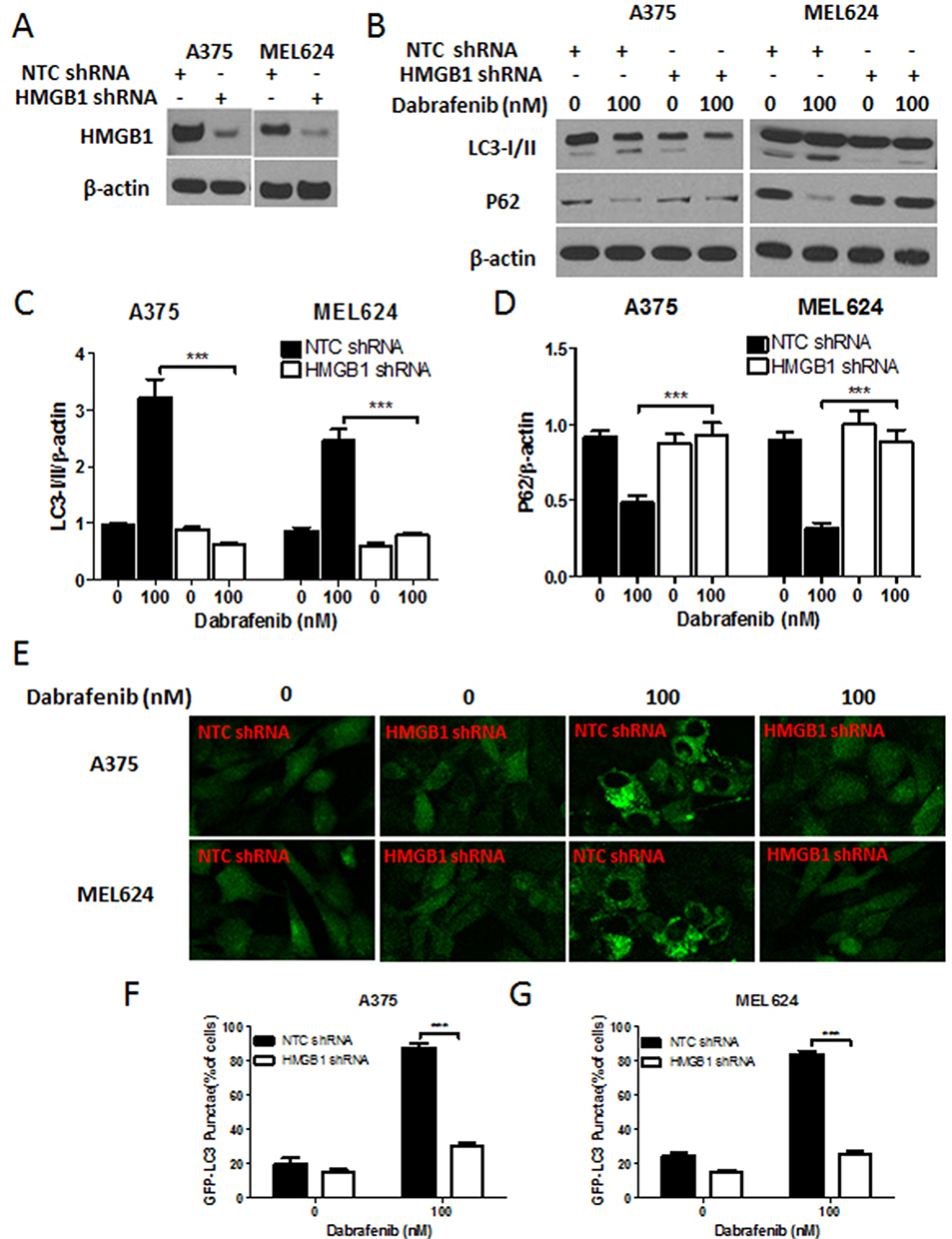 |
Figure 5 HMGB1 deficiency inhibits autophagy induced by dabrafenib in melanoma cells. Melanoma cells were first transfected with NTC shRNA or HMGB1 shRNA. (A) Verification of shRNA-mediated knockdown of HMGB1 by Western blot. (B) Western blot analysis of LC3-I/II, p62, and β-actin proteins after dabrafenib treatment (100nM) for 24 hrs in A375 (left panel) and MEL624 (right panel) cells. (C) LC3-I/II and (D) p62 levels were quantified. (E) Confocal images showing GFP-LC3 puncta formation induced by dabrafenib treatment. The percentage of cells showing accumulation of GFP-LC3 puncta in (F) A375 and (G) MEL624 cells. Results showed that dabrafenib-induced autophagy signaling was diminished in HMGB1-deficient melanoma cells. ***p < 0.001 compared to control group. |
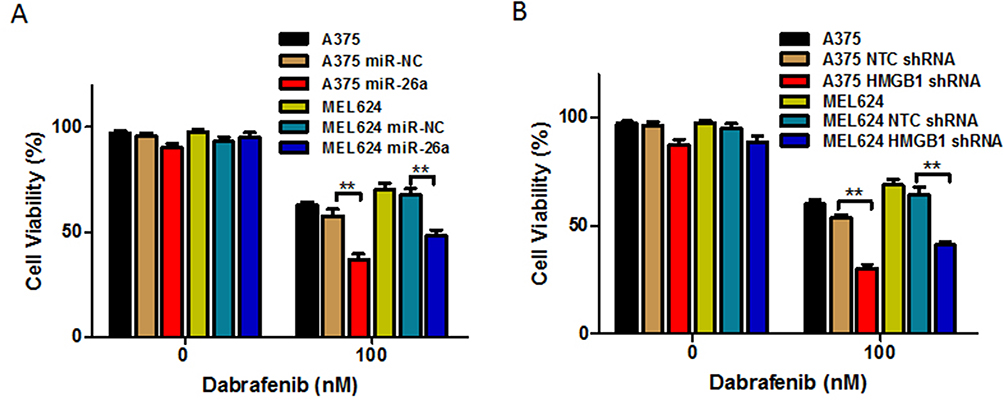 |
Figure 6 Overexpression of miR-26a and HMGB1 deficiency enhances the chemotherapeutic efficacy of dabrafenib in melanoma. A375 and MEL624 cells were transfected with miR-NC, miR-26a mimic, NTC shRNA, or HMGB1 shRNA, respectively. Chemotherapeutic efficacy of dabrafenib in melanoma cells regulated by (A) miR-26a and (B) HMGB1 were measured by CCK-8 test. **p < 0.01 compared with control group. |
Acknowledgments
This research was supported by grants from the National Natural Science Foundation of China (No. 81673066 and No. 81473684), the Natural Science Foundation of Fujian Province (No. 2016J01534), and Joint Funds for the Innovation of Science and Technology of Fujian Province (No. 2016Y91020018).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tsao H, Chin L, Garraway LA, Fisher DE. Melanoma: from mutations to medicine. Genes Dev. 2012;26:1131–1155. doi:10.1101/gad.191999.112
2. Ascierto PA, Marincola FM. 2015: the year of anti-PD-1/PD-L1s against melanoma and beyond. EBioMedicine. 2015;2:92–93. doi:10.1016/j.ebiom.2015.01.011
3. Menzies AM, Long GV. Systemic treatment for BRAF-mutant melanoma: where do we go next? Lancet Oncol. 2014;15:e371–e381. doi:10.1016/S1470-2045(14)70072-5
4. Simeone E, Grimaldi AM, Ascierto PA. Anti-PD1 and anti-PD-L1 in the treatment of metastatic melanoma. Melanoma Manag. 2015;2:41–50. doi:10.2217/mmt.14.30
5. Korn EL, Liu P-Y, Lee SJ, et al. Meta-analysis of phase II cooperative group trials in metastatic stage IV melanoma to determine progression-free and overall survival benchmarks for future phase II trials. J Clin Oncol. 2008;26:527–534. doi:10.1200/JCO.2007.12.7837
6. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi:10.1056/NEJMoa1002011
7. Zhang C, Spevak W, Zhang Y, et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature. 2015;526:583–586. doi:10.1038/nature14982
8. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi:10.1056/NEJMoa1103782
9. Lo RS. Combinatorial therapies to overcome B-RAF inhibitor resistance in melanomas. Pharmacogenomics. 2012;13:125–128. doi:10.2217/pgs.11.166
10. Corcoran RB, Settleman J, Engelman JA. Potential therapeutic strategies to overcome acquired resistance to BRAF or MEK inhibitors in BRAF mutant cancers. Oncotarget. 2011;2:336–346. doi:10.18632/oncotarget.262
11. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi:10.1038/nrc1997
12. Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med. 2014;20:460–469. doi:10.1016/j.molmed.2014.06.005
13. Jansson MD, Lund AH. MicroRNA and cancer. Mol Oncol. 2012;6:590–610. doi:10.1016/j.molonc.2012.09.006
14. Calin GA, Sevignani C, Dumitru CD, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999–3004. doi:10.1073/pnas.0307323101
15. Acunzo M, Romano G, Wernicke D, Croce CM. MicroRNA and cancer – a brief overview. Adv Biol Regul. 2015;57:1–9. doi:10.1016/j.jbior.2014.09.013
16. Jin F, Wang Y, Li M, et al. MiR-26 enhances chemosensitivity and promotes apoptosis of hepatocellular carcinoma cells through inhibiting autophagy. Cell Death Dis. 2017;8:e2540. doi:10.1038/cddis.2016.461
17. Lin G, Liu B, Meng Z, et al. MiR-26a enhances invasive capacity by suppressing GSK3β in human lung cancer cells. Exp Cell Res. 2017;352:364–374. doi:10.1016/j.yexcr.2017.02.033
18. Zhao WT, Lin X-L, Liu Y, et al. miR-26a promotes hepatocellular carcinoma invasion and metastasis by inhibiting PTEN and inhibits cell growth by repressing EZH2. Lab Invest. 2019;99:1484–1500. doi:10.1038/s41374-019-0270-5
19. Coronel-Hernández J, López-Urrutia E, Contreras-Romero C, et al. Cell migration and proliferation are regulated by miR-26a in colorectal cancer via the PTEN-AKT axis. Cancer Cell Int. 2019;19:80. doi:10.1186/s12935-019-0802-5
20. Luzi E, Marini F, Sala SC, Tognarini I, Galli G, Brandi ML. Osteogenic differentiation of human adipose tissue-derived stem cells is modulated by the miR-26a targeting of the SMAD1 transcription factor. J Bone Miner Res. 2008;23:287–295. doi:10.1359/jbmr.071011
21. Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249:158–175. doi:10.1111/j.1600-065X.2012.01146.x
22. Amaravadi RK, Lippincott-Schwartz J, Yin X-M, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17:654–666. doi:10.1158/1078-0432.CCR-10-2634
23. Gong T, Yu Y, Yang B, et al. Celecoxib suppresses cutaneous squamous-cell carcinoma cell migration via inhibition of SDF1-induced endocytosis of CXCR4. Onco Targets Ther. 2018;11:8063–8071. doi:10.2147/OTT.S180472
24. Ji C, Yang B, Huang SY, Huang JW, Cheng B. Salubrinal protects human skin fibroblasts against UVB-induced cell death by blocking endoplasmic reticulum (ER) stress and regulating calcium homeostasis. Biochem Biophys Res Commun. 2017;493:1371–1376. doi:10.1016/j.bbrc.2017.10.012
25. Ghanbari R, Mosakhani N, Asadi J, et al. Downregulation of plasma MiR-142-3p and MiR-26a-5p in patients with colorectal carcinoma. Iran J Cancer Prev. 2015;8:e2329. doi:10.17795/ijcp2329
26. Song Q, Xu K. [MicroRNA-26a and tumor]. Zhongguo Fei Ai Za Zhi. 2017;20:769–774. doi:10.3779/j.issn.1009-3419.2017.11.08
27. Lopez-Urrutia E, Coronel-Hernández J, García-Castillo V, et al. MiR-26a downregulates retinoblastoma in colorectal cancer. Tumour Biol. 2017;39:1010428317695945. doi:10.1177/1010428317695945
28. Wu Z, Lu B, Li X, et al. MicroRNA-26a inhibits proliferation and tumorigenesis via targeting CKS2 in laryngeal squamous cell carcinoma. Clin Exp Pharmacol Physiol. 2018;45:444–451. doi:10.1111/1440-1681.12890
29. Konishi H, Fujiya M, Ueno N, et al. microRNA-26a and -584 inhibit the colorectal cancer progression through inhibition of the binding of hnRNP A1-CDK6 mRNA. Biochem Biophys Res Commun. 2015;467:847–852. doi:10.1016/j.bbrc.2015.10.055
30. Kota J, Chivukula RR, O’Donnell KA, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. doi:10.1016/j.cell.2009.04.021
31. Clague J, Lippman SM, Yang H, et al. Genetic variation in MicroRNA genes and risk of oral premalignant lesions. Mol Carcinog. 2010;49:183–189. doi:10.1002/mc.20588
32. Boni V, Zarate R, Villa JC, et al. Role of primary miRNA polymorphic variants in metastatic colon cancer patients treated with 5-fluorouracil and irinotecan. Pharmacogenomics J. 2011;11:429–436. doi:10.1038/tpj.2010.58
33. Lu J, He M-L, Wang L, et al. MiR-26a inhibits cell growth and tumorigenesis of nasopharyngeal carcinoma through repression of EZH2. Cancer Res. 2011;71:225–233. doi:10.1158/0008-5472.CAN-10-1850
34. Zhang B, Liu -X-X, He J-R, et al. Pathologically decreased miR-26a antagonizes apoptosis and facilitates carcinogenesis by targeting MTDH and EZH2 in breast cancer. Carcinogenesis. 2011;32:2–9. doi:10.1093/carcin/bgq209
35. Reuland SN, Smith SM, Bemis LT, et al. MicroRNA-26a is strongly downregulated in melanoma and induces cell death through repression of silencer of death domains (SODD). J Invest Dermatol. 2013;133:1286–1293. doi:10.1038/jid.2012.400
36. Tormo E, Adam-Artigues A, Ballester S, et al. The role of miR-26a and miR-30b in HER2+ breast cancer trastuzumab resistance and regulation of the CCNE2 gene. Sci Rep. 2017;7:41309. doi:10.1038/srep41309
37. Xu S, Wang T, Yang Z, et al. miR-26a desensitizes non-small cell lung cancer cells to tyrosine kinase inhibitors by targeting PTPN13. Oncotarget. 2016;7:45687–45701. doi:10.18632/oncotarget.9920
38. Wen L, Cheng F, Zhou Y, Yin C. MiR-26a enhances the sensitivity of gastric cancer cells to cisplatin by targeting NRAS and E2F2. Saudi J Gastroenterol. 2015;21:313–319. doi:10.4103/1319-3767.166206
39. Zhang M, Duan W, Sun W. LncRNA SNHG6 promotes the migration, invasion, and epithelial–mesenchymal transition of colorectal cancer cells by miR-26a/EZH2 axis. Onco Targets Ther. 2019;12:3349–3360. doi:10.2147/OTT.S197433
40. Zhang T, Qian H, Hu C, et al. MiR-26a mediates ultraviolet B-induced apoptosis by targeting histone methyltransferase EZH2 depending on Myc expression. Cell Physiol Biochem. 2017;43:1188–1197. doi:10.1159/000481759
41. Yao L, Lv X, Wang X. MicroRNA 26a inhibits HMGB1 expression and attenuates cardiac ischemia-reperfusion injury. J Pharmacol Sci. 2016;131:6–12. doi:10.1016/j.jphs.2015.07.023
42. Jia P, Wu N, Jia D, Sun Y. Downregulation of MALAT1 alleviates saturated fatty acid-induced myocardial inflammatory injury via the miR-26a/HMGB1/TLR4/NF-κB axis. Diabetes Metab Syndr Obes. 2019;12:655–665. doi:10.2147/DMSO.S203151
43. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi:10.1038/nature06639
44. Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi:10.1016/j.molcel.2010.09.023
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.