")
Back to Journals » OncoTargets and Therapy » Volume 11
Mifepristone inhibits proliferation, migration and invasion of HUUA cells and promotes its apoptosis by regulation of FAK and PI3K/AKT signaling pathway
Authors Sang L, Lu D, Zhang J, Du S, Zhao X
Received 2 April 2018
Accepted for publication 4 July 2018
Published 4 September 2018 Volume 2018:11 Pages 5441—5449
DOI https://doi.org/10.2147/OTT.S169947
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr XuYu Yang
Lin Sang,1 Dawei Lu,1 Jun Zhang,2 Shihua Du,1 Xingbo Zhao3
1Department of Obstetrics and Gynecology, The Second People’s Hospital of Hefei City Affiliated to Anhui Medical University, Hefei City, Anhui Province, People’s Republic of China; 2Department of Obstetrics, Tai’an City Central Hospital, Tai’an City, Shandong Province, People’s Republic of China; 3Department of Obstetrics and Gynecology, Shandong Provincial Hospital Affiliated to Shandong University, Ji’nan City, Shandong Province, People’s Republic of China
Purpose: The aim was to investigate mifepristone effects on endometrial carcinoma and the related mechanism.
Methods: HHUA cells were treated with DMEM containing different concentrations of mifepristone. HHUA cells treated with 100 µmol/L mifepristone were named the Mifepristone group. HHUA cells co-transfected with pcDNA3.1-PI3K and pcDNA3.1-AKT overexpression vectors were treated with 100 µmol/L mifepristone and named the Mifepristone + PI3K/AKT group. mRNA expression was detected by quantitative reverse transcription PCR. Protein expression was performed by Western blot. Cell proliferation was conducted by MTT assay. Wound-healing assay was conducted. Transwell was used to detect cells migration and invasion. Apoptosis detection was performed by flow cytometry.
Results: Mifepristone inhibited HHUA cells proliferation in a dose-dependent manner. Compared with HHUA cells treated with 0 µmol/L mifepristone, HHUA cells treated by 50–100 µmol/L mifepristone had a lower wound-healing rate, a greater number of migrating and invasive cells (P<0.01), as well as a higher percentage of apoptotic cells and Caspase-3 expression (P<0.01). When HHUA cells were treated with 50–100 µmol/L of mifepristone, FAK, p-FAK, p-PI3K and p-AKT relative expression was all significantly lower than HHUA cells treated with 0 µmol/L of mifepristone (P<0.01). Compared with the Mifepristone group, HHUA cells of the Mifepristone + PI3K/AKT group had a lower cell growth inhibition rate and percentage of apoptotic cells (P<0.01).
Conclusion: Mifepristone inhibited HUUA cells proliferation, migration and invasion and promoted its apoptosis by regulation of FAK and PI3K/AKT signaling pathway.
Keywords: Mifepristone, HHUA cells, proliferation, FAK, PI3K/AKT signaling pathway
Introduction
Endometrial carcinoma is one of the most common gynecological malignancies that often occurs in perimenopausal women and postmenopausal women.1 The mortality caused by endometrial carcinoma is very high all over the world.2 The incidence of endometrial carcinoma is reported to be closely related to life habits and regions.3,4 And unfortunately, the incidence of endometrial carcinoma has also increased year by year.5,6 Understanding of the occurrence and development mechanism of endometrial carcinoma can fundamentally improve the prevention, diagnosis and treatment of endometrial carcinoma.
Mifepristone, a common progesterone receptor anti-caking agent, is widely used as an anti-pregnancy drug and also can be used during gynecological surgery operations.7,8 In recent studies, researchers have found that mifepristone also has inhibitory effects on a variety of tumor cells, such as breast cancer and prostate cancer, especially for inhibiting gynecological-related tumor cells.9–11 It could effectively inhibit the progesterone receptor contained in uterine fibroids and reduce the size of the uterine fibroids.12 Research studies have also found that mifepristone at doses of 5 mg and 10 mg daily could effectively maintain uterine fibroids treatment for 3–12 months.13,14 One explanation was that mifepristone could reduce progesterone levels as well as progesterone levels around the tumor. Therefore, it could finally achieve the effect of inhibiting the growth of uterine fibroids.15,16 Other explanations were that mifepristone could block ceramide glycosylation and promote cell apoptosis, reduce exocytosis of MDR-associated proteins and P-glycoprotein, enhance DNA repair capacity and regulate tumor suppressor genes expression.11
There were also articles which reported the effect of mifepristone on endometrial carcinoma, while the exact mechanism has not yet been determined. In this research, we explored the effect of mifepristone on endometrial carcinoma cells proliferation, migration, invasion and apoptosis. The relevant mechanism has also been further studied. We hope this study could provide guidance for the clinical treatment of endometrial carcinoma by mifepristone.
Material and methods
Cell culture and treatment
HHUA cells were purchased from China Type Culture Collection Center. They were maintained in DMEM containing 10% FBS in a 5% CO2, 37°C incubator. These HHUA cells at logarithmic growth phase were collected and suspended in DMEM medium (10% FBS) containing 0 μmol/L, 25 μmol/L, 50 μmol/L, 75 μmol/L, and 100 μmol/L mifepristone, respectively. Then they were inoculated in 24-well plates at a density of 1×105 cells per well. All plates were placed in the 5% CO2, 37°C incubator for continued incubation.
Cells transfection and grouping
pcDNA3.1-PI3K and pcDNA3.1-AKT overexpression vectors were constructed to co-transfect normal HHUA cells. These cells were treated with 100 μmol/L mifepristone and were set as the Mifepristone + PI3K/AKT group. In addition, for HHUA cells only treated with 100 μmol/L mifepristone, they were known as the Mifepristone group. Cells of these two groups were inoculated in 24-well plates at a density of 1×105 cells per well. They were incubated in the incubator at 37°C, 5% CO2.
Quantitative reverse transcription PCR
HHUA cells treated with different conditions were collected after being cultured for 48 hours. Total RNA was obtained by using a Trizol reagent kit (Thermo Fisher Scientific, Waltham, MA, USA). PCR amplification reaction was performed in a 20 μL reaction system, including 1 μL of cDNA, 1 μL of forward primer and 1 μL of reverse primer in the system. The sequences of primers used in this research were as follows: FAK, forward, GAGCGTCTAATCCGACAGCAACAG, reverse, GCCCGTCACATTCTCGTACACCT; Caspase-3, forward, GTGGAATTGATGCGTGATG, reverse, GGAATCTGTTTCTTTGCATG; PI3K, forward, GGACCCGATGCGGTTAGA, reverse, GATGATGGTCGTGGAGGC; AKT, forward, ATGGCACCTTCATTGGCTAC, reverse, GGGCCGGACTCGTCATAC; GAPDH, forward, GTCGATGGCTAGTCGTAGCATCGAT, reverse, TGCTAGCTGGCATGCCCGATCGATC. The cycling conditions were as follows: For FAK: 38 cycles of 95°C for 10 seconds, 58°C for 20 seconds and 72°C for 34 seconds. For Caspase-3: 50 cycles of 95°C for 10 seconds, 58°C for 20 seconds and 72°C for 34 seconds. For PI3K: 40 cycles of 95°C for 10 seconds, 58°C for 20 seconds and 72°C for 34 seconds. For AKT: 40 cycles of 95°C for 10 seconds, 58°C for 20 seconds and 72°C for 34 seconds. For GAPDH: 42 cycles of 95°C for 10 seconds, 58°C for 20 seconds and 72°C for 34 seconds. The relative gene expression was calculated by the comparative Ct formula 2−ΔΔCt.
Western blot
After 48 hours of incubation, HHUA cells were collected and lysed in RIPA buffer (Beyotime, Beijing, China) to extract total protein. A Pierce BCA kit (Thermo Fisher Scientific, Waltham, MA, USA) was used to determine protein concentration. Then equal cell lysate (30 μg) from each sample was separated by 10% SDS-PAGE gel. Protein was then transferred onto PVDF membrane. The membrane was blocked with 5% fat-free milk in TBS-T for 2 hours at room temperature. Primary antibody (mouse anti-human FAK and p-FAK antibody, mouse anti-rabbit Caspase-3, PI3K, AKT, p-PI3K and p-AKT antibody, 1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA, USA) was added for 12 hours incubation at 4°C. Secondary antibody (Beijing Zhongshan Jinqiao Biotechnology Co., Ltd., China, 1:5,000) was also added for an extra 1 hour of incubation. After the membrane was washed three times with TBST, the enhanced chemiluminescence kit was used to identify the reactive bands.
MTT assay
In this study, proliferative capacity of HHUA cells was determined using MTT assay. Cells were cultured for 24 hours, 48 hours, 72 hours, and 96 hours, respectively, in 24-well plates. A total of 20 μL MTT solution (5 mg/mL) was added into each well for 4 hours incubation at 37°C. The residual liquid in each well was discarded. DMSO at a dose of 150 μL was added into each well. Ten min shaking was performed to promote the complete dissolution of purple formazan crystals. At last, these 24-well plates were subjected to absorbance measurements by ELISA at 495 nm wavelength (OD495 value). The cell growth inhibition rate was calculated according to the following formula: cell growth inhibition rate=−(OD495 value of treatment group/OD495 value of control group)×100%. In this research, HHUA cells treated with 0 μmol/L mifepristone were used as a control group.
Wound-healing assay
HHUA cells were prepared as cell suspensions at a density of 2×104 cells/mL. Six-well plates with 1 mL cell suspensions per well were incubated in the incubator for 12 hours at 37°C, 5% CO2. A scratch was made by using a 100 μL sterile pipette tip. Then the residual liquid in each well was removed and 1 mL DMEM (10% FBS) was added. Six-well plates were placed under an inverted microscope and photographed. All 6-well plates were returned to the incubator for 48 hours incubation at 37°C, 5% CO2. Afterward, they were placed under the inverted microscope again for observation and photographing. The wound-healing rate was calculated according to the following formula: (initial scratch width− scratch width at 48 hours)×100%/initial scratch width.
Transwell assay
Cells were harvested and prepared as serum-free cell suspensions after 48 hours incubation in 24-well plates. They were seeded onto the upper chamber of Transwell at a density of 2×104 cells per well. DMEM containing 10% FBS was then added into the lower chamber of Transwell. After 24 hours incubation in the 5% CO2, 37°C incubator, the penetrated cells were fixed with formaldehyde for 5 minutes. Crystal violet was used to dye for 10 minutes. Cells were placed under an inverted microscope. Five fields were randomly selected to count the number of cells that passed through the membrane. The number of cells that passed through the membrane was defined as migrating cell numbers.
Furthermore, invasion ability was detected using the same operating procedure. However, the membrane on the upper chamber was precoated with Matrigel. Also, the number of cells that passed through the membrane was considered as the number of invading cells.
Apoptosis detection by flow cytometry
Cells were collected after 48 hours incubation and fixed with pre-cooled 70% ethanol for 2 hours at 4°C. Then 0.2 mL of PI solution (50 mg/mL) was added for 30 minutes incubation at 4°C in darkness. Flow cytometry was used to detect apoptosis and apoptosis percentage was also analyzed.
Statistical analysis
All data were expressed as mean±SD. Student t-test was used for the comparison between two groups. Comparison among three or more groups was detected by one-way ANOVA test. SPSS 17.0 and GraphPad Prism 5.0 were used for statistical analysis of data. P<0.05 was considered statistically significant. In this research, all experiments were repeated three times.
Results
Mifepristone inhibited HHUA cells proliferation in a dose-dependent manner
This study examined the effects of different concentrations of mifepristone on HHUA cells proliferation by MTT assay. The results demonstrated that mifepristone inhibited HHUA cells proliferation in a dose-dependent manner. The cell growth inhibition rate was gradually increased with the increase of mifepristone concentration. When the concentration of mifepristone was at 100 μmol/L, cell growth inhibition rate was the highest (Figure 1).
 | Figure 1 Mifepristone inhibited HHUA cells proliferation in a dose-dependent manner. |
Mifepristone inhibited HHUA cells migration and invasion in a dose-dependent manner
Cell migration ability was detected by wound-healing assay and Transwell assay, respectively. According to the results, we noted that, for HHUA cells treated with 50–100 μmol/L mifepristone, their wound-healing rate and number of migrating cells was both markedly decreased when compared with HHUA cells treated with 0 μmol/L mifepristone (P<0.01) (Figure 2A and B). We also noticed that, compared with HUA cells treated with 0 μmol/L mifepristone, those treated with 50–100 μmol/L mifepristone had dramatically lower invasive cell numbers (P<0.01) (Figure 2C).
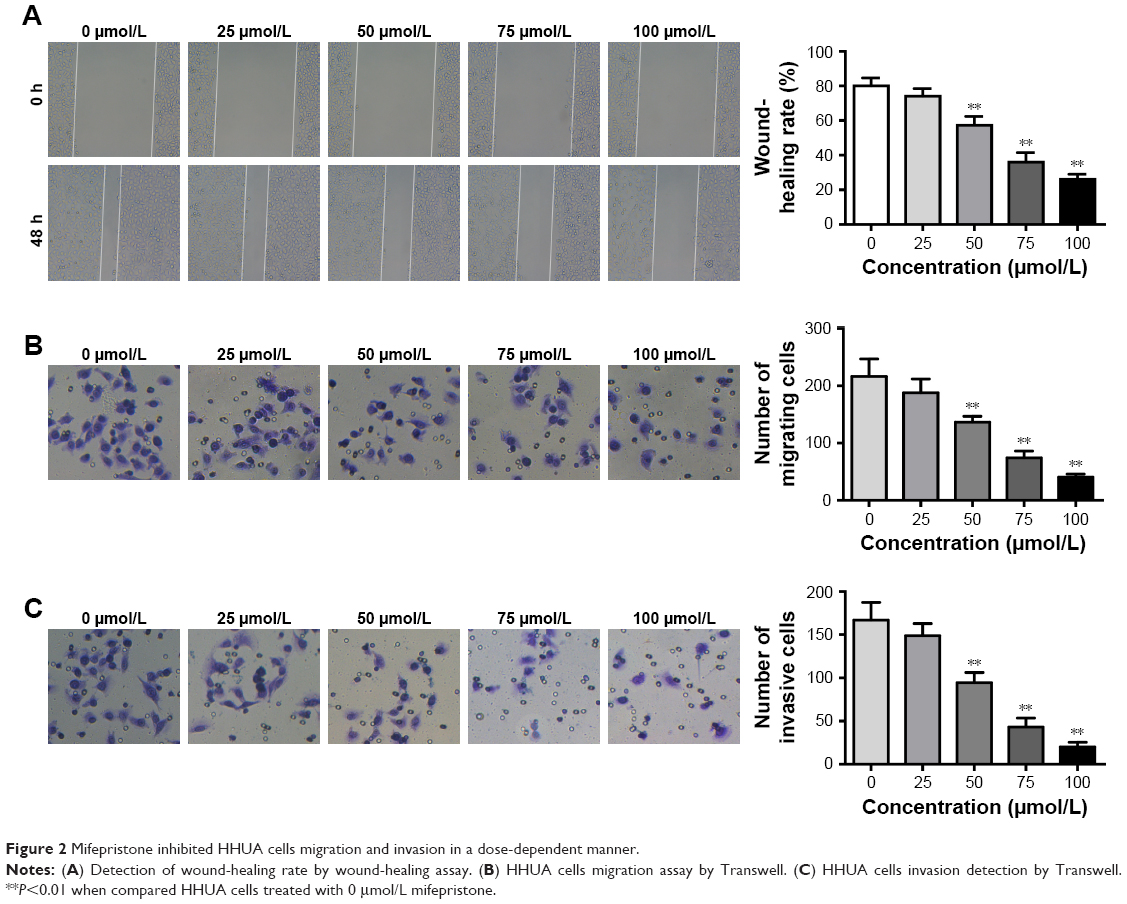 | Figure 2 Mifepristone inhibited HHUA cells migration and invasion in a dose-dependent manner. |
Mifepristone promoted HHUA cells apoptosis in a dose-dependent manner
Mifepristone promoted apoptosis of HHUA cells in a dose-dependent manner. Compared with the percentage of apoptotic HHUA cells treated with 0 μmol/L mifepristone, significantly increased apoptotic cells percentage was found when they were treated with 25–100 μmol/L mifepristone (P<0.01) (Figure 3A). Meanwhile, we also found that mifepristone could promote Caspase-3 expression in a dose-dependent manner. Caspase-3 mRNA and protein were both significantly increased after they were treated by 50–100 μmol/L of mifepristone (P<0.01) (Figure 3B and C).
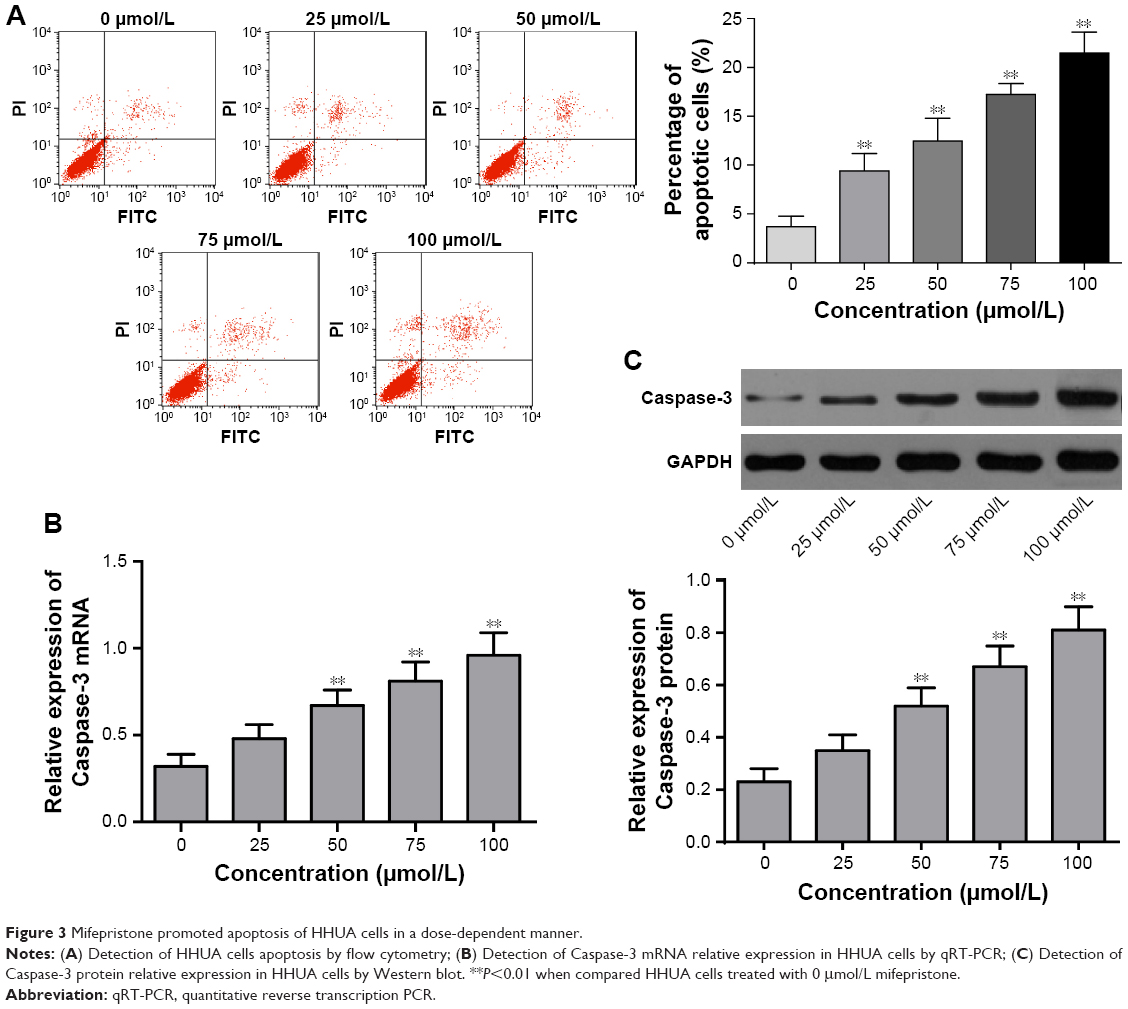 | Figure 3 Mifepristone promoted apoptosis of HHUA cells in a dose-dependent manner. |
Mifepristone inhibited FAK and p-FAK expression in a dose-dependent manner
We further explored the effect of mifepristone on the FAK pathway in HHUA cells. The results revealed that mifepristone could inhibit FAK mRNA and protein relative expression in a dose-dependent manner. When HHUA cells were treated with 50–100 μmol/L of mifepristone, FAK mRNA and protein relative expression was significantly lower than those treated with 0 μmol/L of mifepristone (P<0.01). Similar trend was also found in p-FAK expression (Figure 4A and B). These results indicated that mifepristone could inhibit the activity of the FAK pathway in HHUA cells in a dose-dependent manner.
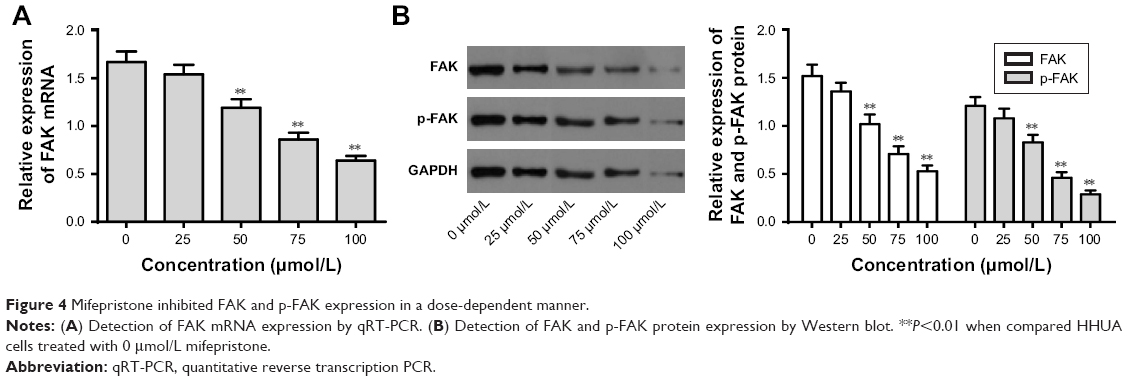 | Figure 4 Mifepristone inhibited FAK and p-FAK expression in a dose-dependent manner. |
Mifepristone inhibited p-PI3K and p-AKT expression in a dose-dependent manner
PI3K/AKT signaling pathway has shown to be involved in tumorigenesis and development. Our findings indicated that different concentrations of mifepristone did not obviously affect PI3K and AKT expression. However, HHUA cells treated with 50–100 μmol/L mifepristone were with significantly lower p-PI3K and p-AKT expression than those treated with 0 μmol/L mifepristone (P<0.01) (Figure 5A–D). The results indicated that, in HHUA cells, mifepristone could inhibit p-PI3K and p-AKT expression in a dose-dependent manner.
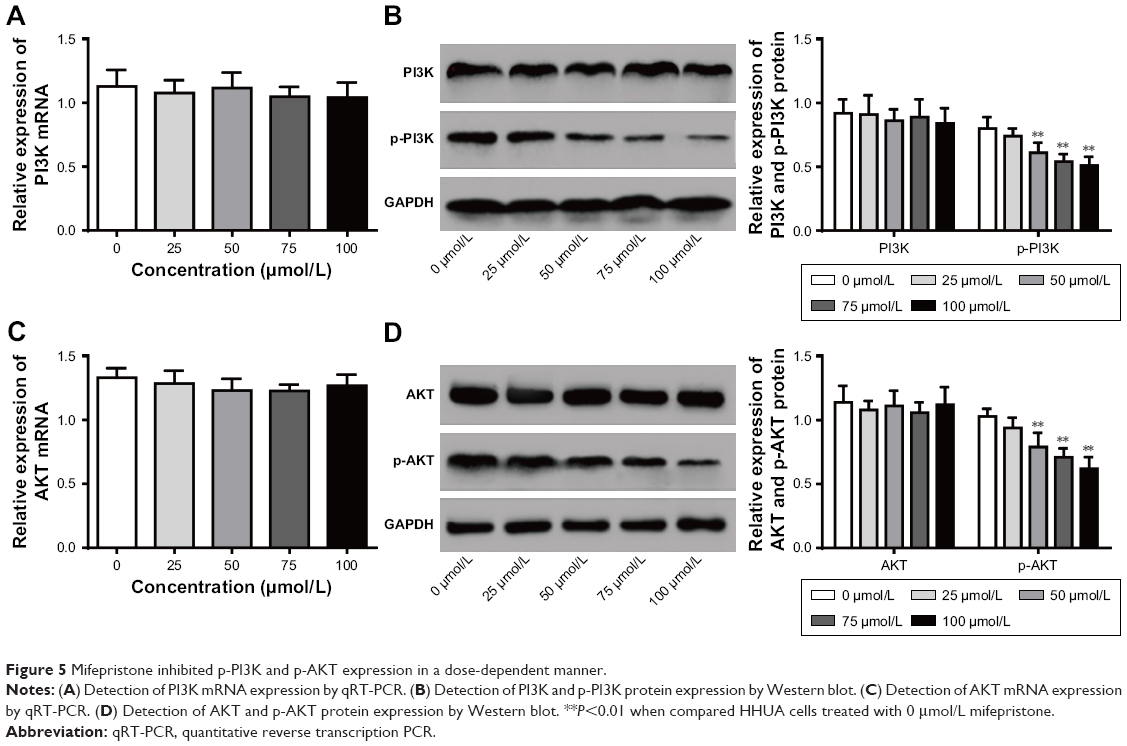 | Figure 5 Mifepristone inhibited p-PI3K and p-AKT expression in a dose-dependent manner. |
Mifepristone inhibited HHUA cells proliferation and promoted its apoptosis by suppressing PI3K/AKT signaling pathway activity
We further investigated whether mifepristone affected HHUA cells proliferation and apoptosis by affecting PI3K/AKT signaling pathway activity. Compared with Mifepristone group, the relative expression of PI3K, p-PI3K, AKT and p-AKT was all significantly upregulated in HHUA cells of Mifepristone + PI3K/AKT group (P<0.01) (Figure 6A). Meanwhile, the cell growth inhibition rate and percentage of apoptotic cells in HHUA cells of Mifepristone + PI3K/AKT group was both markedly lower than those of Mifepristone group (P<0.01) (Figure 6B and C).
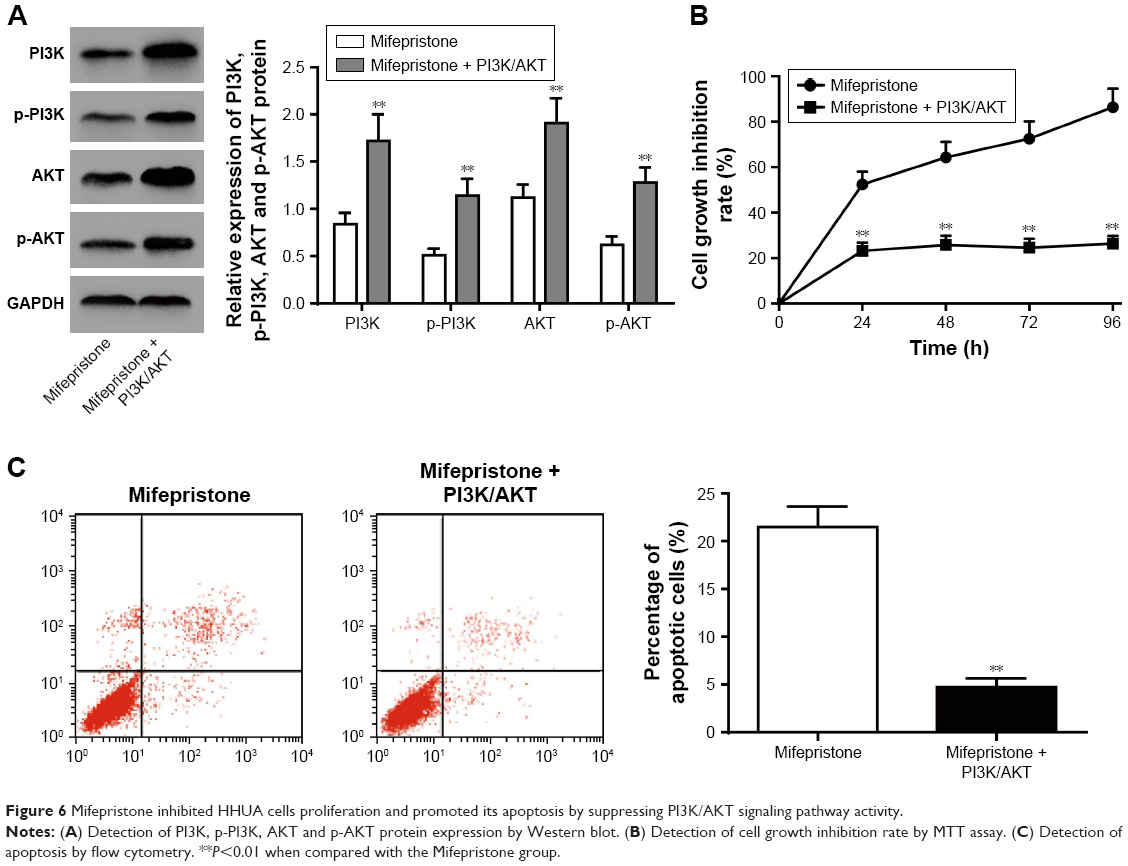 | Figure 6 Mifepristone inhibited HHUA cells proliferation and promoted its apoptosis by suppressing PI3K/AKT signaling pathway activity. |
Discussion
The present study investigated the effects of mifepristone on HHUA cells. The results indicated that mifepristone could inhibit HHUA cells proliferation, migration and invasion and promote its apoptosis in a dose-dependent manner. The mechanism of mifepristone on affecting HHUA cells was through inhibiting the activity of FAK pathway and PI3K/AKT signaling pathway.
Previous researches have proven that mifepristone could exert an anti-tumor effect, which could selectively inhibit the formation and development of multiple tumors.17–19 Liu et al20 proved that mifepristone could be used as a therapeutic drug for treating triple negative breast cancer. By in vivo and in vitro studies, they found that mifepristone significantly inhibited triple negative breast cancer cells proliferation and promote its apoptosis. The mechanism was through down-regulation of KLF5. It is well known that KLF5 is a key transcription factor, which is able to maintain triple negative breast cancer cancer stem cells.21,22 Jurado et al23 discovered that mifepristone could greatly improve the therapeutic effect of cisplatin on cervical cancer. They recommend that anti-hormonal drugs combined with anti-cancer drugs could be used in the treatment of cervical cancer as well as other cancers. However, in-depth studies of the relevant mechanisms have not been conducted. Lu et al24 used different concentrations of mifepristone to investigate its effect on human endometrial carcinoma cells migration. They found that mifepristone could inhibit H19 transcriptional levels by promoting the methylation of the H19 promoter, which eventually resulted in the up-regulation of E-cadherin expression and led to an inhibitory effect on the migration of human endometrial carcinoma cells.
Further studies indicated that mifepristone suppressed the activity of the FAK pathway in HHUA cells. It could inhibit the expression of FAK as well as p-FAK in a dose-dependent manner. FAK, a regulator of focal adhesion dynamics, is involved in signal transduction events between cells and their extracellular matrix.25,26 FAK plays a significant role in adhesion and cooperative signaling of tumor cells’ growth factors. It increases tumor cells’ motility, invasiveness and proliferation, and viability.27 FAK has been found to be up-regulated in a variety of tumors, including breast cancer, pancreatic neuroendocrine tumors and gastric cancer.28–30 Inhibition of FAK phosphorylation could significantly inhibit tumor cell growth and metastasis.31 There were also studies demonstrated that high expression of FAK was found in both endometrial hyperplasia and endometrial carcinoma. It was participated in epithelial-mesenchymal transition and migration during the development and progression of endometrial carcinoma.32–37 Alowayed et al38 suggested that endometrial carcinoma cell proliferation and migration ability were both impaired after FAK activity was inhibited. We also validated that mifepristone inhibits the development of endometrial carcinoma by inhibiting the activity of FAK. In addition, our study also found that mifepristone could suppress the activity of PI3K/AKT signal pathway in a dose-dependent manner. It impaired HHUA cell proliferation by suppressing the phosphorylation of PI3K and AKT. Accumulated studies showed that dysregulated phosphorylation of PI3K/AKT played an important role in the development of cancers.39–41 Lian et al42 thought that activated PI3K/AKT signaling pathway has a positive effect on the development of laryngeal squamous cell carcinoma. Liu et al43 illustrated that PI3K/AKT pathway could be used as a potential target of colorectal cancer. Previous research has proven that, FAK was one of the key genes regulating the PI3K/AKT signaling pathway.44 Activated FAK could phosphorylate PI3K, which further led to the phosphorylation of AKT.45 AKT participated in many tumor cellular processes and tumor development after it was activated.46–48 Therefore, we speculated that, mifepristone might suppress the activity of the PI3K/AKT signaling pathway by impairing the activation of FAK. This mechanism will be one of the focuses of our future research.
Conclusions
This research demonstrated that mifepristone inhibited HUUA cells proliferation, migration, invasion and promoted its apoptosis by inhibition of the FAK and PI3K/AKT signaling pathways. It provided a new theoretical basis for the treatment of endometrial carcinoma with mifepristone, and also provided a new potential target for the treatment of endometrial carcinoma.
Author contributions
Lin Sang: Substantial contributions to conception and design, data acquisition; Dawei Lu: Substantial contributions to conception and design, data analysis and interpretation; Jun Zhang: Drafting the article or critically revising it for important intellectual content; Shihua Du: Drafting the article or critically revising it for important intellectual content; Xingbo Zhao: Final approval of the version to be published. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Heng S, Evans J, Salamonsen LA, Jobling TW, Nie G. The significance of post-translational removal of α-DG-N in early stage endometrial cancer development. Oncotarget. 2017;8(47):81942–81952. | ||
Tejerizo-García A, Álvarez-Conejo C, Muñoz-Hernando L, et al. Tumor recurrence and tumor-related mortality in endometrial cancer: analysis in 276 patients. Indian J Cancer. 2015;52(4):682–684. | ||
Lee NK, Mills KA, Lyman PS, et al. Promoting community-based lifestyle modification and weight management in African American endometrial cancer survivors and their female social network. Gynecologic Oncology. 2015;137:119–120. | ||
Kraetschell RW, Fotopoulou C, Dowdy SC, et al. Regional differences in therapy and clinical management of endometrial cancer: findings of an international survey by the North-eastern German Society of Gynaecological Oncology (NOGGO). J Clin Oncol. 2013;31(15 Suppl):5593. | ||
Trabert B, Wentzensen N, Felix AS, Yang HP, Sherman ME, Brinton LA. Metabolic syndrome and risk of endometrial cancer in the United States: a study in the SEER-medicare linked database. Cancer Epidemiol Biomarkers Prev. 2015;24(1):261–267. | ||
Cohen CJ, Rahaman J. Endometrial cancer. Management of high risk and recurrence including the tamoxifen controversy. Cancer. 1995;76(10 Suppl):2044–2052. | ||
Check JH, Diantonio G, Diantonio A, Duroseau M. The progesterone receptor antagonist mifepristone does not lower serum progesterone induced blocking factor (PIBF) in the presence of progesterone. Clin Exp Obstet Gynecol. 2016;43(2):189–191. | ||
Heikinheimo O, Leminen A, Cacciatore B, Rutanen EM, Kajanoja P. Advanced cervical pregnancy: uterus-sparing therapy initiated with a combination of methotrexate and mifepristone followed by evacuation and local hemostatic measures. Acta Obstet Gynecol Scand. 2004;83(2):211–212. | ||
Liguori M, Lanari C, Gass H, et al. Abstract OT1-04-02: Mifepristone treatment for breast cancer patients expressing levels of progesterone receptor isoform A (PRA) higher than those of isoform B (PRB). Cancer Res. 2018;78(4 Suppl):OT1-04-02. | ||
Zhou H, Jachan N, Singh M, et al. Abstract 4172: Activation of AR signaling by mifepristone enhances prostate cancer growth and impairs enzalutamide response. Cancer Res. 2017;77(13 Suppl):4172. | ||
Chen H, Duan J, Zuo F. Mechanism of the reversal effect of mifepristone on drug resistance of the human cervical cancer cell line HeLa/MMC. Genet Mol Res. 2014;13(1):1288–1295. | ||
Liu C, Lu Q, Qu H, et al. Different dosages of mifepristone versus enantone to treat uterine fibroids: a multicenter randomized controlled trial. Medicine. 2017;96(7):e6124. | ||
Carbonell Esteve JL, Riverón AM, Cano M, et al. Mifepristone 2.5 mg versus 5 mg daily in the treatment of leiomyoma before surgery. Int J Womens Health. 2012;4:75–84. | ||
Esteve JL, Acosta R, Pérez Y, Campos R, Hernández AV, Texidó CS. Treatment of uterine myoma with 5 or 10 mg mifepristone daily during 6 months, post-treatment evolution over 12 months: double-blind randomised clinical trial. Eur J Obstet Gynecol Reprod Biol. 2012;161(2):202–208. | ||
Check JH, Check D, Cohen R, Sarumi M. Mifepristone causing complete remission of rapidly advancing leukemia with measurement of progesterone-induced blocking factor. Anticancer Res. 2014;34(5):2413–2416. | ||
Rekawiecki R, Kowalik MK, Kotwica J. Onapristone (ZK299) and mifepristone (RU486) regulate the messenger RNA and protein expression levels of the progesterone receptor isoforms A and B in the bovine endometrium. Theriogenology. 2015;84(3):348–357. | ||
de Bruin C, Hofland LJ, Nieman LK, et al. Mifepristone effects on tumor somatostatin receptor expression in two patients with Cushing’s syndrome due to ectopic adrenocorticotropin secretion. J Clin Endocrinol Metab. 2012;97(2):455–462. | ||
Wang Y, Yang D, Song L, et al. Mifepristone-inducible caspase-1 expression in mouse embryonic stem cells eliminates tumor formation but spares differentiated cells in vitro and in vivo. Stem Cells. 2012;30(2):169–179. | ||
Cerliani JP, Giulianelli S, Sahores A, et al. Mifepristone inhibits MPA-and FGF2-induced mammary tumor growth but not FGF2-induced mammary hyperplasia. Medicina. 2010;70(6):529–532. | ||
Liu R, Shi P, Nie Z, et al. Mifepristone suppresses basal triple-negative breast cancer stem cells by down-regulating KLF5 expression. Theranostics. 2016;6(4):533–544. | ||
Shi P, Liu W, Tala, et al. Metformin suppresses triple-negative breast cancer stem cells by targeting KLF5 for degradation. Cell Discov. 2017;3:17010. | ||
Chen C, Qin J, Zhou Z. Abstract 4967: the BAP1 deubiquitinase promotes triple-negative breast cancer partially by stabilizing the KLF5 transcription factor. Cancer Res. 2015;75(15 Suppl):4967. | ||
Jurado R, López-Flores A, Álvarez-Figueroa A, García-López P. Synergistic effects of mifepristone on the cytotoxicity of cisplatin in cervical carcinoma cell lines and tumors grown in athymic mice. BMC Cancer. 2007;7(Suppl 1):A18-1. | ||
Lu ZZ, Yan L, Zhang H, Li MJ, Zhang XH, Zhao XX. Mifepristone inhibits the migration of endometrial cancer cells through regulating H19 methylation. Zhonghua Zhong Liu Za Zhi. 2016;38(6):411. | ||
Parsons JT, Martin KH, Slack JK, Taylor JM, Weed SA. Focal adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene. 2000;19(49):5606–5613. | ||
Deramaudt TB, Dujardin D, Noulet F, et al. Altering FAK-paxillin interactions reduces adhesion, migration and invasion processes. PLoS One. 2014;9(3):e92059. | ||
Dy GK, Ylagan L, Pokharel S, et al. The prognostic significance of focal adhesion kinase expression in stage I non-small-cell lung cancer. J Thorac Oncol. 2014;9(9):1278–1284. | ||
Tancioni I, Miller NL, Uryu S, et al. FAK activity protects nucleostemin in facilitating breast cancer spheroid and tumor growth. Breast Cancer Res. 2015;17(1):47. | ||
Moen I, Gebre M, Alonso-Camino V, Chen D, Epstein D, McDonald DM. Anti-metastatic action of FAK inhibitor OXA-11 in combination with VEGFR-2 signaling blockade in pancreatic neuroendocrine tumors. Clin Exp Metastasis. 2015;32(8):799–817. | ||
Cheng Z, Liu F, Zhang H, et al. miR-135a inhibits tumor metastasis and angiogenesis by targeting FAK pathway. Oncotarget. 2017;8(19):31153. | ||
Shi J, Guo B, Hui Q, Chang P, Tao K. Fangchinoline suppresses growth and metastasis of melanoma cells by inhibiting the phosphorylation of FAK. Oncol Rep. 2017;38(1):63–70. | ||
Zhao X, Guan JL. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev. 2011;63(8):610–615. | ||
Ho JN, Jun W, Choue R, Lee J. I3C and ICZ inhibit migration by suppressing the EMT process and FAK expression in breast cancer cells. Mol Med Rep. 2013;7(2):384–388. | ||
Taliaferro-Smith L, Oberlick E, Liu T, et al. FAK activation is required for IGF1R-mediated regulation of EMT, migration, and invasion in mesenchymal triple negative breast cancer cells. Oncotarget. 2015;6(7):4757. | ||
Wilson C, Nicholes K, Bustos D, et al. Overcoming EMT-associated resistance to anti-cancer drugs via Src/FAK pathway inhibition. Oncotarget. 2014;5(17):7328. | ||
Zhang L, Li Z, Fan Y, Li H, Li Z, Li Y. Overexpressed GRP78 affects EMT and cell-matrix adhesion via autocrine TGF-β/Smad2/3 signaling. Int J Biochem Cell Biol. 2015;64:202–211. | ||
Zhang P, Bai H, Liu G, et al. MicroRNA-33b, upregulated by EF24, a curcumin analog, suppresses the epithelial-to-mesenchymal transition (EMT) and migratory potential of melanoma cells by targeting HMGA2. Toxicol Lett. 2015;234(3):151–161. | ||
Alowayed N, Salker MS, Zeng N, Singh Y, Lang F. LEFTY2 controls migration of human endometrial cancer cells via focal adhesion kinase activity (FAK) and miRNA-200a. Cell Physiol Biochem. 2016;39(3):815–826. | ||
Goler-Baron V, Sladkevich I, Assaraf YG. Inhibition of the PI3K-AKT signaling pathway disrupts ABCG2-rich extracellular vesicles and overcomes multidrug resistance in breast cancer cells. Biochem Pharmacol. 2012;83(10):1340–1348. | ||
Cai Q, Wang ZQ, Wang SH, et al. Upregulation of long non-coding RNA LINC00152 by SP1 contributes to gallbladder cancer cell growth and tumor metastasis via PI3K/AKT pathway. Am J Transl Res. 2016;8(10):4068. | ||
Yang SX, Polley E, Lipkowitz S. New insights on PI3K/AKT pathway alterations and clinical outcomes in breast cancer. Cancer Treat Rev. 2016;45:87–96. | ||
Lian R, Lu B, Jiao L, et al. MiR-132 plays an oncogenic role in laryngeal squamous cell carcinoma by targeting FOXO1 and activating the PI3K/AKT pathway. Eur J Pharmacol. 2016;792:1–6. | ||
Liu G, Song Y, Cui L, Wen Z, Lu X. Inositol hexaphosphate suppresses growth and induces apoptosis in HT-29 colorectal cancer cells in culture: PI3K/Akt pathway as a potential target. Int J Clin Exp Pathol. 2015;8(2):1402. | ||
Casar B, Rimann I, Kato H, Shattil SJ, Quigley JP, Deryugina EI. In vivo cleaved CDCP1 promotes early tumor dissemination via complexing with activated β1 integrin and induction of FAK/PI3K/Akt motility signaling. Oncogene. 2014;33(2):255–268. | ||
Hers I, Vincent EE, Tavaré JM. Akt signalling in health and disease. Cell Signal. 2011;23(10):1515–1527. | ||
Ponnusamy M, Li PF, Wang K. Understanding cardiomyocyte proliferation: an insight into cell cycle activity. Cell Mol Life Sci. 2017;74(6):1019–1034. | ||
Zhang Y, Wang B, Chen X, Li W, Dong P. AGO2 involves the malignant phenotypes and FAK/PI3K/AKT signaling pathway in hypopharyngeal-derived FaDu cells. Oncotarget. 2017;8(33):54735–54746. | ||
Oudart JB, Doué M, Vautrin A, et al. The anti-tumor NC1 domain of collagen XIX inhibits the FAK/PI3K/Akt/mTOR signaling pathway through αvβ3 integrin interaction. Oncotarget. 2016;7(2):1516–1528. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.