")
Back to Journals » Cancer Management and Research » Volume 11
Midostaurin In Acute Myeloid Leukemia: An Evidence-Based Review And Patient Selection
Authors Abbas HA, Alfayez M, Kadia T, Ravandi-Kashani F, Daver N
Received 28 May 2019
Accepted for publication 12 September 2019
Published 4 October 2019 Volume 2019:11 Pages 8817—8828
DOI https://doi.org/10.2147/CMAR.S177894
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Rudolph Navari
Hussein A Abbas,* Mansour Alfayez,* Tapan Kadia, Farhad Ravandi-Kashani, Naval Daver
Department of Leukemia, University of Texas M. D. Anderson Cancer Center, Houston, TX, USA
*These authors contributed equally to this work
Correspondence: Naval Daver
Department of Leukemia, University of Texas M. D. Anderson Cancer Center, 1400 Holcombe Boulevard, Unit FC 4.2012, Houston, TX 77030, USA
Tel +1713-794-4392
Email [email protected]
Abstract: Fms-related-tyrosine kinase 3 (FLT3) mutations occur in approximately a third of acute myeloid leukemia (AML) patients and confer an adverse prognosis. Numerous studies have evaluated FLT3 targeting as single agent and in combination approaches in frontline and relapsed AML. At this time, midostaurin, a multikinase inhibitor, is the only FLT3-inhibitor that is US FDA approved to be used in combination with induction therapy in the frontline FLT3-mutated AML setting based on improved overall survival noted in the RATIFY Phase III trial. The utility of midostaurin in maintenance post stem cell transplantation has shown promising results and further studies are still ongoing. In this review, we discuss the studies that led to the inception of midostaurin as a targeted kinase inhibitor, its evaluation in AML, the early clinical trials and the large Phase III clinical trial that led to its eventual US FDA-approval in FLT3-mutated AML. Our review also discusses data on midostaurin adverse effects, mechanisms of resistance and limitations of its utility. We further discuss emerging second-generation FLT3 inhibitors, with a focus on quizartinib and gilteritinib and future directions to enhance FLT3-inhibitor efficacy and overcome mechanisms of resistance.
Keywords: acute myeloid leukemia, FLT3, midostaurin
Introduction
Acute myeloid leukemia (AML) represents a malignant clonal disorder of myeloid cells that impairs normal hematopoiesis. Age, history of pre-leukemic hematologic disorders, karyotype and mutation profile provide significant prognostic information that dictate therapeutic decisions in AML.1,2 Treatment of AML traditionally encompasses induction with high-dose anthracyclines, such as idarubicin (or daunorubicin), and cytarabine arabinoside-based regimens. Consolidation therapy traditionally includes high-dose chemotherapy (cytarabine) alone (HiDAC) or in combination with anthracycline (also referred to as “2+5”) depending on regional and institutional preferences in low-risk patients, or allogeneic transplantation in the majority of intermediate and in high-risk patients, respectively.3
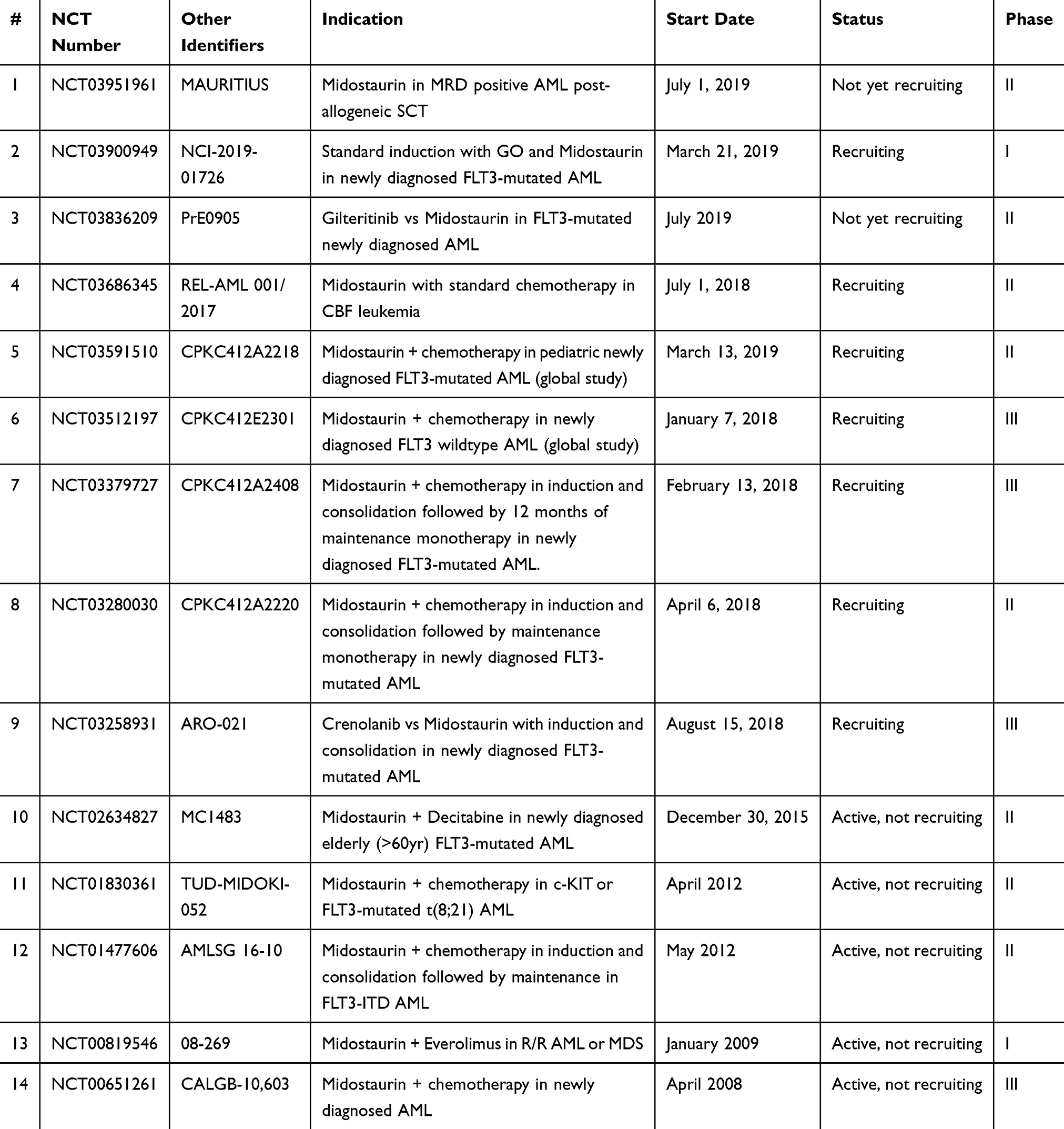 |
Table 1 Ongoing Or Planned Clinical Trials Of Midostaurin In AML |
Traditionally, normal karyotype in AML conferred intermediate-risk disease. However, with the advent of mutational analysis, it became evident that there were certain clonal mutations that could significantly alter the AML pathogenesis and prognosis. Some mutations such as nucleophosmin 1 (NPM1) and CCAAT/enhancer-binding protein-α (CEBPA) confer favorable risk. On the other hand, mutations in genes such as Fms-related-tyrosine kinase 3 (FLT3), runt-related transcription factor 1 (RUNX1), DNA methyl transferase 3 alpha (DNMT3A), tumor protein p53 (TP53) and others confer unfavorable risk.2 An internal tandem duplication (ITD) mutation in FLT3 gene on chromosome 13q12 is one of the most common mutations noted in AML, occurring in approximately a third of newly diagnosed adults with AML.4–6 FLT3 encodes a class III receptor tyrosine kinase (RTK), which is normally expressed in CD34+ hematopoietic stem cells and promotes diverse downstream pathways depending on the impact of co-occurring signals.7 For instance, binding of the FLT3-ligand (FL) to FLT3 receptor in the absence of other growth factors induces monocytic differentiation of hematopoietic progenitors.8 However, FLT3 activation induces proliferation and maintenance of progenitors when interleukin-3, stem cell factor and FL are engaged.8–10
Mutations in FLT3 commonly occur in patients with AML who have diploid cytogenetics indicating that the mutated FLT3 is the driver for leukemogenesis.11 There are two major classes of activating FLT3 mutations reported in AML patients. The first class of mutations is 3–400 base pair in-frame duplications detected in 20% to 25% of patients with AML and referred to as internal-tandem duplications (FLT3-ITD).12 FLT3-ITD mutations lead to constitutive activation of the FLT3-signaling cascade with subsequent stimulation of downstream signaling pathways including signal transducer and activator of transcription 5 (STAT5), phosphatidyl-inositol 3-kinase (PI3K) and protein kinase B (AKT) pathways.13,14 The second class of FLT3 mutations occurs as point mutations, most commonly a substitution of tyrosine for aspartic acid at codon 835 (D835Y), in the tyrosine kinase domain (FLT3-TKD).15 These occur in 5% to 10% of patients with AML. Similar to FLT3-ITD, these point mutations lead to downstream activation of proliferative pathways.11 Patients with FLT3-ITD mutations have similar complete remission (CR) rates compared with non-FLT3 mutated patients, but inferior outcomes due to shorter CR duration, high relapse rates, and inferior overall survival when treated with induction therapy alone, without the addition of a TKI.4,11 FLT3-TKD mutations on the other hand have been generally noted to have a neutral impact on overall survival (OS).16 The significance of these mutations in clinical practice has been further fortified by the emergence of effective targeted therapies to these mutations and their implications on survival.
FLT3 Inhibitors In AML
AML patients with FLT3-ITD mutations have shorter CR durations, higher rates of recurrence, and inferior OS compared to patients without FLT3 mutations.11,17,18 Of significance, the FLT3 allelic burden is also important and has a prognostic impact.19 Polymerase chain reaction (PCR) technique is the most commonly used tool to assay for FLT3 allelic burden. However, because of the competition from wildtype allele, the sensitivity of the PCR assay is low which may be overcome by using patient-specific primers.20 A more recent assay detects FLT3-positive minimal residual disease AML with higher sensitivity and specificity. The proposed approach starts with a PCR amplification step, followed by next-generation sequencing, and uses a unique software program to quantify the findings.21 Further, developing assays to measure the inhibitory effects of an oral drug is also important. Plasma inhibitory activity is the first assay to measure the efficacy of target inhibition of FLT3.22
Given the significance of FLT3 mutations in AML, there has been significant interest in developing and applying targeted therapies for FLT3-mutated AML patients in the induction, consolidation, and/or maintenance phases, to decrease the risk of relapse and improve OS, as well as in relapsed FLT3-mutated AML.23 Thomas and Campbell (2019) recently reviewed four agents that showed promising results in AML FLT3 inhibition.24 Briefly, ponatinib, sunitinib, and sorafenib are non-specific tyrosine kinase inhibitors approved in multiple solid tumor malignancies with known FLT3-inhibitory activity.25 Ponatinib showed modest activity in a small cohort of AML patients with overall response rate of 25% but had significant adverse events.26 Sunitinib showed activity in inhibiting FLT3 in AML patients but the duration of response was short lived.25,27 Both ponatinib and sunitinib have not been widely incorporated in AML therapy. Sorafenib, while not approved in AML, has been used effectively for many years as a maintenance post stem cell transplantation in FLT3-mutated AML patients,28 as well as in combination with induction chemotherapy in newly diagnosed FLT3-ITD mutated AML and in combination with azacitidine in frontline and relapsed older FLT3-mutated AML.29,30 Interestingly, sorafenib improved event-free survival (EFS), but not OS when added to 3+7 induction regimen even among FLT3-wildtype patients, likely more through its multi-kinase inhibitory activity rather than its direct FLT3-inhibitory activity,31 and this study is ongoing with follow-up data eagerly awaited. Of note, gilteritinib a potent second-generation FLT3-inhibitor was also recently approved as single-agent therapy for patients with relapsed/refractory FLT3 mutated (both ITD and TKD) AML based on improved OS and response rates compared to conventional cytotoxic or low-intensity therapies in a randomized Phase III setting.32 The only agent that is currently FDA approved in combination with induction and consolidation therapy for newly diagnosed FLT3-mutated AML patients is midostaurin, and will be the focus of this review article.
Midostaurin Development
Midostaurin, formerly known as PKC412, is an orally administered multi-targeted tyrosine kinase inhibitor that inhibits multiple kinases including proto-oncogene c-Kit (KIT), platelet-derived growth factor (PDGFR)-α/-β, protein kinase C (PKC), spleen associated tyrosine kinase (SYK), cellular Src kinase (SRC), and vascular endothelial growth factor receptor (VEGFR)-1/-2.23 Midostaurin was developed as a therapeutic alternative to the naturally available staurosporine in order to inhibit PKC activity.33,34 Several in vitro and animal studies demonstrated the efficacy of midostaurin in halting cellular proliferation.35–38 These findings led to a Phase I study in 32 patients with advanced solid tumors whereby midostaurin demonstrated a relatively safe profile with low-grade gastrointestinal and hematological toxicities.39 A treatment dose of 150 mg/day was considered adequate for further investigation although subsequent studies used different regimens. Subsequent clinical trials investigated the utility of midostaurin in solid cancers and lymphomas but failed to replicate the preclinical findings. Specifically, midostaurin was tested in lymphoproliferative disorders as a single agent,40 different solid tumors in combination with 5-fluorouracil,41 non-small cell lung cancer in combination with gemcitabine and cisplatin,42 and metastatic melanoma as a single agent,43 without demonstrating significant activity but notably with a maintained low toxicity profile. Further, the anti-angiogenic activity of midostaurin via inhibition of vascular endothelial growth factor (VEGF) in animal models led to clinical studies that examined its role in diabetic retinopathy and macular edema.44,45 Midostaurin at a lower dose of 100 mg/day demonstrated activity in reducing diabetic retinopathy and macular edema but the gastrointestinal toxicity profile noted in these patients with chronic use of midostaurin limited its further clinical development in diabetic patients.46
Preclinical Studies Of Midostaurin In AML
The first evidence of the activity of midostaurin in AML was in 2002. In a drug screening assay, Weisberg et al demonstrated the efficacy of midostaurin in inducing G1 arrest and apoptosis in FLT3-mutated Ba/F3 leukemia cell lines and mouse models.47 In a subsequent study, Grundler et al demonstrated that midostaurin can inhibit FLT3-ITD encoded protein, which by then was known to be one of the most common and aggressive mutations in AML.48 Combining midostaurin with the histone deacetylase inhibitor LAQ824, now known as dacinostat, demonstrated synergistic activity in inhibiting FLT3-mutated AML cell lines, which was one of the first preclinical experiments suggesting a role for combining midostaurin with other established leukemia drugs.49 Also, midostaurin demonstrated more potent synergism when combined with conventional anti-leukemic agents such as cytarabine, doxorubicin and idarubicin in inhibiting FLT3 mutated, compared with FLT3 wildtype leukemia cell lines in vitro.50,51 Interestingly, midostaurin elicited apoptotic cell death in FLT3-mutated AML cell lines, while it induced cell cycle arrest in FLT3-wildtype cell lines.52 Hence, the context in which midostaurin was used likely mattered in dictating its response. These preclinical studies in AML and the relatively safe profile of midostaurin in solid tumor clinical trials supported the development of clinical trials to evaluate midostaurin in the treatment of patients with AML.
Early Phase Clinical Trials Of Midostaurin In AML
The relatively safe profile of midostaurin in prior clinical trials in non-leukemia patients paved the way for the initiation of a Phase II clinical trial in AML. To that end, a total of 20 patients with relapsed/refractory AML with an FLT3-ITD or FLT3-D835Y mutation received midostaurin at a dose of 75 mg orally for 3 times daily (TID).53 The drug was relatively well tolerated. The most common toxicities were gastrointestinal side effects including nausea, vomiting and diarrhea. Grade 1/2 gastrointestinal toxicities occurred in 65% of the patients on trial. Of note, 3 patients developed fatal pulmonary events in the context of progressive leukocytosis, pulmonary infiltrates of unclear etiology and pneumonia, respectively. Midostaurin demonstrated measurable responses. Specifically, 14/20 (70%) and 6/20 (30%) of treated patients had 50% reduction in peripheral blood and bone marrow blasts, respectively. However, none of the patient attained CR, despite the significant blast reductions, although 1 patient had <5% blasts in a hypocellular bone marrow and was documented as a partial remission. A subsequent Phase IIB trial treated 95 patients with relapsed/refractory AML and myelodysplastic syndrome (MDS) with mutant or wildtype FLT3 with midostaurin as a single agent.54 Similar to the prior study, there was a reduction in blasts on treatment in 71% of FLT3-mutant and 42% of FLT3 wildtype-treated patients.54 Blast reduction of 50% of more was observed in 42% of treated patients. However, only one patient experienced a partial remission and no patients experienced a CR or CR with incomplete hematologic recovery (CRi) suggesting that midostaurin would likely not be sufficient as a monotherapy agent. Subsequent studies explored combining midostaurin with other agents. In a Phase I/II trial, the combination of midostaurin with the hypomethylating agent 5-azacitidine in 54 untreated and relapsed/refractory AML and high-risk MDS patients showed a modest overall response rate of 26% (1/54 CR, 6/54 CRi), 6/54 morphologic leukemia-free state (MLFS) and 1/54 partial remission).55 The median response duration was 20 weeks and median overall survival was 22 weeks at a median follow-up of 15 weeks (range, 1–85 weeks). The longest response duration was noted in patients without prior exposure to FLT3 inhibitors and patients who did not have a previous bone marrow transplantation.55
With the emergence of greater understanding of potential efficacy requisites with FLT3 inhibition in AML, Levis et al demonstrated that the lack of response to midostaurin may be due to the lack of a sufficient inhibitory effect on FLT3 as demonstrated by plasma inhibitory levels.22 This led to considerations that utilizing midostaurin in an FLT3-inhibitor naïve population, ensuring adequate inhibition of FLT3 by monitoring plasma inhibitory levels, and combining midostaurin with other agents in frontline setting may be a better approach to use this agent in patients with AML. In a Phase Ib study, midostaurin at different dosing schedules in combination with chemotherapy in 79 younger adults (<60 years of age) newly diagnosed AML patients with mutant or wild-type FLT3 demonstrated CR rates of 80% in the 50 mg twice a day dosing schedule cohort (40 patients).56 The OS probabilities at 1 and 2 years were 85% and 62% in patients with FLT3-mutated AML, and 78% and 52% in patients with FLT3-wildtype AML, respectively. Interestingly, the median OS of FLT3-mutated patients was similar to that of the FLT3 wildtype patients, leading to the hypothesis that the addition of TKI midostaurin could potentially neutralize the adverse impact of the FLT3 mutation and improve the outcomes of these patients. Midostaurin was not well tolerated when administered at a dose of 50 mg twice a day or 100mg twice a day starting from Day 1 of induction due to significant gastrointestinal side effects during days 1–7 when patients received midostaurin concomitantly with the cytotoxic therapy (3+7). The tolerance improved significantly when patient received 3+7 alone on Days 1 to 7 and the midostaurin was introduced from Day 8 onwards, especially with the 50 mg twice a day dose. Collectively, these data supported the potential benefit of midostaurin in combination with induction therapy in younger patients with FLT3-mutated AML, and was the basis for the Phase III RATIFY trial.
RATIFY Trial
The RATIFY trial enrolled FLT3-ITD or FLT3-TKD patients with newly diagnosed AML 18–60 years of age from 13 AML cooperative groups (225 sites). A total of 3277 patients were screened and 717 were randomized patients to receive either midostaurin or placebo with 3+7 (daunorubicin with cytarabine) induction and high-dose cytarabine consolidation therapy (up to 4 consolidations), followed by 12 months of maintenance with either midostaurin or placebo.57 Midostaurin was administered at a dose of 50 mg twice a day on Days 8–21 in induction and each consolidation cycle. During maintenance midostaurin 50 mg twice a day was administered continuously from Cycle 1 Day 1 onwards, without interruption. In order to ensure rapid FLT3 mutational testing and support enrollment to the trial across multiple sites in different countries, a large-scale cooperative effort established an efficient polymerase chain reaction-based FLT3 mutation assay with turnaround time of less than 48 hrs.58 This milestone allowed rapid accrual of patients into the trial, frequently within 4 to 5 days of AML presentation.
Patients were stratified based on the type and frequency of FLT3 mutation into 3 groups: FLT3-TKD, high allelic ratio FLT3-ITD (>0.70) and low allelic ratio FLT3-ITD (</=0.70). The primary end point of the trial was overall survival uncensored for transplant. The RATIFY trial demonstrated a significant improvement in the 4-year overall survival in patients who received midostaurin compared to placebo with induction therapy (51.4% versus 44.3%, hazard ratio (HR), 0.78, p=0.009). On per protocol response assessment (up to 60 days from start of induction) the rates of complete remission were not significantly different between the two groups (58.9% versus 53.5%, p=0.15). However, there were an additional 14% of all patients who had achieved CR after the protocol-specified 60 days timepoint of assessment: 32 additional patients on midostaurin arm and 25 additional patients on control arm, leading to intent-to-treat complete remission rates of 68% with midostaurin compared with 61% on the control arm (p=0.04). Of note, the median duration of midostaurin exposure for all patients on study was 3 months, because a number of patients who achieved remission went to allogeneic stem cell transplant (ASCT) and received only 2 to 3 cycles of therapy. Post-ASCT maintenance with midostaurin was not a part of the RATIFY trial. The cut-off for entry into the RATIFY study was an FLT3 allelic ratio of >/=0.05. On a post-hoc analysis using an arbitrarily selected FLT3 allelic ratio cut-off of 0.7 (to separate what the RATIFY authors called high >/=0.7 versus low allelic ratio <0.7) it was noted that statistically, a similar degree of benefit appeared to be noted in both “high” and “low” allelic ratio. At this time what we can conclude from the RATIFY data is that there is no data regarding the use of midostaurin in patients with an FLT3 allelic ratio <0.05 at diagnosis, and that addition of midostaurin to induction therapy is recommended in all patients with an FLT3 allelic ration >0.05, irrespective of the allelic ratio.
RATIFY represented a landmark study as this was the first-ever study to demonstrate the effectiveness of targeted biomarker-driven therapy in patients with AML, a critical proof of concept success that has galvanized the further development and approval of numerous additional-targeted therapies in AML. The RATIFY findings led to US Food and Drug Administration (FDA) approval of midostaurin in combination with cytarabine and daunorubicin induction and cytarabine consolidation in newly diagnosed adult AML patients of all ages with FLT3-ITD or FLT3-TKD mutations in April 2017. Of significance, midostaurin was the first drug to be approved for AML in 15 years. A companion FLT3 diagnostic testing was developed and also US FDA approved through a partnership between Invivoscribe and Novartis.58 Accordingly, FLT3 mutational analysis and incorporation of midostaurin into frontline therapy have been integrated into the standard of care work up and treatment for patients with AML in US and Europe. The recommended dosage of midostaurin is 50 mg twice a day with food on Days 8 to 21 of each induction therapy cycle with cytarabine and daunorubicin, and on Days 8 to 21 of each consolidation cycle with high-dose cytarabine.
The Role Of Midostaurin In Maintenance
We discuss maintenance in this section using currently available (and unfortunately insufficient) data. We agree that the efficacy of midostaurin in maintenance therapy based on currently evaluable data is unclear and unfortunately no matter how we dissect and analyze the RATIFY data this is a topic on which we (or others) will not be able to make any definitive conclusions or recommendations,59 until post-consolidation randomized trials of maintenance that include pre-maintenance and sequential MRD assessment (ideally both flow and NGS based) both prior to and post stem cell transplant are conducted and analyzed. Such trials should ideally help establish not only whether maintenance is beneficial or not in a general fashion, but more importantly help identify patients who benefits most from maintenance versus those who do not benefit or benefit marginally: is it MRD-negative patients who benefit from maintenance by further suppressing emergence of a resistance clone or is it in fact MRD+ patients who can be converted to MRD-negative status by maintenance improving their survival with or without a subsequent SCT. Similarly, could maintenance obviate the need for SCT in a subset of patients versus conversely would maintenance not obviate SCT but actually serve as a bridge to make SCT better by reducing preSCT disease burden? These are questions being studied in Ph+ ALL, a disease with similar development history to FLT3, but with a longer history and more advanced longitudinal datasets. An example of such an ongoing well-designed trial with rich correlative analysis including sequential MRD assessment is the MORPHO-trial of gilteritinib versus observation in the post-SCT setting in FLT3 (ITD or D835) mutated patients who under allogeneic SCT. At this time with the currently available-limited randomized data in FLT3 AML maintenance and based on our experience with TKIs in other disease such as CML, Ph+ ALL, sorafenib in AML our groups recommendation has been to continue maintenance and furthermore to consider prolonged maintenance rather than 1 year maintenance given that late relapses have been seen at 2 and 3 years post-induction and consolidation and these may be prevented by prolonged maintenance. At this time this is a recommendation without available randomized clinical trial data.
Recently, the German-Austrian AML Study Group investigated the efficacy of midostaurin plus intensive chemotherapy, followed by allogeneic hematopoietic stem cell transplantation and single-agent midostaurin maintenance therapy in FLT3-ITD-mutated AML patients.60 In this Phase II study, 284 AML patients received induction therapy and 76.4% (217/284) attained CR/CRi at first or second induction, of which 134/217 (61.7%) underwent allogeneic hematopoietic stem cell transplantation as consolidation. Seventy-five of 134 patients (56%) eventually received maintenance midostaurin. The median overall survival was 26 months (95% CI, 18.8–36 months). In univariable analysis, patients who started maintenance therapy with midostaurin within 100 days of consolidation had better OS compared to those who did not. While older patients (61–70 years) also benefited from this regimen, there were more frequent cardiac toxicities (22%) and induction death rate (10.5%) in this population. This Phase II study demonstrated an efficacious use of midostaurin in maintenance therapy which also extended to older patients, albeit in a non-randomized fashion. However, a randomized controlled study is needed to further validate these findings.
Adverse Events And Pharmacokinetic Considerations
The most common adverse effects of midostaurin were nausea, vomiting, diarrhea, fatigue and headaches. Given the gastrointestinal side effect profile of midostaurin, prophylactic anti-emetics, such as ondansetron, olanzapine or lorazepam, are recommended prior to its administration. While midostaurin was not associated with QTc prolongation in healthy individuals, 10.1% of AML patients on midostaurin had QTc prolongation, compared to 5.7% on placebo.61 No clinical events related to QTc prolongation were noted. It is recommended to assess QTc intervals in AML patients receiving midostaurin, especially concomitantly with other QTc-prolonging medications such as some of the anti-emetics, quinolones, and azoles and if possible to avoid concomitant QTc prolonging medications or replace them with suitable alternatives, when feasible.
Midostaurin is metabolized to its active metabolites GGP6221 and CGP52421 via CYP3A4 in the liver, and then excreted through feces.24 Midostaurin levels significantly increased when concomitantly administered with ketoconazole, posaconazole or voriconazole, and levels decreased when rifampicin was co-administered.24,62 Since most AML patients are usually on second-generation anti-fungals as a prophylaxis or as treatment, dose adjustments and monitoring for toxicities are warranted. In patients taking a concomitant strong CYP3A4 inhibitor (such as posaconazole or voriconazole), it is recommended that the midostaurin dose be reduced to 25 mg twice a day. Some experts may recommend using isavuconazole as the preferred azole in this scenario as it is considered a moderate inhibitor of CYP3A4 and is still quite an effective anti-fungal.63 While this may be a reasonable option there are no clear consensus guidelines on this topic.
Mechanisms Of Resistance
Further correlative analysis on midostaurin-treated patients and subset analysis of the RATIFY study set have attempted to elucidate the mechanisms of primary and secondary resistance to midostaurin in patients with AML. Upregulation of myeloid cell leukemia 1 protein (MCL-1) was shown to be an important mechanism of primary resistance to midostaurin in AML.64 Similarly, upregulation of anti-apoptotic genes and down-regulation of proapoptotic genes were demonstrated to be associated with acquired resistance to midostaurin in AML.65 A single amino acid substitution at position 676 (N676K) within the FLT3 kinase domain was identified to be the cause of resistance in 1 of 6 evaluable relapsed/refractory AML patients who relapsed while on midostaurin treatment in the Phase II trial of single-agent midostaurin.53,66 Subsequent work demonstrated that the allelic burden of mutant FLT3 dictated responses and established the basis for assessing the allelic frequency in AML.67 These findings were the impetus for attempting to combine midostaurin with other drugs and to generate other, potentially more potent, specific and better tolerated FLT3 inhibitors.
Limitation And Future Direction
Better understanding of the underlying AML biology, clonal evolution and selection, emergence of a number of effective-targeted therapies and combinations with FLT3 and isocitrate dehydrogenase (IDH) inhibitors have transformed the way we treat AML. Recently the US FDA approved second-generation FLT3 inhibitor gilteritinib as a single agent in patients with FLT3-mutated relapsed/refractory AML (ITD or D835) based on impressive CR/CRi and OS compared with conventional chemotherapy (cytotoxic combination chemotherapy or hypomethylating agent therapy) in a randomized Phase III (ADMIRAL trial).68 Another highly potent and very selective FLT3 inhibitor quizartinib improved CR/CRi and OS in a randomized Phase III trial in relapsed/refractory FLT3-ITD mutated AML and is anticipated to have US FDA approval in the near future.69,70 A third second-generation FLT3-inhibitor is crenolanib and this agent targets both FLT3-ITD and -D835 potently (Figure 1).71,72 These FLT3 inhibitors have a high FLT3-specificity and high response rates as single agents compared with first-generation FLT3-inhibitors such as midostaurin and sorafenib with a relatively good tolerability profile, and as such, are being evaluated in frontline combinations with induction therapy and hypomethylating agent-based therapy in newly diagnosed FLT3-mutated AML. Clinical trials in newly diagnosed AML with quizartinib (NCT01390337, NCT03723681, NCT02668653, NCT02834390), gilteritinib (NCT03836209, NCT02752035, NCT02236013, NCT02310321) and crenolanib (NCT03258931, NCT02283177) in combination with induction therapy (3+7 or HMA) are ongoing, and preliminary-reported data appear encouraging.73–75 An emerging question is regarding how these more selective, potent TKIs such as quizartinib, gilteritinib and crenolanib will compare to broad, non-selective multikinase inhibitors such as midostaurin and sorafenib when combined with frontline induction therapy in FLT3-mutated AML. It has been postulated that newly diagnosed FLT3-mutated AML is less addicted to FLT3 signaling but more dependent on multiple kinase pathways for growth and proliferation suggesting that broad kinase inhibitors may be of benefit and potentially preferable over selective TKIs. This is in contrast to the time of relapse wherein FLT3 is often selected out or emerges as the resistance driver clone and more specific, potent FLT3 inhibitors may be more effective as single agents or even more so in combinations. This assumption is being explored in ongoing-randomized studies of frontline induction with second-generation FLT3 TKIs (including crenolanib and gilteritinib) versus frontline induction with midostaurin (NCT03258931, NCT03836209).
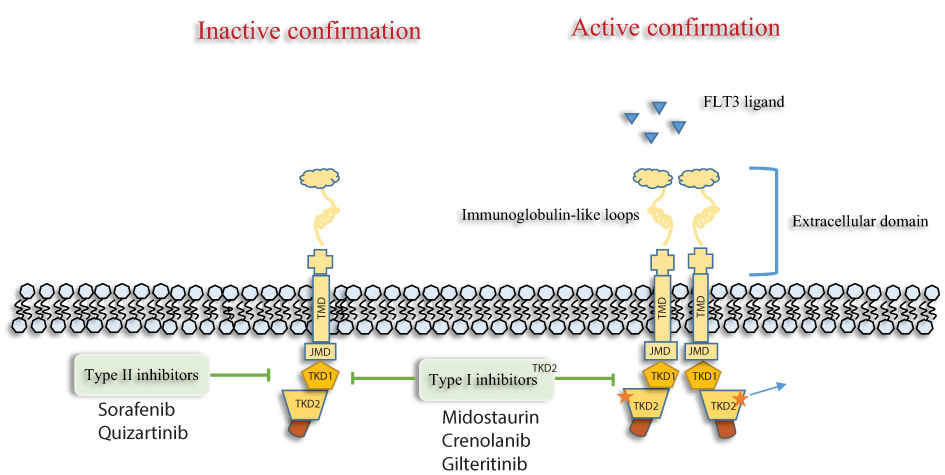 |
Figure 1 Type I FLT3 inhibitors (Midostaurin, Gilteritinib and crenolanib) bind the FLT3 receptor in the active as well as inactive conformation, while Type II FLT3 inhibitors (Sorafenib, Quizartinib) bind the FLT3 receptor in the inactive conformation. As a result of this affinity, type I acts on both FLT3-ITD and TKD mutations, whereas type II act only on FLT3-ITD. Abbreviations: FLT3, FMS-like tyrosine kinase; TMD, transmembrane domain; JMD, juxtamembrane domain; TKD, tyrosine kinase domain. |
Given the nature of midostaurin as a broad, multikinase inhibitor, and the recognition that it inhibits number of kinases and pro-survival pathways that are essential to leukemic cell growth, survival, and proliferation, and taking into account the clinical activity seen with midostaurin in patients with wildtype FLT3,54 it is possible that the on-leukemia, off-FLT3 effects of midostaurin afford benefit regardless of the FLT3 status. The impact of midostaurin in FLT3 wildtype AML is currently being explored in an ongoing Phase III clinical trial (NCT03512197).
Midostaurin was approved in young patients fit for high-intensity chemotherapy who received 3+7 regimen based on the RATIFY trial results. The impact of adding midostaurin to low-intensity therapies such as hypomethylating agents (azacitidine or decitabine) or to low dose cytarabine in older patients with AML who are not fit for high-intensity chemotherapy is yet to be defined. In vitro the combination of decitabine and midostaurin was synergistically active against FLT3-ITD mutation expressing AML cells, advocating for such combinations.76 In a Phase I/II trial of midostaurin combined with azacitidine the combination appeared to be effective and safe in patients with AML and high-risk MDS.55 A number of midostaurin containing low-intensity combinations in elderly unfit population are actively been investigated (NCT01130662, NCT01093573, NCT01093573, NCT02634827, NCT01846624).
Another potential use of midostaurin in AML is in core binding leukemia (CBF), with or without FLT3 mutations, as KIT mutations are commonly seen and thought to impact prognosis in CBFs.77,78 This is currently being explored in two clinical trials (NCT03686345, NCT01830361). Post-transplant maintenance with midostaurin is also of interest in FLT3-mutated AML and is being evaluated in an ongoing-randomized Phase II clinical trial (NCT01883362; RADIUS trial) with preliminary data suggesting a trend for improved EFS and OS with post-allogeneic SCT midostaurin maintenance,79 similar to what was noted with sorafenib in the SORMAIN trial, but not yet achieving statistical significance in the RADIUS trial. It must be noted though that midostaurin and sorafenib maintenance, particularly post-allogeneic SCT, are associated with more toxicities and require frequent and liberal dose adjustments to allow patients to stay on therapy. As FLT3 inhibitors are more frequently being used in FLT3-mutated AML, the role of stem cell transplant needs to be better defined. In the RATIFY trial, patients who received midostaurin with induction/consolidation and then underwent subsequent SCT in first remission had the best outcomes, supporting the choice of transplant in the frontline setting once remission was achieved. Of note, midostaurin was recommended to be used as a maintenance for 1 year in this clinical trial preASCT or in patients who did not got to ASCT, but post-ASCT midostaurin maintenance was not part of the RATIFY trial. The duration of midostaurin and other TKI usage (such as BCR-ABL inhibitors in Ph+ ALL) is not yet definitely defined, both post-transplant and in patients who do not go for transplant. It is possible that longer FLT3 inhibitor maintenance beyond 1 year may change the prognostic significance of FLT3-ITD mutation and potentially even the need for SCT (especially is specific molecularly select, favorable groups) and this is being evaluated in ongoing Phase II studies.
Additional expected developments in the usage of midostaurin, particularly as we better understand the underlying mechanisms of resistance to TKIs in general and FLT3 inhibitors specifically, are rationally designed treatment combinations with agents targeting known resistance pathways. One expected combination is with Bcl-2 inhibitor venetoclax and HMA or LDAC (triple combination). As was discussed earlier, up-regulation of anti-apoptotic and down-regulation of pro-apoptotic proteins is a known major mechanism of resistance to FLT3 inhibitor therapy that may be circumvented with the addition of venetoclax. One report showed that FLT3-ITD cells have higher level of Bcl-2 compared to FLT3 wildtype cells.80 In addition, venetoclax primary and secondary resistance appears to be driven by FLT3-ITD mutation.81 MCL-1 has been reported to be an essential effector of FLT3-ITD-mediated drug resistance.82 A number of FLT3 inhibitors down-regulate MCL1, and may thus reduce resistance to BCL2 inhibitors.83 Hence, in addition to synergism with HMAs, the triple combination of midostaurin plus venetoclax with HMA (or LDAC) in FLT3-mutated AML is rational and its evaluation in clinical trial is anticipated in the near future.
In summary, a number of active trials combining midostaurin with other agents are ongoing (Table 1). We hope to identify and further refine in the coming years the patient subgroups with the greatest potential benefit from midostaurin, disease biomarkers of response and resistance, the optimal time of usage, and rationally designed treatment combinations that will result in improved efficacy and survival.
Disclosure
MA and HAA have no conflicts of interest in this work. TK has received honoraria and consulting from Novartis. FRK has received research support from Astellas, and honoraria from Novartis and Astellas. ND has received honoraria and research support from Jazz, Pfizer, Daiichi-Sankyo, BMS, Astellas, Novartis, Celgene, Abbvie, Daiichi-Sankyo and Genentech. The authors report no other conflicts of interest in this work.
References
1. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid Leukemia. N Engl J Med. 2016;374(23):2209–2221. doi:10.1056/NEJMoa1516192
2. Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447. doi:10.1182/blood-2016-08-733196
3. Available from: https://www.nccn.org/professionals/physician_gls/pdf/aml.pdf.
4. Kottaridis PD, Gale RE, Frew ME, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98(6):1752–1759. doi:10.1182/blood.v98.6.1752
5. Levis M. FLT3 mutations in acute myeloid leukemia: what is the best approach in 2013? Hematology Am Soc Hematol Educ Program. 2013;2013:220–226. doi:10.1182/asheducation-2013.1.220
6. Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99(12):4326–4335. doi:10.1182/blood.v99.12.4326
7. Rosnet O, Birnbaum D. Hematopoietic receptors of class III receptor-type tyrosine kinases. Crit Rev Oncog. 1993;4(6):595–613.
8. Gabbianelli M, Pelosi E, Montesoro E, et al. Multi-level effects of flt3 ligand on human hematopoiesis: expansion of putative stem cells and proliferation of granulomonocytic progenitors/monocytic precursors. Blood. 1995;86(5):1661–1670.
9. Rusten LS, Lyman SD, Veiby OP, Jacobsen SE. The FLT3 ligand is a direct and potent stimulator of the growth of primitive and committed human CD34+ bone marrow progenitor cells in vitro. Blood. 1996;87(4):1317–1325.
10. Shah AJ, Smogorzewska EM, Hannum C, Crooks GM. Flt3 ligand induces proliferation of quiescent human bone marrow CD34+CD38- cells and maintains progenitor cells in vitro. Blood. 1996;87(9):3563–3570.
11. Frohling S, Scholl C, Levine RL, et al. Identification of driver and passenger mutations of FLT3 by high-throughput DNA sequence analysis and functional assessment of candidate alleles. Cancer Cell. 2007;12(6):501–513. doi:10.1016/j.ccr.2007.11.005
12. Nakao M, Yokota S, Iwai T, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10(12):1911–1918.
13. Brandts CH, Sargin B, Rode M, et al. Constitutive activation of Akt by Flt3 internal tandem duplications is necessary for increased survival, proliferation, and myeloid transformation. Cancer Res. 2005;65(21):9643–9650. doi:10.1158/0008-5472.CAN-05-0422
14. Mizuki M, Fenski R, Halfter H, et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood. 2000;96(12):3907–3914.
15. Yamamoto Y, Kiyoi H, Nakano Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97(8):2434–2439. doi:10.1182/blood.v97.8.2434
16. Chou SC, Tang JL, Hou HA, et al. Prognostic implication of gene mutations on overall survival in the adult acute myeloid leukemia patients receiving or not receiving allogeneic hematopoietic stem cell transplantations. Leuk Res. 2014;38(11):1278–1284. doi:10.1016/j.leukres.2014.08.012
17. Ravandi F, Kantarjian H, Faderl S, et al. Outcome of patients with FLT3-mutated acute myeloid leukemia in first relapse. Leuk Res. 2010;34(6):752–756. doi:10.1016/j.leukres.2009.10.001
18. Bienz M, Ludwig M, Leibundgut EO, et al. Risk assessment in patients with acute myeloid leukemia and a normal karyotype. Clin Cancer Res. 2005;11(4):1416–1424. doi:10.1158/1078-0432.CCR-04-1552
19. Levis M, Small D. FLT3: ITDoes matter in leukemia. Leukemia. 2003;17(9):1738–1752. doi:10.1038/sj.leu.2403099
20. Levis M. FLT3 as a marker of minimal residual disease: time to re-think? Am J Hematol. 2017;92(4):329–330. doi:10.1002/ajh.24667
21. Levis MJ, Perl AE, Altman JK, et al. A next-generation sequencing-based assay for minimal residual disease assessment in AML patients with FLT3-ITD mutations. Blood Adv. 2018;2(8):825–831. doi:10.1182/bloodadvances.2018015925
22. Levis M, Brown P, Smith BD, et al. Plasma inhibitory activity (PIA): a pharmacodynamic assay reveals insights into the basis for cytotoxic response to FLT3 inhibitors. Blood. 2006;108(10):3477–3483. doi:10.1182/blood-2006-04-015743
23. Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33(2):299–312. doi:10.1038/s41375-018-0357-9
24. Thomas CM, Campbell P. FLT3 inhibitors in acute myeloid leukemia: current and future. J Oncol Pharm Pract. 2019;25(1):163–171. doi:10.1177/1078155218802620
25. O’Farrell AM, Foran JM, Fiedler W, et al. An innovative phase I clinical study demonstrates inhibition of FLT3 phosphorylation by SU11248 in acute myeloid leukemia patients. Clin Cancer Res. 2003;9(15):5465–5476.
26. Shah NP, Talpaz M, Deininger MW, et al. Ponatinib in patients with refractory acute myeloid leukaemia: findings from a phase 1 study. Br J Haematol. 2013;162(4):548–552. doi:10.1111/bjh.12382
27. Fiedler W, Serve H, Dohner H, et al. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood. 2005;105(3):986–993. doi:10.1182/blood-2004-05-1846
28. Burchert A. Sorafenib as maintenance therapy post allogeneic stem cell transplantation for FLT3-ITD positive AML: results from the randomized, double-blind, placebo-controlled multicentre sormain trial. Blood. 2018;132:661.
29. Antar A, Kharfan-Dabaja MA, Mahfouz R, Bazarbachi A. Sorafenib maintenance appears safe and improves clinical outcomes in FLT3-ITD acute myeloid leukemia after allogeneic hematopoietic cell transplantation. Clin Lymphoma Myeloma Leuk. 2015;15(5):298–302. doi:10.1016/j.clml.2014.12.005
30. Ravandi F, Cortes JE, Jones D, et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol. 2010;28(11):1856–1862. doi:10.1200/JCO.2009.25.4888
31. Rollig C, Serve H, Huttmann A, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015;16(16):1691–1699. doi:10.1016/S1470-2045(15)00362-9
32. Perl A. Gilteritinib significantly prolongs overall survival in patients with FLT3-mutated (FLT3mut+) relapsed/refractory (R/R) acute myeloid leukemia (AML): results from the phase III ADMIRAL trial.
33. Fabbro D, Ruetz S, Bodis S, et al. PKC412 – a protein kinase inhibitor with a broad therapeutic potential. Anticancer Drug Des. 2000;15(1):17–28.
34. Meyer T, Regenass U, Fabbro D, et al. A derivative of staurosporine (CGP 41 251) shows selectivity for protein kinase C inhibition and in vitro anti-proliferative as well as in vivo anti-tumor activity. Int J Cancer. 1989;43(5):851–856. doi:10.1002/ijc.2910430519
35. Bahlis NJ, Miao Y, Koc ON, Lee K, Boise LH, Gerson SL. N-benzoylstaurosporine (PKC412) inhibits Akt kinase inducing apoptosis in multiple myeloma cells. Leuk Lymphoma. 2005;46(6):899–908. doi:10.1080/10428190500080595
36. Ganeshaguru K, Wickremasinghe RG, Jones DT, et al. Actions of the selective protein kinase C inhibitor PKC412 on B-chronic lymphocytic leukemia cells in vitro. Haematologica. 2002;87(2):167–176.
37. Nakamura K, Yoshikawa N, Yamaguchi Y, Kagota S, Shinozuka K, Kunitomo M. Effect of PKC412, an inhibitor of protein kinase C, on spontaneous metastatic model mice. Anticancer Res. 2003;23(2B):1395–1399.
38. Yoshikawa N, Nakamura K, Yamaguchi Y, Kagota S, Shinozuka K, Kunitomo M. Effect of PKC412, a selective inhibitor of protein kinase C, on lung metastasis in mice injected with B16 melanoma cells. Life Sci. 2003;72(12):1377–1387. doi:10.1016/s0024-3205(02)02407-4
39. Propper DJ, McDonald AC, Man A, et al. Phase I and pharmacokinetic study of PKC412, an inhibitor of protein kinase C. J Clin Oncol. 2001;19(5):1485–1492. doi:10.1200/JCO.2001.19.5.1485
40. Virchis A, Ganeshaguru K, Hart S, et al. A novel treatment approach for low grade lymphoproliferative disorders using PKC412 (CGP41251), an inhibitor of protein kinase C. Hematol J. 2002;3(3):131–136. doi:10.1038/sj.thj.6200165
41. Eder JP
42. Monnerat C, Henriksson R, Le Chevalier T, et al. Phase I study of PKC412 (N-benzoyl-staurosporine), a novel oral protein kinase C inhibitor, combined with gemcitabine and cisplatin in patients with non-small-cell lung cancer. Ann Oncol. 2004;15(2):316–323. doi:10.1093/annonc/mdh052
43. Millward MJ, House C, Bowtell D, et al. The multikinase inhibitor midostaurin (PKC412A) lacks activity in metastatic melanoma: a phase IIA clinical and biologic study. Br J Cancer. 2006;95(7):829–834. doi:10.1038/sj.bjc.6603331
44. Saishin Y, Saishin Y, Takahashi K, Seo MS, Melia M, Campochiaro PA. The kinase inhibitor PKC412 suppresses epiretinal membrane formation and retinal detachment in mice with proliferative retinopathies. Invest Ophthalmol Vis Sci. 2003;44(8):3656–3662. doi:10.1167/iovs.02-1143
45. Saishin Y, Silva RL, Saishin Y, et al. Periocular injection of microspheres containing PKC412 inhibits choroidal neovascularization in a porcine model. Invest Ophthalmol Vis Sci. 2003;44(11):4989–4993. doi:10.1167/iovs.03-0600
46. Campochiaro PA, Group CPS. Reduction of diabetic macular edema by oral administration of the kinase inhibitor PKC412. Invest Ophthalmol Vis Sci. 2004;45(3):922–931. doi:10.1167/iovs.03-0955
47. Weisberg E, Boulton C, Kelly LM, et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. 2002;1(5):433–443.
48. Grundler R, Thiede C, Miething C, Steudel C, Peschel C, Duyster J. Sensitivity toward tyrosine kinase inhibitors varies between different activating mutations of the FLT3 receptor. Blood. 2003;102(2):646–651. doi:10.1182/blood-2002-11-3441
49. Bali P, George P, Cohen P, et al. Superior activity of the combination of histone deacetylase inhibitor LAQ824 and the FLT-3 kinase inhibitor PKC412 against human acute myelogenous leukemia cells with mutant FLT-3. Clin Cancer Res. 2004;10(15):4991–4997. doi:10.1158/1078-0432.CCR-04-0210
50. Furukawa Y, Vu HA, Akutsu M, et al. Divergent cytotoxic effects of PKC412 in combination with conventional antileukemic agents in FLT3 mutation-positive versus -negative leukemia cell lines. Leukemia. 2007;21(5):1005–1014. doi:10.1038/sj.leu.2404593
51. Mollgard L, Deneberg S, Nahi H, et al. The FLT3 inhibitor PKC412 in combination with cytostatic drugs in vitro in acute myeloid leukemia. Cancer Chemother Pharmacol. 2008;62(3):439–448. doi:10.1007/s00280-007-0623-4
52. Odgerel T, Kikuchi J, Wada T, et al. The FLT3 inhibitor PKC412 exerts differential cell cycle effects on leukemic cells depending on the presence of FLT3 mutations. Oncogene. 2008;27(22):3102–3110. doi:10.1038/sj.onc.1210980
53. Stone RM, DeAngelo DJ, Klimek V, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105(1):54–60. doi:10.1182/blood-2004-03-0891
54. Fischer T, Stone RM, Deangelo DJ, et al. Phase IIB trial of oral midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28(28):4339–4345. doi:10.1200/JCO.2010.28.9678
55. Strati P, Kantarjian H, Ravandi F, et al. Phase I/II trial of the combination of midostaurin (PKC412) and 5-azacytidine for patients with acute myeloid leukemia and myelodysplastic syndrome. Am J Hematol. 2015;90(4):276–281. doi:10.1002/ajh.23924
56. Stone RM, Fischer T, Paquette R, et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia. 2012;26(9):2061–2068. doi:10.1038/leu.2012.115
57. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454–464. doi:10.1056/NEJMoa1614359
58. Stone RM, Manley PW, Larson RA, Capdeville R. Midostaurin: its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv. 2018;2(4):444–453. doi:10.1182/bloodadvances.2017011080
59. Larson RA, Mandrekar SJ, Sanford BL, et al. An analysis of maintenance therapy and post-midostaurin outcomes in the international prospective randomized, placebo-controlled, double-blind trial (CALGB 10603/RATIFY [Alliance]) for newly diagnosed Acute Myeloid Leukemia (AML) Patients with FLT3 mutations. Blood. 2017;130:145.
60. Schlenk RF, Weber D, Fiedler W, et al. Midostaurin added to chemotherapy and continued single-agent maintenance therapy in acute myeloid leukemia with FLT3-ITD. Blood. 2019;133(8):840–851. doi:10.1182/blood-2018-08-869453
61. Kim ES. Midostaurin: first global approval. Drugs. 2017;77(11):1251–1259. doi:10.1007/s40265-017-0779-0
62. Dutreix C, Munarini F, Lorenzo S, Roesel J, Wang Y. Investigation into CYP3A4-mediated drug-drug interactions on midostaurin in healthy volunteers. Cancer Chemother Pharmacol. 2013;72(6):1223–1234. doi:10.1007/s00280-013-2287-6
63. Outas T, Duval V, Sinclair K, Berkowitz N. Concomitant use of midostaurin with strong CYP3A4 inhibitors: an analysis from the ratify trial. Blood. 2017;130:3814.
64. Breitenbuecher F, Markova B, Kasper S, et al. A novel molecular mechanism of primary resistance to FLT3-kinase inhibitors in AML. Blood. 2009;113(17):4063–4073. doi:10.1182/blood-2007-11-126664
65. Stolzel F, Steudel C, Oelschlagel U, et al. Mechanisms of resistance against PKC412 in resistant FLT3-ITD positive human acute myeloid leukemia cells. Ann Hematol. 2010;89(7):653–662. doi:10.1007/s00277-009-0889-1
66. Heidel F, Solem FK, Breitenbuecher F, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107(1):293–300. doi:10.1182/blood-2005-06-2469
67. Pratz KW, Sato T, Murphy KM, Stine A, Rajkhowa T, Levis M. FLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood. 2010;115(7):1425–1432. doi:10.1182/blood-2009-09-242859
68. Perl AE. Gilteritinib significantly prolongs overall survival in patients with FLT3-mutated (FLT3mut+) relapsed/refractory (R/R) acute myeloid leukemia (AML): results from the phase III ADMIRAL trial.
69. Cortes JE, Khaled SK, Martinelli G, et al. Efficacy and safety of single-agent Quizartinib (Q), a potent and selective FLT3 Inhibitor (FLT3i), in patients (pts) with FLT3-Internal Tandem Duplication (FLT3-ITD)-mutated Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML) enrolled in the global, phase 3, randomized controlled quantum-R trial. Blood. 2018;132(Suppl 1):563.
70. Cortes J, Perl AE, Döhner H, et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: an open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2018;19(7):889–903. doi:10.1016/S1470-2045(18)30240-7
71. Smith CC, Lasater EA, Lin KC, et al. Crenolanib is a selective type I pan-FLT3 inhibitor. Proc Natl Acad Sci U S A. 2014;111(14):5319–5324. doi:10.1073/pnas.1320661111
72. Randhawa JK, Kantarjian HM, Borthakur G, et al. Results of a phase II study of crenolanib in relapsed/refractory acute myeloid leukemia Patients (Pts) with activating FLT3 mutations. Blood. 2014;124(21):389.
73. Altman JK, Foran JM, Pratz KW, Trone D, Cortes JE, Tallman MS. Phase 1 study of quizartinib in combination with induction and consolidation chemotherapy in patients with newly diagnosed acute myeloid leukemia. Am J Hematol. 2018;93(2):213–221. doi:10.1002/ajh.24974
74. Pratz KW, Cherry M, Altman JK, et al. Updated results from a phase 1 study of gilteritinib in combination with induction and consolidation chemotherapy in subjects with newly diagnosed Acute Myeloid Leukemia (AML). Blood. 2018;132(Suppl 1):564.
75. Wang ES, Stone RM, Tallman MS, Walter RB, Eckardt JR, Collins R. Crenolanib, a type I FLT3 TKI, can be safely combined with cytarabine and anthracycline induction chemotherapy and results in high response rates in patients with newly diagnosed FLT3 Mutant Acute Myeloid Leukemia (AML). Blood. 2016;128(22):1071.
76. Williams CB, Kambhampati S, Fiskus W, et al. Preclinical and phase I results of decitabine in combination with midostaurin (PKC412) for newly diagnosed elderly or relapsed/refractory adult patients with acute myeloid leukemia. Pharmacotherapy. 2013;33(12):1341–1352. doi:10.1002/phar.1316
77. Chen W, Xie H, Wang H, et al. Prognostic significance of KIT mutations in core-binding factor acute myeloid leukemia: a systematic review and meta-analysis. PLoS One. 2016;11(1):e0146614. doi:10.1371/journal.pone.0146614
78. Qin Y-Z, Zhu H, Jiang Q, et al. Prevalence and prognostic significance of c-KIT mutations in core binding factor acute myeloid leukemia: a comprehensive large-scale study from a single chinese center. Blood. 2014;124(21):1000.
79. Maziarz RTT, Patnaik MM, Scott BL, et al. Radius: a phase 2 randomized trial investigating standard of care ± midostaurin after allogeneic stem cell transplant in FLT3-ITD-mutated AML. Blood. 2018;132(Suppl 1):662. doi:10.1182/blood-2018-05-846428
80. Sobhanifar MA, Mashkani B, Saadatmandzadeh M, Sadeghnia HR, Mousavi SH. Induction of cytotoxicity and apoptosis in FLT3 mutant expressing cells using novel pyrimido cyanoacrylates and quinoline derivatives. Biomed Pharmacother. 2018;108:893–905. doi:10.1016/j.biopha.2018.09.001
81. Chyla B, Daver N, Doyle K, et al. Genetic biomarkers of sensitivity and resistance to venetoclax monotherapy in patients with relapsed acute myeloid leukemia. Am J Hematol. 2018. doi:10.1002/ajh.25146
82. Kasper S, Breitenbuecher F, Heidel F, et al. Targeting MCL-1 sensitizes FLT3-ITD-positive leukemias to cytotoxic therapies. Blood Cancer J. 2012;2(3):e60. doi:10.1038/bcj.2012.5
83. Kohl TM, Hellinger C, Ahmed F, et al. BH3 mimetic ABT-737 neutralizes resistance to FLT3 inhibitor treatment mediated by FLT3-independent expression of BCL2 in primary AML blasts. Leukemia. 2007;21(8):1763–1772. doi:10.1038/sj.leu.2404776
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.