")
Back to Journals » OncoTargets and Therapy » Volume 7
Midazolam induces apoptosis in MA-10 mouse Leydig tumor cells through caspase activation and the involvement of MAPK signaling pathway
Authors So EC , Lin Y, Tseng C, Pan B, Cheng K , Wong K, Hao L, Wang Y, Huang B , Huang M
Received 16 October 2013
Accepted for publication 16 December 2013
Published 13 February 2014 Volume 2014:7 Pages 211—221
DOI https://doi.org/10.2147/OTT.S56084
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Edmund Cheung So,1,2 Yu-Xuan Lin,3 Chi Hao Tseng,1 Bo-Syong Pan,3 Ka-Shun Cheng,2 Kar-Lok Wong,2 Lyh-Jyh Hao,4 Yang-Kao Wang,5 Bu-Miin Huang2
1Department of Anesthesia, Tainan Municipal An Nan Hospital, China Medical University, Tainan, Taiwan; 2Department of Anesthesia, China Medical University, Taichung, Taiwan; 3Department of Cell Biology and Anatomy, National Cheng Kung University, Tainan, Taiwan; 4Department of Internal Medicine, Division of Endocrinology and Metabolism, Kaohsiung Veteran General Hospital Tainan Branch Tainan, Taiwan; 5Graduate Institute of Biomedical Materials and Tissue Engineering, Taipei Medical University, Taipei, Taiwan
Purpose: The present study aims to investigate how midazolam, a sedative drug for clinical use with cytotoxicity on neuronal and peripheral tissues, induced apoptosis in MA-10 mouse Leydig tumor cells.
Methods: The apoptotic effect and underlying mechanism of midazolam to MA-10 cells were investigated by flow cytometry assay and Western blotting methods.
Results: Data showed that midazolam induced the accumulation of the MA-10 cell population in the sub-G1 phase and a reduction in the G2/M phase in a time- and dose-dependent manner, suggesting an apoptotic phenomenon. Midazolam could also induce the activation of caspase-8, -9, and -3 and poly (ADP-ribose) polymerase proteins. There were no changes in the levels of Bax and cytochrome-c, whereas Bid was significantly decreased after midazolam treatment. Moreover, midazolam decreased both pAkt and Akt expression. In addition, midazolam stimulated the phosphorylation of p38 and c-Jun NH2-terminal kinase but not extracellular signal-regulated kinase.
Conclusion: Midazolam could induce MA-10 cell apoptosis through the activation of caspase cascade, the inhibition of pAkt pathway, and the induction of p38 and c-Jun NH2-terminal kinase pathways.
Keywords: midazolam, apoptosis, MA-10 cell, caspase, Akt, MAPKs
Introduction
Midazolam (Dormicum®; F. Hoffmann-La Roche Ltd, Basel, Switzerland), a benzodiazepine-derivative drug, has powerful anxiolytic, amnestic, hypnotic, and sedative properties by modulating the γ-aminobutyric acid (GABAA) receptor in the central nervous system.1,2 The putative receptor of midazolam, the peripheral-type benzodiazepine receptor (PBR), as a small 18 kDa protein, is organized in clusters of four to six molecules on the outer mitochondrial membrane.3,4 Studies have illustrated that the binding of PBR ligand to PBR results in the cholesterol movement from the outer mitochondrial membrane to the inner mitochondrial membrane, which could stimulate steroidogenesis.5 In fact, we have previously demonstrated that midazolam could significantly stimulate steroidogenesis in MA-10 mouse Leydig tumor cells by activating protein kinase A and protein kinase C pathways with the expression of PBR and steroidogenic acute regulatory proteins.6 Interestingly, we also observed that higher dosage with long treatment of midazolam could significantly induce MA-10 cell apoptosis.
Apoptosis is a process of program cell death, and plays an important role in physiological processes such as embryonic development and tissue homeostasis.7,8 Apoptosis can be induced by various stimuli, and two major signaling pathways leading cell apoptosis have been studied intensively: the extrinsic and intrinsic pathways. The extrinsic pathway is initiated through death ligands binding to death receptors, and subsequently activates downstream death-inducing signaling complex (DISC).9–12 DISC then activates caspase-8 and -3 through the cleavage of these enzymes from proenzymes and results in the cleavage of poly (ADP-ribose) polymerase (PARP), which induces apoptosis.11,12 In the other way, the intrinsic pathway is initiated by mitochondrial damage where it releases cytochrome-c and activates caspase-9 to associate with Apaf-1 to form apoptosome, and activates caspase-3 to induce apoptosis.10,13 Extensive evidence indicates that during apoptosis, mitochondrial outer membrane becomes permeable, and this permeability transition of mitochondrial membrane is regulated by the Bcl-2 family.14 The Bcl-2 family includes two groups, antiapoptotic and proapoptotic proteins, which share one or more homologous domains called BH domains. The antiapoptotic family members include Bcl-xl, Mcl-1, and Bcl-2, which contain BH1 to BH4 domains. The proapoptotic family members, such as Bax and Bak, are redundant promoters of cell death.15 The BH3-only proteins, such as Bid, however, are usually kept inactivated by different mechanisms, and these proteins are activated to function as effectors of apoptosis upon various death stimuli.16,17 The activation of caspase cascade is required in both extrinsic and intrinsic pathways.
Besides caspase cascades, mitogen-activated protein kinases (MAPKs) are also involved in apoptosis regulation.18 MAPKs consist of three family members: extracellular signal-regulated kinase (ERK), c-Jun NH2-terminal kinase (JNK), and p38 proteins.18 Studies have reported that stress signals can activate the SAPK/JNK protein kinases to mediate cellular steps in apoptosis on some cell types.18,19 It has been shown that ERK is responsive to growth stimuli as the important signal for antiapoptosis.18,19 However, the involvement of p38 in apoptosis is diverse. Phosphorylation of p38 can be initiated by MKK3 and MKK6 at threonine and tyrosine regions, which can control many transcriptional factors and kinases to enhance cell survival or prompt apoptosis.18,19 In fact, studies have also shown that the PI3K/Akt/mTOR signaling pathway could promote cell growth and survival.20 Akt, a serine–threonine kinase that is directly activated in response to PI3K, is a major effector of PI3K and leads to increased cellular growth and survival in cancers.21 Accordingly, caspase and Akt and MAPKs pathways may play important roles in apoptosis of tumor cells activated by chemotherapy agents.
There are reports indicating that general anesthetic drugs could regulate apoptosis in the developing brain in rats and rabbits.22,23 We have previously demonstrated that midazolam could induce apoptosis in MA-10 mouse Leydig tumor cells but not in normal mouse Leydig cells.6 In the present study, we investigated the mechanism of midazolam-induced MA-10 Leydig tumor cell apoptosis. In fact, this study first demonstrated the cytotoxic/apoptotic effect of midazolam in the reproductive system.
Materials and methods
Chemicals
Midazolam (Dormicum) was purchased from Roche Products Ltd (Auckland, New Zealand). Waymouth MB 752/1 medium, fetal bovine serum, Dulbecco’s phosphate buffered saline (PBS), and lyophilized trypsin-ethylenediaminetetraacetic acid were purchased from Life Technologies (Carlsbad, CA, USA). Bovine serum albumin, propidium iodide (PI), and ribonuclease A were purchased from Sigma-Aldrich (St Louis, MO, USA). Tissue culture grade sodium bicarbonate (NaHCO3), sodium carbonate (Na2CO3), sodium chloride (NaCl), sodium dihydrogen phosphate (NaH2PO4), and potassium chloride (KCl) were purchased from Riedel-deHaen (Seelze, Germany). Sodium hydroxide (NaOH), hydrochloric acid (HCl), sodium dodecyl sulfate (SDS), sucrose, and Tween-20® were purchased from Merck KGaA (Darmstadt, Germany). Tris base was purchased from Calbiochem (San Diego, CA, USA). HEPES was purchased from Mallinckrodt Baker, Inc. (Paris, KY, USA). Acrylamide was purchased from JT Baker (Phillipsburg, NJ, USA). The enhanced chemiluminescence detection kit was purchased from Amersham Biosciences (Piscataway, NJ, USA). Donkey antirabbit immunoglobulin G conjugated with horseradish peroxidase and sheep antimouse immunoglobulin G conjugated with horseradish peroxidase were purchased from Amersham Life Science Inc. (Arlington Heights, IL, USA). Polyclonal antibody against β-actin, cleaved caspase-3, cleaved caspase-8, cleaved caspase-9, phosphor-ERK and phosphor-JNK, and monoclonal antibody against Bid, cytochrome-c, Akt, phosphor-Akt, and phosphor-p38 were purchased from Cell Signaling (Beverly, MA, USA). PARP was purchased from Oncogene (San Diego, CA, USA).
Cell line and cell culture
The MA-10 mouse Leydig tumor cell line was a gift from Dr Mario Ascoli (University of Iowa, Iowa City, IA, USA), and was maintained in Waymouth medium (Life Technologies) containing 15% fetal bovine serum and incubated in a humidified atmosphere containing 95% air and 5% CO2 at 37°C. Cells were starved for 24 hours to be synchronized before the incubations of midazolam for various experiments.
Flow cytometry analysis
To determine whether midazolam could induce apoptosis in MA-10 cells, different phase distribution of the cell cycle was assessed by flow cytometry analysis using PI staining. MA-10 cells (5 × 105) were seeded in a 6 cm dish (Techno Plastic Products AG, Trasadingen, Switzerland) with 3 mL serum-containing medium. After 70%–80% confluence, cells were treated with serum-free medium containing various concentrations of midazolam for 1 hour, 3 hours, 6 hours, 12 hours, and 24 hours, respectively. Midazolam-treated cells were harvested by trypsinization, washed with PBS, and fixed in 75% ethanol for 2 hours at −20°C. After fixation, cells were washed with cold PBS, spun down, mixed with 100 μg/mL RNase, and stained with PI (40 μg/mL). The stained cells were analyzed using a fluorescence activated cell sorter (FACScan, Becton-Dickinson, Mountain View, CA, USA) at λ=488 nm using Cell-Quest™ software (Becton-Dickinson). The DNA content distribution of normal growing cells is characterized by two peaks, G1/G0 and G2/M phases. The hypodiploid cells were defined as apoptotic cells, where hypoploid DNA content represents DNA fragmentation.
Immunoblotting analysis
Cells (3 × 105) were seeded in a six-well plate. After treatment, cells were rinsed with cold PBS and then harvested by 40 μL lysis buffer (50 mM Tris-base, 150 mM NaCl, 1% NP40, 0.1% SDS, 0.5% deoxychloride acid, and 1 mM phenylmethylsulfonyl fluoride). Cell lysate was centrifuged at 32,000 rpm for 12 minutes at 4°C. The pellet was collected by 20 μL lysis buffer and was centrifuged again at 12,000× g. The supernatant, which contained total protein, was collected and stored at −20°C. Protein concentration of the cell lysate was determined by the Lowry method.24 Lysate (30~50 μg) was separated in 12% SDS–polyacrylamide gel and electrophoretically transferred to a polyvinylidene difluoride membrane. The membranes were blocked with 5% nonfat milk, washed by Tris-buffered saline and Tween-20®, and subsequently incubated with primary antibodies against cleaved caspase-3, cleaved caspase-8, cleaved caspase-9, cleaved PARP, Bax, cytochrome-c, Akt, phosphor-Akt, phosphor-ERK, phosphor-p38, and phosphor-JNK. After washing, the membrane was incubated with horseradish peroxidase-conjugated sheep antimouse antibody or donkey antirabbit antibody, and the immunocomplex was then visualized by an enhanced chemiluminescence detection kit. The optical density of each protein band was quantified by a Quantity One (PDI, Huntington Station, NY, USA) computer-assisted image analysis system. Due to the huge number of samples, two gels were exploited to hold 16 protein samples (one gel for eight samples) for electrophoresis, simultaneously. The amount of β-actin in each lane was performed as an internal control to normalize the expression of each protein.
Statistics
Each result represents mean ± standard error of the mean (SEM) of at least three separate experiments. Statistically significant differences between treatments and controls were determined by analysis of variance and then a least significant difference comparison procedure. Statistical significance was set at P<0.05.
Results
Midazolam induced apoptosis in MA-10 mouse Leydig tumor cells
In the present study, MA-10 cells were treated without or with midazolam (0 μM, 6 μM, 30 μM, and 150 μM) for different time points, and the cell cycle distribution was then analyzed by flow cytometry to confirm the apoptotic phenomenon. Results showed that there was no difference in sub-G1 or G2/M phase between control and 6 μM and 30 μM midazolam treatments before 12 hours (Figure 1A–E, P>0.05), whereas the G2/M phase significantly decreased at 6 hours with 150 μM midazolam treatment (Figure 1A–D and F, P<0.05). After 12-hour and 24-hour treatments, the cell population in the sub-G1 phase significantly increased with 150 μM midazolam (Figure 1A–E, P<0.01), whereas the G2/M phase significantly decreased with 150 μM midazolam (Figure 1A–D and F, P<0.001). The increasing sub-G1 phase percentage with the decreasing G2/M phase percentage in 12-hour and 24-hour treatments strongly suggests midazolam-induced apoptosis in MA-10 mouse Leydig tumor cells.
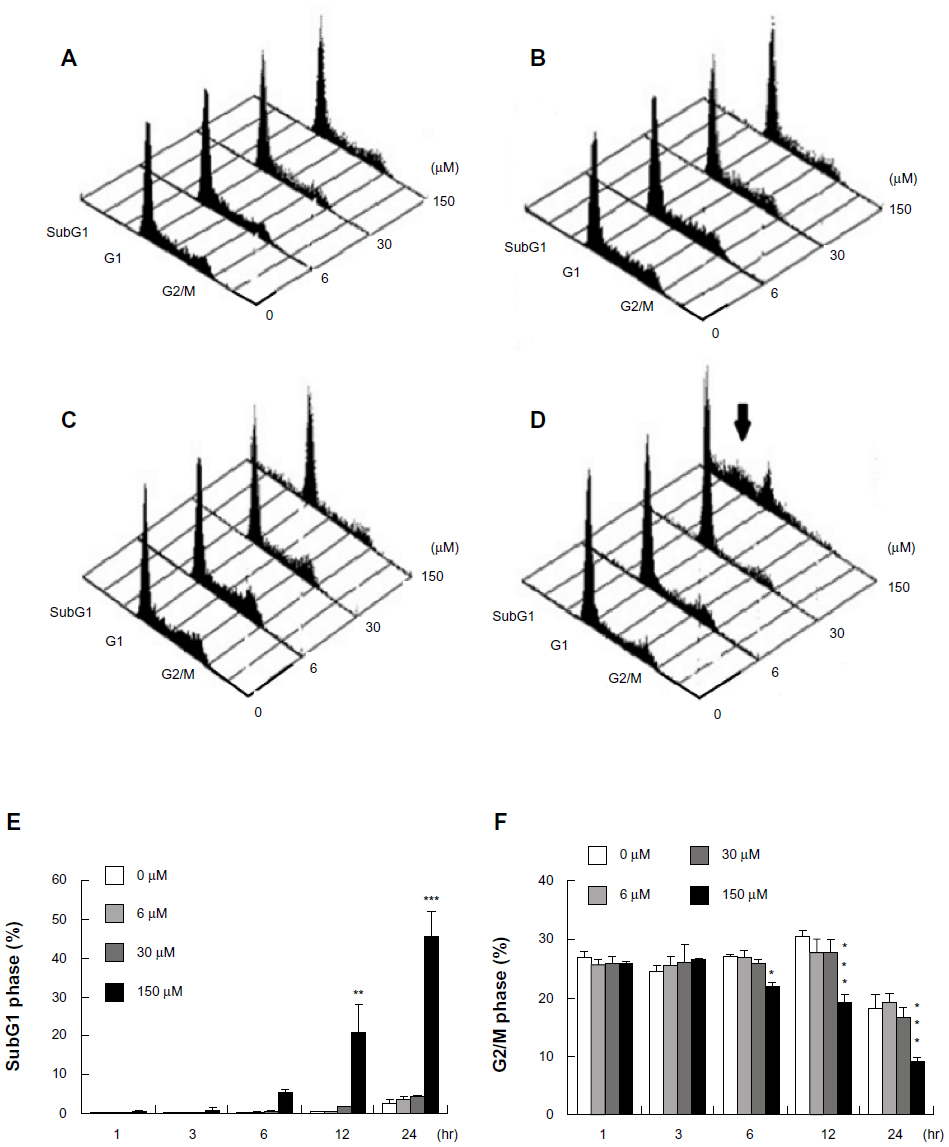 | Figure 1 Midazolam induced apoptosis in MA-10 mouse Leydig tumor cells. MA-10 cells (6 × 105) were treated without (control) or with different concentrations of midazolam (6 μM, 30 μM, and 150 μM) for 3 hours (A), 6 hours (B), 12 hours (C), and 24 hours (D), respectively. Cell population in the subG1 (E) and G2/M (F) phase was then determined by FACScan (Becton-Dickinson, Mountain View, CA, USA) analysis after propidium iodide staining. The three-dimensional graphs of cell number in subG1, G1, and G2/M phases under different dosages and time treatments of midazolam are illustrated. The arrow indicates the subG1 phase. Each point in (E and F) represents the mean ± standard error of the mean of three independent experiments. |
Midazolam activated caspase cascade in MA-10 mouse Leydig tumor cells for apoptosis
To further elucidate the mechanism of apoptosis induced by midazolam in MA-10 cells, caspase cascade was investigated by Western blotting. Results showed that procaspase-8 significantly decreased with 150 μM midazolam after 12-hour and 24-hour treatments (Figure 2A and B, P<0.05), whereas cleaved caspase-8 significantly increased after 12-hour treatment with 150 μM midazolam (Figure 2A and C, P<0.05). In addition, midazolam significantly induced caspase-9 cleavage at a dosage of 150 μM after 24-hour treatment (Figure 2A and D, P<0.05). Moreover, cleaved caspase-3 significantly increased after 12-hour treatment with 150 μM midazolam (Figure 2A and E, P<0.01). It is well known that PARP is one of the downstream substrates of activated caspase-3, which could be cleaved into 85 KDa degraded product of PARP.8 We also examined the cleavage of PARP upon midazolam treatment, and the results showed that cleavage of PARP activated by caspase-3 significantly increased after 6-hour treatment with 150 μM midazolam, and this phenomenon was sustained for 24 hours (Figure 2A, P<0.05). These results suggested that midazolam could stimulate the cleavage of caspase-8, -9, and -3 and PARP to induce apoptosis in MA-10 mouse Leydig tumor cells.
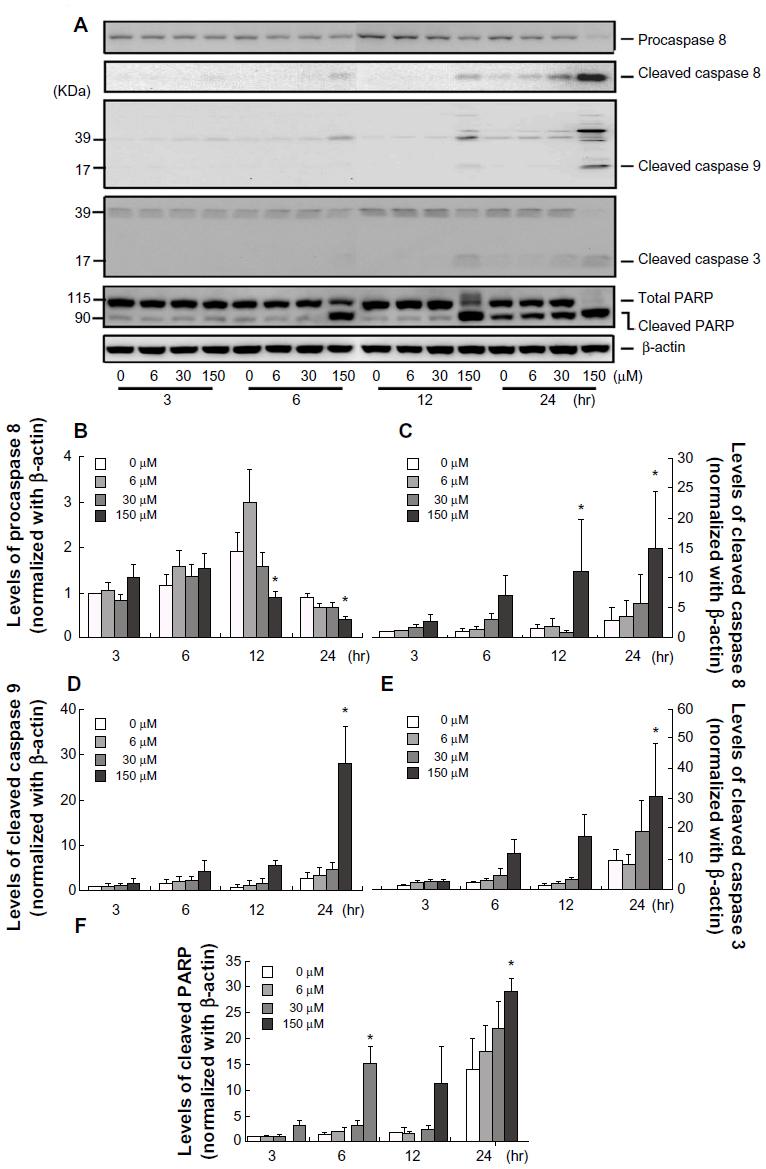 | Figure 2 Midazolam activated caspase cascade in MA-10 mouse Leydig tumor cells. MA-10 cells were treated without or with different concentrations of midazolam (6 μM, 30 μM, and 150 μM) for 3 hours, 6 hours, 12 hours, and 24 hours, respectively. The levels of procaspase-8 protein (57 KDa), cleaved caspase-8 (47 KDa), cleaved caspase-9 (17 KDa), cleaved caspase-3 (19 KDa), and cleaved poly (ADP-ribose) polymerase (PARP) (90 KDa) were analyzed by Western blot. Immunoblots represent the observations from one single experiment repeated three times (A). The integrated optical densities of procaspase-8 (B), cleaved caspase-8 (C), cleaved caspase-9 (D), cleaved caspase-3 (E), and cleaved PARP (F) proteins were analyzed after normalization with β-actin (43 kDa) in each lane. Data in (B–F) represent the mean ± standard error of the mean of three separate experiments. |
Midazolam modulated mitochondria pathways in MA-10 mouse Leydig tumor cells for apoptosis
We next investigated the role of mitochondrial pathway in midazolam-induced MA-10 cell apoptosis. Studies have demonstrated that while cells undergo apoptosis through mitochondrial pathway, activation of the proapoptotic BH3-only protein, Bid, will induce the release of cytochrome-c from mitochondria to form apoptosome with caspase-9, which could link the interaction between intrinsic and extrinsic caspase pathways.25,26 Our results showed that total Bid was significantly decreased by 150 μM midazolam after 24-hour treatment (Figure 3A and B, P<0.05), which implied the increase of t-Bit that might be associated with caspase-9. However, midazolam did not change the cytosolic levels of cytochrome-c, which was expected to be released from mitochondria due to the activation of Bid (Figure 3A and D, P>0.05). Furthermore, we also examined the proapoptotic protein Bax protein, and the results showed that midazolam did not change levels of Bax (Figure 3A and C, P>0.05). These results suggested that midazolam might activate caspase-8 to downregulate Bid without affecting Bax and cytochrome-c expressions in MA-10 mouse Leydig tumor cells.
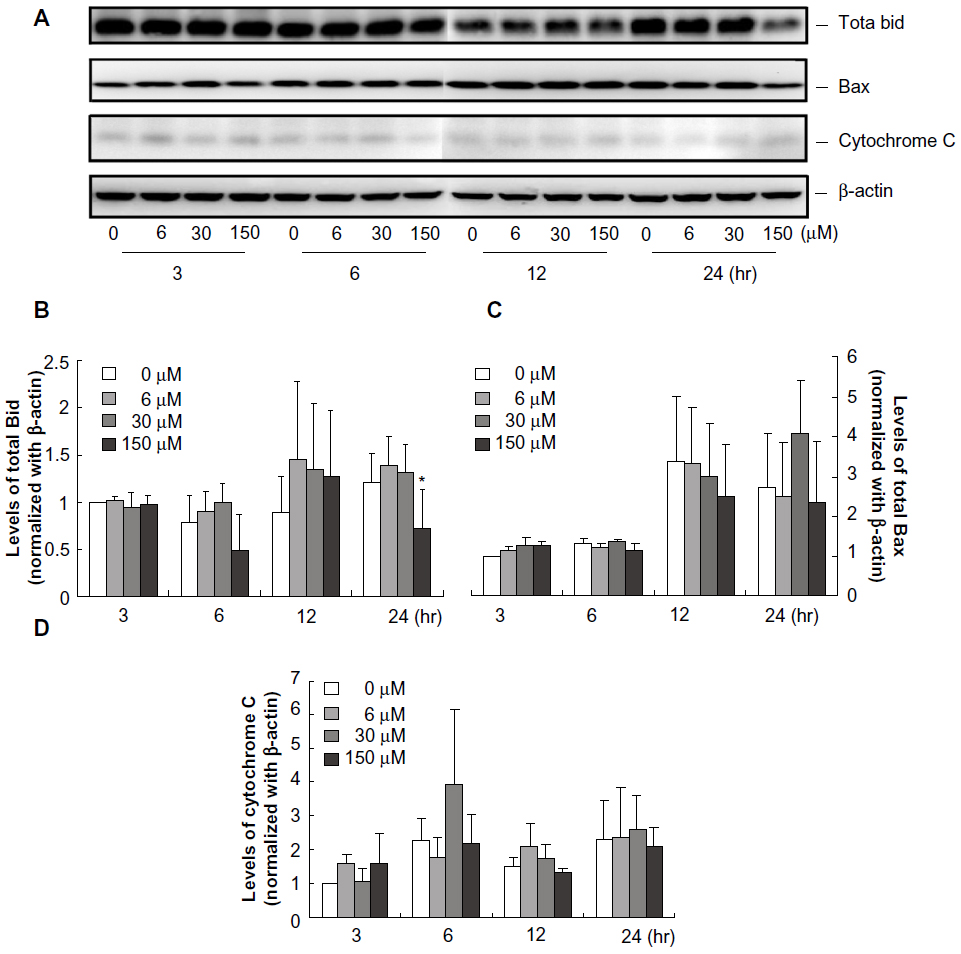 | Figure 3 Midazolam regulated mitochondria pathways in MA-10 mouse Leydig tumor cells. MA-10 cells were treated without or with different concentrations of midazolam (6 μM, 30 μM, and 150 μM) for 3 hours, 6 hours, 12 hours, and 24 hours, respectively. Protein levels of Bid (22 KDa), Bax (20 KDa), and cytochrome-c (14 KDa) were analyzed by Western blot. Immunoblots represent the observations from one single experiment repeated three times (A). The integrated optical densities of Bid (B), Bax (C), and cytochrome-c (D) proteins were analyzed after normalization with β-actin (43 kDa) in each lane. Data in (B–D) represent the mean ± standard error of the mean of three separate experiments. |
Midazolam downregulated Akt pathway in MA-10 mouse Leydig tumor cells for apoptosis
We further investigated whether midazolam would affect the expression of Akt and phosphor-Akt in MA-10 mouse Leydig tumor cells. Results showed that midazolam (6 μM, 30 μM, and 150 μM for 3 hours and 6 hours, respectively) did not have any effect on total Akt protein expressions (Figure 4A and B, P>0.05). However, 150 μM midazolam at 12 hours and 24 hours did significantly reduce total Akt protein levels (Figure 4A and B, P<0.05). In addition, 150 μM midazolam from 6 hours to 24 hours significantly reduced phosphor-Akt (Figure 4A and C, P<0.01). These results suggest that midazolam could downregulate Akt pathway to induce apoptosis in MA-10 mouse Leydig tumor cells.
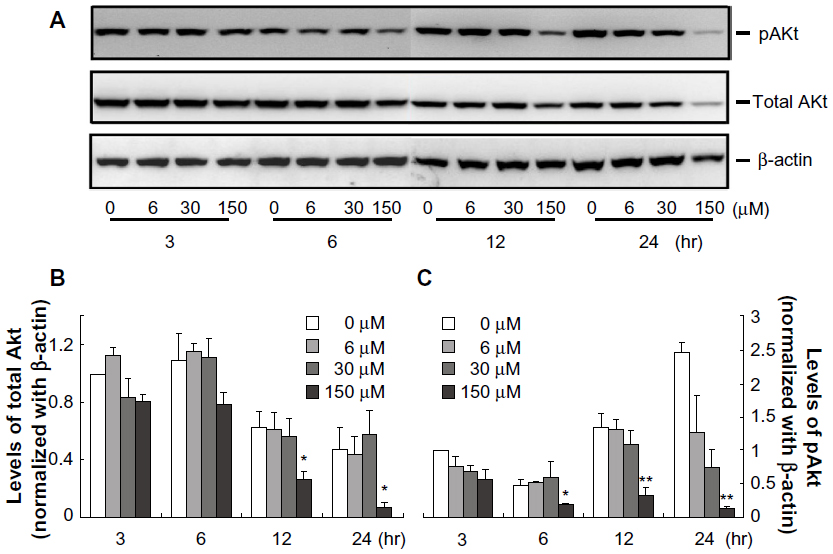 | Figure 4 Midazolam downregulated Akt pathway in MA-10 mouse Leydig tumor cells. MA-10 cells were treated without or with different concentrations of midazolam (6 μM, 30 μM, and 150 μM) for 3 hours, 6 hours, 12 hours, and 24 hours, respectively. The protein levels of total Akt protein (60 KDa) and phosphor-Akt (60 KDa) were analyzed by Western blot. Immunoblots represent the observations from one single experiment repeated three times (A). The integrated optical densities of total Akt protein (B) and phosphor-Akt (C) proteins were analyzed after normalization with β-actin (43 kDa) in each lane. Data in (B and C) represent the mean ± standard error of the mean of three separate experiments. |
Midazolam regulated MAPK pathway in MA-10 mouse Leydig tumor cells for apoptosis
Recent studies have illustrated that MAPKs pathway could affect cell proliferation, differentiation, migration, and apoptosis.18,19,27 Thus, we sought to investigate the possible regulation of MAPKs pathway activated by midazolam in MA-10 cell apoptosis. Data showed that the expressions of phosphor-JNK1 and phosphor-p38 were significantly increased by 150 μM midazolam after 24-hour treatment (Figure 5A, B, and D, P<0.05). However, the protein levels of phosphor-ERK and phosphor-JNK2/3 were not changed by various midazolam treatments (Figure 5A, C, and E, P>0.05). These results suggest that midazolam could activate JNK1 and p38, but not ERK and JNK2/3, to induce MA-10 cell apoptosis.
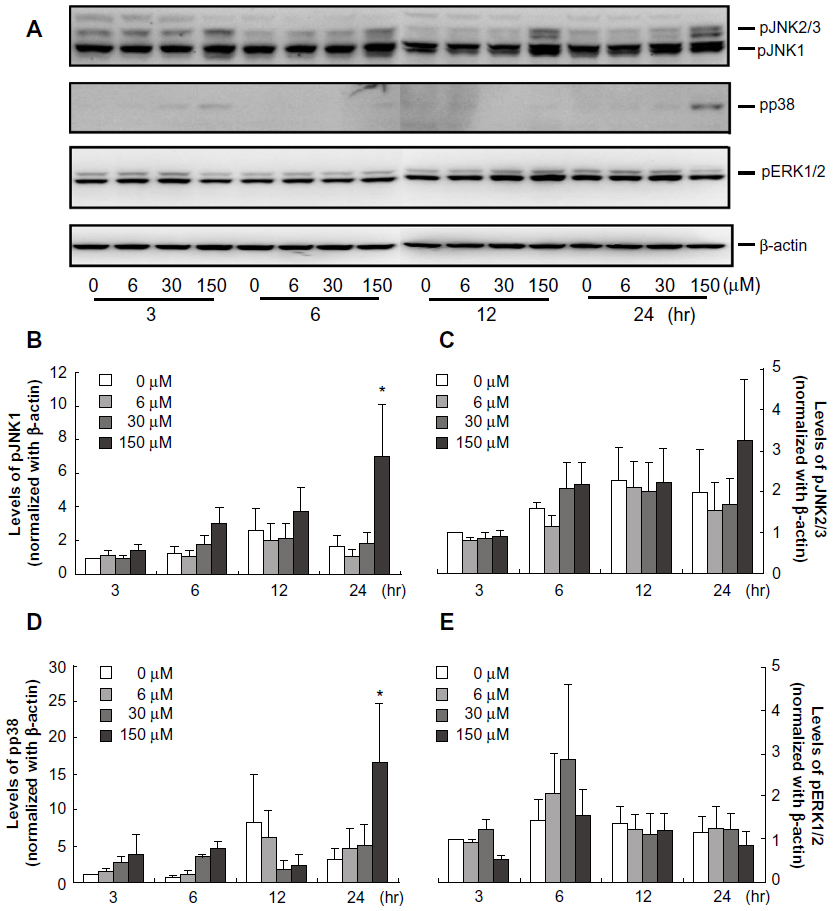 | Figure 5 Midazolam regulated mitogen-activated protein kinase (MAPK) cascade in MA-10 mouse Leydig tumor cells. MA-10 cells were treated without or with different concentrations of midazolam (6 μM, 30 μM, and 150 μM) for 3 hours, 6 hours, 12 hours, and 24 hours, respectively. The levels of phosphor-p38 protein (43 KDa), phosphor-c-Jun NH2-terminal kinase (JNK) (54 KDa and 46 KDa), and phosphor-extracellular signal-regulated kinase (ERK) (42 KDa and 44 KDa) were analyzed by Western blot. Immunoblots represent the observations from one single experiment repeated three times (A). The integrated optical densities of cleaved phosphor-JNK1 (B), phosphor-JNK2/3 (C), phosphor-p38 (D), and phosphor-ERK (E) proteins were analyzed after normalization with β-actin (43 kDa) in each lane. Data in (B–E) represent the mean ± standard error of the mean of three separate experiments. |
Discussion
Early studies on sedative drug effects mainly focus on its clinical dosages and pharmacokinetic properties.28 Previous studies have indicated that midazolam has a possible neurotoxic effect in young animals and rabbits.22,23,29–31 One study did illustrate that midazolam could activate the intrinsic pathway of apoptosis independent of benzodiazepine and death receptor signaling in human lymphoma and neuroblastoma cell lines.31 Our earlier study showed that midazolam had cytotoxic effects on MA-10 cells.6 However, the underlying mechanism remains unknown. In the present study, we observed that midazolam could induce apoptosis in MA-10 cells in a dose- and time-dependent manner through the activation of caspase and MAPKs pathways. Thus, our findings are parallel to other studies,31,32 suggesting a similar mechanism of the cytotoxic side effect of midazolam upon neural, immune, and reproductive cells.
In general, there are extrinsic and intrinsic pathways for cells undergoing apoptosis.5 The extrinsic pathway is initiated through death receptor to activate downstream DISC and caspase-8. In the other way, intrinsic pathway is initiated at the mitochondrial outer membrane permeability to activate caspase-9 through cytochrome-c and Bcl-2 proteins.33 Caspase-8 and -9 will then stimulate caspase-3 cleavage.10,13 In the present study, we found that midazolam could activate extrinsic and intrinsic caspase cascades to induce apoptosis in MA-10 cells. The cleavage of PAPR after midazolam treatment further confirmed this phenomenon. In fact, studies have also illustrated that galectin-1 could simultaneously activate caspase-8 and -9 pathways to induce apoptosis in MA-10 cells and rat isolated Leydig cells.34,35 Thus, our findings, that midazolam induced MA-10 cell apoptosis through the activation of caspase cascades, are parallel to other studies in different cell types.
It has been shown that the induction of caspase-9 could be mediated by converging at the mitochondria, which can be activated by cytochrome-c, Bax, reactive oxygen species, and JNK pathways to induce cell apoptosis. Release of cytochrome-c from the mitochondria to the cytoplasm initiates a caspase-9 cascade.36 Our data showed that the expression of Bax and cytochrome-c did not significantly change with midazolam treatment. Orser et al37 have found that sedative drugs such as midazolam and propofol could bind to different types of GABA receptors to achieve sedative effects. It is possible that midazolam could also bind to GABA receptors to activate specific signal pathway, inducing MA-10 cell apoptosis without activating cytochrome-c release. The report has also shown that factors could induce apoptosis without the existence of Bax.38 Thus, midazolam could induce MA-10 cell apoptosis without the expression of Bax and cytochrome-c. In fact, the increasing trends of Bax expression at 24 hours and cytochrome-c release at 6 hours by 30 μM midazolam treatments could be observed in the present study. Further investigation will be executed to dissect the changes of Bax and cytochrome-c between cytoplasmic and mitochondrial portions under midazolam treatments, which could truly reveal whether or not midazolam could induce both proteins to activate caspase-9 and then apoptosis in MA-10 cells.
A previous study indicated that mitochondrial intrinsic pathway and extrinsic pathway could be linked by Bid, a proapoptotic protein, which could be activated by caspase-8 in the cytoplasm to regulate the mitochondrial membrane potential by forming a complex with Bax.15 In the present study, the amount of total Bid significantly decreased by 150 μM midazolam at 4-hour treatment, which indicates that t-Bit was significantly stimulated. This observation suggests that midazolam might activate caspase-8 and -9 pathways through the linkage of t-Bit. However, our study also illustrated that the level of total Bax did not significantly change after midazolam treatment on MA-10 mouse Leydig tumor cells. This phenomenon that upregulation in t-Bit without significant change of Bax protein will be further investigated to understand the detail mechanism.
It is well documented that induction of cell apoptosis by different cellular stresses is involved with JNK, ERK, and p38 MAPK pathways.18,19,27,39 Mitogen-activated protein kinases are signaling components that are important in converting extracellular stimuli into a wide range of cellular responses.18 It is reported that JNK signaling pathway is essential for neuronal apoptosis in response to excitotoxic stress, but the role of JNK in the apoptotic responses of other cell types is unclear.40 On the other hand, many studies demonstrate that JNK and p38 pathways are activated to promote cell survival.18,19,27,41 In addition, the ERK signaling pathway plays a key role in several steps of tumorigenesis, including cancer cell proliferation, migration, invasion, and apoptosis.18,19 As well, PI3K/Akt pathway has been reported to crosstalk with MAPK signaling pathway by autocrine signaling through Ras for cell survival.42 In fact, a study has demonstrated that constitutive activation of PI3K/Akt could be observed in many cancers, and the targeting of the PI3K/Akt pathway is of great importance in treating prostate cancer.43 In the present study, we observed that midazolam significantly activated p38 and pJNK1. However, midazolam had no effect on ERK activity. Moreover, midazolam significantly decreased the levels of Akt and phosphor-Akt proteins. These results demonstrate that midazolam could upregulate MAPK pathway and downregulate Akt survival signal to induce MA-10 cell apoptosis.
Midazolam is a common sedative used by patients who are on mechanical ventilation in the intensive care unit (ICU) or during procedure sedation.44 In general, dosages used in the ICU are much higher than during anesthesia or procedure sedations. Daily doses as high as 120 mg/day or 8~10 mg/kg/hour have been reported.45,46 High mortality and morbidity rates have also been reported related to ICU patients after heavy sedation.44 Despite all these reports, studies exploring the effects of midazolam on reproductive tissue could hardly be found, and we did find that a low dosage of midazolam (150 μM midazolam is equal to 0.8 mg/kg/hour) could have a significant apoptotic effect on MA-10 mouse Leydig tumor cells. Thus, a low dosage of midazolam could possibly be used as a novel therapeutic agent against testicular cancer, which would not have a paradoxical impact on clinical guidance.
Conclusion
In summary, midazolam could inhibit cell viability and arrest cell cycle to induce apoptosis in MA-10 mouse Leydig tumor cells. Midazolam did induce t-Bit, JNK, and p38, but not ERK, with the activation of caspase-8, -9, and -3 and PARP plus downregulation of Akt, to induce apoptosis. The current observations regarding the apoptotic effect of midazolam on MA-10 mouse Leydig tumor cells could highlight a potential antitumor therapeutic event in clinical application upon the reproductive system. Taken together, midazolam induced MA-10 cell apoptosis through the activation of caspase cascade, the inhibition of Akt pathway, and the stimulation of p38 and JNK pathways.
Acknowledgments
This study was supported by the National Science Council (Taiwan) to BMH (NSC101-2320-B-006-005-MY3) and to YKW (NSC101-2320-B-006-016-MY3) and the Chi Mei Medical Center Fund to ECS (CMFHR 9940).
Disclosure
The authors report no conflicts of interest in this work.
References
García-Pedrajas F, Arroyo J. Midazolam in anesthesiology. Rev Med Univ Navarra. 1989;33:211–221. | |
Olkkola KT, Ahonen J. Midazolam and other benzodiazepines. Handb Exp Pharmacol. 2008;182:335–360. | |
Boujrad N, Vidic B, Papadopoulos V. Acute action of choriogonadotropin on MA-10 tumor cells: changes in the topography of the mitochondrial peripheral-type benzodiazepine receptor. Endocrinology. 1996;137:5727–5730. | |
Walser A, Zenchoff G, Fryer RI. Quinazolines and 1,4-benzodiazepines. 75. 7-Hydroxyaminobenzodiazepines and derivatives. J Med Chem. 1976;19:1378–1381. | |
Miller WL. Steroidogenic acute regulatory protein (StAR), a novel mitochondrial cholesterol transporter. Biochim Biophys Acta. 2007;1771:663–676. | |
So EC, Chang YT, Hsing CH, Poon PW, Leu SF, Huang BM. The effect of midazolam on mouse Leydig cell steroidogenesis and apoptosis. Toxicol Lett. 2010;192:169–178. | |
Evan G, Littlewood T. A matter of life and cell death. Science. 1998;281:1317–1322. | |
Zimmermann KC, Bonzon C, Green DR. The machinery of programmed cell death. Pharmacol Ther. 2001;92:57–70. | |
Abdulghani J, El-Deiry WS. TRAIL receptor signaling and therapeutics. Expert Opin Ther Targets. 2010;14:1091–1108. | |
Arnoult D. Mitochondrial fragmentation in apoptosis. Trends Cell Biol. 2007;17:6–12. | |
Decker P, Muller S. Modulating poly (ADP-ribose) polymerase activity: potential for the prevention and therapy of pathogenic situations involving DNA damage and oxidative stress. Curr Pharm Biotechnol. 2002;3:275–283. | |
Virág L. Structure and function of poly (ADP-ribose) polymerase-1: role in oxidative stress-related pathologies. Curr Vasc Pharmacol. 2005;3:209–214. | |
Ozawa T. Oxidative damage and fragmentation of mitochondrial DNA in cellular apoptosis. Biosci Rep. 1997;17:237–250. | |
Harris MH, Thompson CB. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ. 2000;7:1182–1191. | |
Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21:92–101. | |
Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–656. | |
Lindsten T, Ross AJ, King A, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. | |
Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802:396–405. | |
Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–549. | |
Efeyan A, Zoncu R, Chang S, et al. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature. 2013;493:679–683. | |
Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. | |
Loepke AW, Soriano SG. An assessment of the effects of general anesthetics on developing brain structure and neurocognitive function. Anesth Analg. 2008;106:1681–1707. | |
Lunardi N, Ori C, Erisir A, Jevtovic-Todorovic V. General anesthesia causes long-lasting disturbances in the ultrastructural properties of developing synapses in young rats. Neurotox Res. 2010;17:179–188. | |
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. | |
Jourdain A, Martinou JC. Mitochondrial outer-membrane permeabilization and remodelling in apoptosis. Int J Biochem Cell Biol. 2009;41:1884–1889. | |
Parsons MJ, Green DR. Mitochondria in cell death. Essays Biochem. 2010;47:99–114. | |
Cuenda A, Rousseau S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta. 2007;1773:1358–1375. | |
Harris CE, Grounds RM, Murray AM, Lumley J, Royston D, Morgan M. Propofol for long-term sedation in the intensive care unit. A comparison with papaveretum and midazolam. Anaesthesia. 1990;45:366–372. | |
Demirel E, Ugur HC, Dolgun H, et al. The neurotoxic effects of intrathecal midazolam and neostigmine in rabbits. Anaesth Intensive Care. 2006;34:218–223. | |
Erdine S, Yücel A, Ozyalçin S, et al. Neurotoxicity of midazolam in the rabbit. Pain. 1999;80:419–423. | |
Stevens MF, Werdehausen R, Gaza N, et al. Midazolam activates the intrinsic pathway of apoptosis independent of benzodiazepine and death receptor signaling. Reg Anesth Pain Med. 2011;36:343–349. | |
Saha B, Mukherjee A, Samanta S, et al. Caffeine augments alprazolam induced cytotoxicity in human cell lines. Toxicol In Vitro. 2009;23:1100–1109. | |
Gupta S. Molecular steps of death receptor and mitochondrial pathways of apoptosis [review]. Life Sci. 2001;69:2957–2964. | |
Biron VA, Iglesias MM, Troncoso MF, et al. Galectin-1: biphasic growth regulation of Leydig tumor cells. Glycobiology. 2006;16:810–821. | |
Martinez VG, Pellizzari EH, Díaz ES, et al. Galectin-1, a cell adhesion modulator, induces apoptosis of rat Leydig cells in vitro. Glycobiology. 2004;14:127–137. | |
Guerrero AD, Chen M, Wang J. Delineation of the caspase-9 signaling cascade. Apoptosis. 2008;13:177–186. | |
Orser BA, McAdam LC, Roder S, MacDonald JF. General anaesthetics and their effects on GABA(A) receptor desensitization. Toxicol Lett. 1998;100–101:217–224. | |
He Q, Montalbano J, Corcoran C, Jin W, Huang Y, Sheikh MS. Effect of Bax deficiency on death receptor 5 and mitochondrial pathways during endoplasmic reticulum calcium pool depletion-induced apoptosis. Oncogene. 2003;22:2674–2679. | |
Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death: apoptosis, autophagy and senescence. FEBS J. 2010;277:2–21. | |
Yang DD, Kuan CY, Whitmarsh AJ, et al. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. | |
Gupta S, Barrett T, Whitmarsh AJ, et al. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15:2760–2770. | |
Sakata K, Kato S, Fox JC, Shigemori M, Morimatsu M. Autocrine signaling through Ras regulates cell survival activity in human glioma cells: potential cross-talk between Ras and the phosphatidylinositol 3-kinase-Akt pathway. J Neuropathol Exp Neurol. 2002;61:975–983. | |
Vlietstra RJ, van Alewijk DC, Hermans KG, van Steenbrugge GJ, Trapman J. Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res. 1998;58:2720–2723. | |
Shehabi Y, Chan L, Kadiman S, et al. Sedation depth and long-term mortality in mechanically ventilated critically ill adults: a prospective longitudinal multicentre cohort study. Intensive Care Med. 2013;39:910–918. | |
Roberts DJ, Hall RI, Kramer AH, Robertson HL, Gallagher CN, Zygun DA. Sedation for critically ill adults with severe traumatic brain injury: a systematic review of randomized controlled trials. Crit Care Med. 2011;39:2743–2751. | |
Olafson K, Ramsey CD, Ariano RE, et al. Sedation and analgesia usage in severe pandemic H1N1 (2009) infection: a comparison to respiratory failure secondary to other infectious pneumonias. Ann Pharmacother. 2012;46:9–20. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.