")
Back to Journals » Cancer Management and Research » Volume 12
MicroRNAs Targeting MYC Expression: Trace of Hope for Pancreatic Cancer Therapy. A Systematic Review
Authors Shams R , Asadzadeh Aghdaei H , Behmanesh A , Sadeghi A , Zali M, Salari S, Padrón JM
Received 13 January 2020
Accepted for publication 13 March 2020
Published 1 April 2020 Volume 2020:12 Pages 2393—2404
DOI https://doi.org/10.2147/CMAR.S245872
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Eileen O'Reilly
Roshanak Shams,1 Hamid Asadzadeh Aghdaei,1 Ali Behmanesh,2 Amir Sadeghi,1 Mohammadareza Zali,1 Sina Salari,3 José M Padrón4
1Research Institute for Gastroenterology and Liver Diseases, Shahid Beheshti University of Medical Sciences, Tehran, Iran; 2Student Research Committee, School of Health Management and Information Sciences, Iran University of Medical Sciences, Tehran, Iran; 3Taleghani Hospital, Shahid Beheshti University of Medical Sciences, Tehran, Iran; 4BioLab, Instituto Universitario de Bio-Orgánica “Antonio González” (IUBO-AG), Universidad de la Laguna, La Laguna, Spain
Correspondence: Roshanak Shams; Ali Behmanesh
Email [email protected]; [email protected]
Background: Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest malignancies and a major health problem worldwide. There were no major advances in conventional treatments in inhibiting tumor progression and increasing patient survival time. In order to suppress mechanisms responsible for tumor cell development such as those with oncogenic roles, more advanced therapeutic strategies should be sought. One of the most important oncogenes of pancreatic cancer is the MYC gene. The overexpression of MYC can activate many tumorigenic processes such as cell proliferation and pancreatic cancer cell invasion. MiRNAs are important molecules that are confirmed by targeting mRNA transcripts to regulate the expression of the MYC gene. Therefore, restoring MYC-repressing miRNAs expression tends to be an effective method of treating MYC-driven cancers.
Objective: The purpose of this study was to identify all validated microRNAs targeting C-MYC expression to inhibit PDAC progression by conducting a systematic review.
Methods: In this systematic review study, the papers published between 2000 and 2020 in major online scientific databases including PubMed, Scopus, and Web of Science were screened, following inclusion and exclusion criteria. We extracted all the experimental studies that showed miRNAs could target the expression of the MYC gene in PDAC.
Results: Eight papers were selected from a total of 89 papers. We found that six miRNAs (Let-7a, miR-145, miR-34a, miR-375, miR-494, and miR-148a) among the selected studies were validated for targeting MYC gene and three of them confirmed Let-7a as a direct MYC expression regulator in PC cells. Finally, we summarized the latest shreds of evidence of experimentally validated miRNAs targeting the MYC gene with respect to PDAC’s therapeutic potential.
Conclusion: Restoring the expression of MYC-repressing miRNAs tends to be an effective way to treat MYC-driven cancers such as PDAC. Several miRNAs have been proposed to target this oncogene via bioinformatics tools, but only a few have been experimentally validated for pancreatic cancer cells and models. Further studies should be conducted to find the interaction network of miRNA-MYC to develop more successful therapeutic strategies for PC, using the synergistic effects of these miRNAs.
Keywords: micro RNA, pancreatic cancer, MYC, cancer therapy
Introduction
Many recent advances in therapeutic medicine and immunotherapy have given a glimmer of hope for many patients with solid tumors. Unfortunately, these therapeutic strategies for patients with pancreatic adenocarcinoma were not successful in reducing unfavorable conditions. Only 8% of patients get treated for this malignancy which is the lowest rate among all solid tumors.1 While targeted therapies have been well defined for many types of solid tumors, multiple tests of targeted PC therapies have not yet been successful.2 Efforts in early diagnosis and PDAC care may be the most effective approach to improve the survival of patients. Therefore, it is crucial to search for more promising and sensitive diagnostic approaches as well as effective medicines, particularly those aimed at cancer progression processes. Moreover, it should be considered that genetic heterogeneity is the main cause of the failure of current PC therapies,3 while genetic mutations in genes such as KRAS, TP53, CDKN2A, and SMAD4 are recurrent in these tumors.4 Therefore, there is an urgent need for an alternative strategy to target some other important signaling hubs crucial to the initiation and progression of PDAC. Based on the current data, myelocytomatosis (C-MYC) oncogene amplification is associated with poor prognosis and decreased PDAC survival.5 In addition, through the integration of oncogenic KRAS signals, MYC is a non-redundant signaling core gene in this disease.6
The purpose of this study was to identify all validated microRNAs targeting C-MYC expression to inhibit PDAC progression through a systematic review. We provided data about the most important aspects of MYC-oncogene, its relevance to pancreatic cancer, and the opportunities of targeting this oncogene to suppress the tumor progression of pancreatic cancer. However, to the best of our knowledge, there is no systematic study gathering all the published data in this regard. Hence, using an organized search strategy via 3 databases (PubMed, SCOPUS, and Web of Science), a systematic search was done to find all the articles related to the subject.
MYC: An Important Hub Gene for PDAC
C-MYC (MYC) is an oncogenic transcription factor that many studies have reported its aberrant expression and involvement in the tumorigenesis of almost one-third of all human cancers.7–9 The MYC proto-oncogene is the main mediator of many signal transduction pathways to critical cellular processes.10 For example, MYC expression can be regulated by numerous mitogenic signal transduction pathways such as Wnt, b-Catenin, Ras, and Jak/Stat.11 MYC activation can lead to induction or repression of transcription of many other genes downstream which may promote multiple tumorigenesis processes such as cell cycle, differentiation, cell growth, cell adhesion, angiogenesis, chromosomal instability, and cell transformation.7,12-17 Figure 1 demonstrates schematically MYC-regulated activities in tumorigenesis and associated gene targets. To be considered as a marker, a gene or protein must hold some basic characteristics. The key point is that in a rare subgroup of patients, the gene/protein must be expressed differently.18 The family of MYC proto-oncogenes (c-Myc, N-Myc, and L-Myc) is an oncogenic transcription factor that many studies have reported its aberrant expression and involvement in the tumorigenesis of almost one-third of all human cancers.4,19 The most widely studied gene in this family is C-MYC, expressed in cells with a higher rate of proliferation. The MYC gene with a high level of Copy Number Variations (CNVs) at the 8q24 chromosomal position has recently been shown to be specifically related to poor prognosis in PDAC patients.20,21 MYC actually plays its role in tumorigenesis by increasing the expression of some other oncogenes or by repressing the expression of a number of tumor suppressor genes.22 In promoters of various genes, MYC can bind to E-box sequences by heterodimerizing with MAX. Furthermore, genes such as zinc-finger transcription factor MIZ-1 can be repressed. (18). In addition, strong evidence suggests that MYC is a key downstream effector of oncogenic KRAS in pancreas18,23 and multi-layer regulation of MYC expression in PDAC.24 Such results introduce MYC as a key driver in PDAC and question the applicability of MYC targeting strategies.
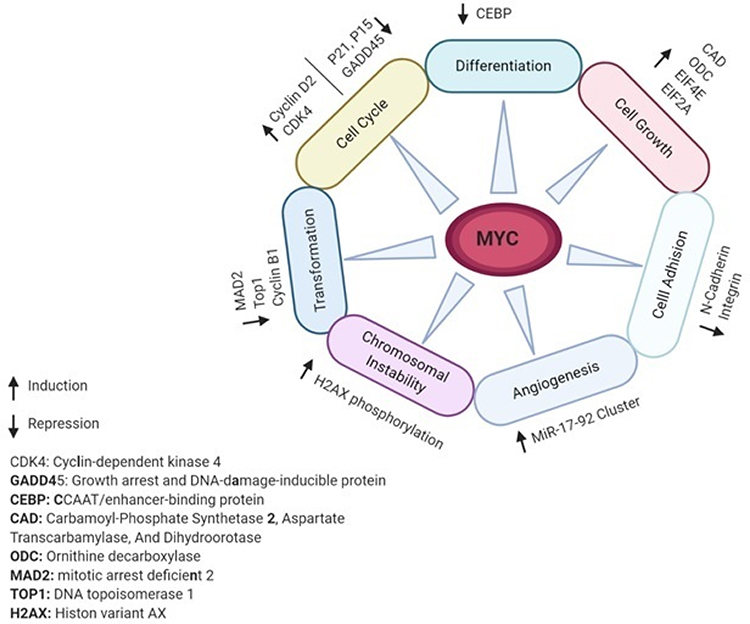 |
Figure 1 MYC-regulated activities and gene targets associated with tumorigenesis. Notes: MYC either as a transcription factor or transcription inhibitor targets various target genes downstream. Based on the type of the target genes activity, MYC can impact on different cell pathways and processes. |
Targeting MYC
A major hurdle in clinical cancer therapeutics is that MYC oncogene family of transcription factors is an undruggable gene product, ie, not easily accessible for inhibition by small drug molecules. Therefore, other strategies are necessary. In general, direct or indirect inhibitors can be used to target the function of MYC. Direct strategies include inhibition of downstream gene transcription of MYC with MYC-associated factor X (MAX).25 For example, Omomyc is a dominant-negative variant of MYC with four different amino acids that makes it able to form heterodimers with wild-type MYC.26 Such molecules interfere with the binding of MYC to MAX and impede downstream transcription of the E-box.27 Indirect inhibitors of MYC can be broken down into two classes. First, molecules that post-transcriptionally suppress MYC; second, inhibitors that hamper gene activation based on MYC.14 In recent decades, post-transcription modulation of gene expression has provided another way to regulate the expression of MYC. In this regard, attention has been paid to a class of small non-coding RNAs, microRNAs (miRNAs), which can regulate the expression of multiple target genes by binding a seed sequence in its target’s 3′-untranslated region.
MicroRNAs Therapeutics Potential
Numerous studies demonstrate that miRNAs have a fundamental role in the progression of several cancer types.28 Since some miRNA groups act as tumor suppressor genes and can prevent tumorigenesis by downregulating target oncogenes; they have the potential to treat cancer as a heterogeneous disease.29 One of the benefits of miRNAs is to simultaneously target multiple associated oncogenes or molecular pathways and bring about a synergistic therapeutic effect in cancer.30 In addition, miRNAs as endogenous antisense nucleotides showed significantly lower immune response and toxicity in contrast to plasmid DNA-based gene therapy and protein-based drug molecules.31 Hence, miRNAs are able to fulfill a substantial impact on cancer therapy.
MicroRNAs Targeting MYC
MYC studies have shown that a number of important signaling pathways including Janus kinase/signal transducers and transcription activators (JAK/STAT), (PI3K/AKT/mTOR), RAS/MAPK, and WNT can function and regulate it. All these paths are explicitly deregulated in the PDAC, and many of them are directly or indirectly dependent on the MYC function.32 Inactivation of miRNAs with tumor suppressor roles, on the other hand, frequently leads to the subsequent overexpression of important proto-oncogenes such as MYC.33 In addition, downregulation of miRNAs with tumor suppressor functions also leads to overexpression of critical proto-oncogenes like MYC. For example, miR-145 and miR-34 are under-expressed in many cancers and the loss of 1q36 miR-34a locus has also been discovered in PDAC cell lines with overexpression of major oncogenes such as MYC, p53, and members of the E2F family.34,35 Ultimately, research seeks for multiple interactions between miRNA networks and oncogenic signaling cascades to control the level of MYC expression and function comprehensively during PDAC progression. Therefore, it appears possible to inhibit the activity of this oncogene with the goal of PDAC therapy by using miRNAs as the main regulators of MYC expression.
Methods
We conducted a systematic review based on our objectives to find the relevant papers. We applied the criteria of the Preferred Reporting Items Statement for Systematic Reviews and Meta-Analysis (PRISMA)36 to conduct our study. In order to perform the PRISMA guideline, this study follows four main sections including, inclusion and exclusion criteria, search and data sources, selection of studies, and data extraction.
Inclusion and Exclusion Criteria
The inclusion criteria for the selection of articles were: (1) The study focused on pancreatic cancer cells or clinical specimens; (2) MYC gene was included in the study; and (3) A miRNA was associated with MYC.
The exclusion criteria were as follows: (1) Publications focusing on other cancers; (2) Publications focusing on other genes; and (3) MiRNAs were not included in publications. After reviewing the abstract and full-text manuscript, the process of evaluating the suitability of articles to be selected was carried out independently by two researchers (RS and JP). All of the authors made the final decision.
Search and Data Sources
Search for relevant papers consisted of identifying the keywords, formulating the search strategy, and selecting data sources. Keywords have been identified based on our objectives, and searches have been made in MeSH to find synonymous keywords. The literature searches have been carried out as online electronic databases through PubMed, Web of Science, and Scopus. Table 1 shows the search strategies applied to each database. In addition, we searched through Google Scholar for other relevant articles published. The articles published between 2000 and 2020 were included in the current research.
 |
Table 1 The Search Strategy Keywords |
Selection of the Studies
After searching process, 89 articles were extracted. EndNote X9 was used as a reference management software for removing duplicate articles. After the removal of the duplicates, 56 papers remained. Selection of relevant articles was made in the two steps. In the first step, the titles and the abstracts of the articles were evaluated based on the inclusion and exclusion criteria, and 42 papers were excluded from the first screening. In the second step, the full texts of the 14 papers remained in the first step were read. Finally, 8 articles were included to extract data (Figure 2).
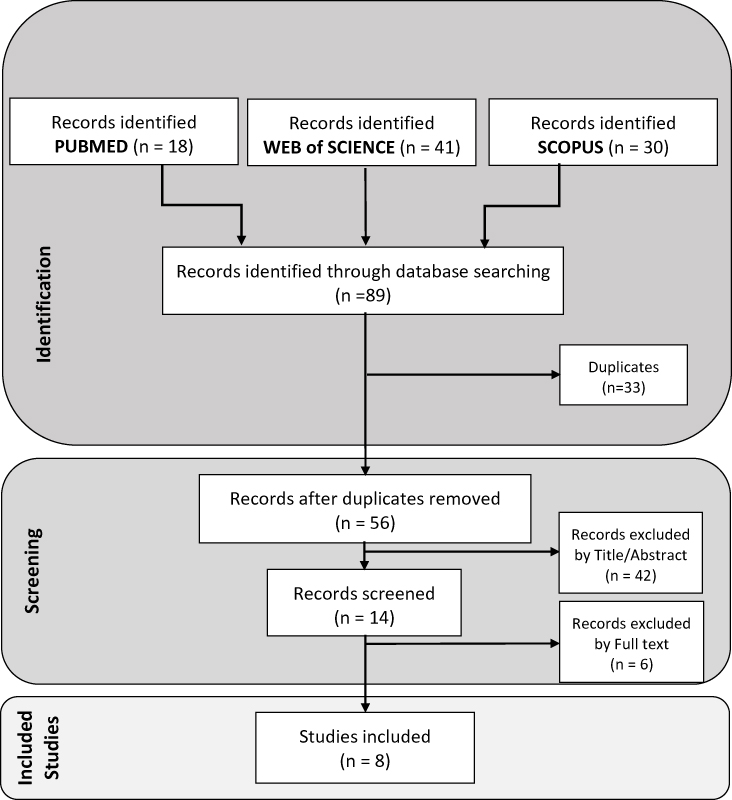 |
Figure 2 The flowchart of selecting articles. |
Data Extraction
In this section, the data were extracted by reading the full texts of the eight papers selected.37–44 They investigated the impact and interaction of some different miRNAs on the expression of the MYC gene in PDAC. The extracted data were collected by two of the authors and were inserted into the extraction form.
Results
The selected papers, containing experimental studies, were published from 2011 to 2019. PDAC was the type of cancer considered. The studies, either directly or as part of their research, focused on the effect of a miRNA on MYC gene expression and examined the effect on the expression of MYC gene in PDAC cell lines of induction or inhibition of certain unique miRNAs. Three studies have shown that the MYC gene is the direct target of their miRNAs, using the luciferase assay process. Three studies examined the effect on inhibition of the expression of certain other genes or drugs plus miRNAs by the MYC gene.
miRNAs Targeting MYC
The findings of the reviewed papers showed that let-7a was confirmed by 3 studies and 2 independent investigators as the direct regulator of the MYC gene. Table 2 systematically summarizes the specifics of the miRNAs screened and the study statistics. Also, the other 5 miRNAs, including miR-145, miR-34a, miR-375, miR-494, and miR-148a were validated as the MYC gene expression regulators.
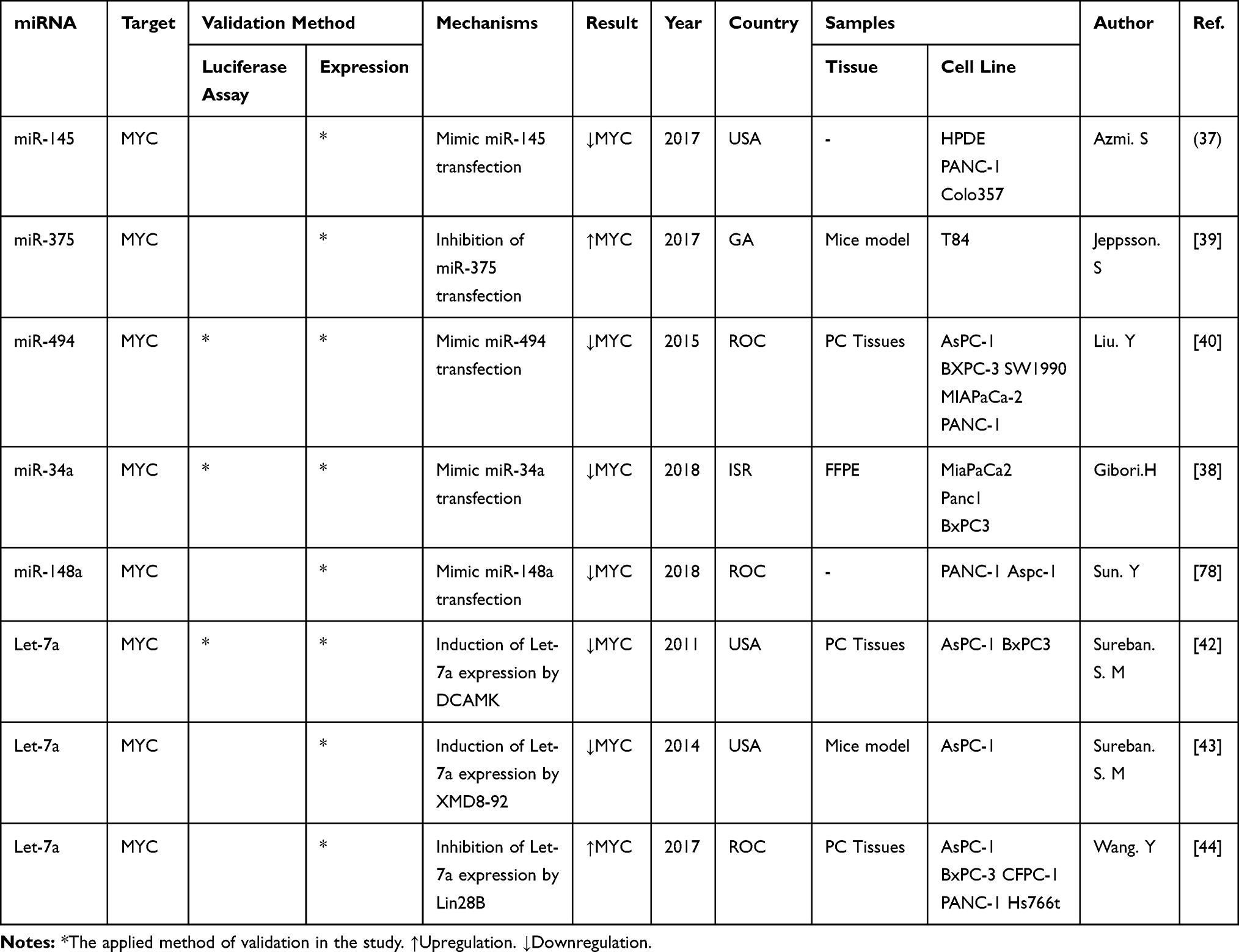 |
Table 2 The Information of All Eight Studies That Represented Association of miRNAs to MYC Expression in Pancreatic Cancer |
Let-7a
Let-7a is a member of let-7 miRNA family and commonly is downregulated in several human cancers, including PDAC.45,46 In a variety of human cancers, let-7 is commonly shown to interact with RAS oncogenes,47 and these results confirmed the notion that let-7 can function as a tumor suppressor gene.48 As a result, many other evidence strongly demonstrated that let-7 acts as a tumor suppressor by targeting multiple processes of tumorigenesis. For instance, let-7 has been shown to be capable of repressing cell cycle regulators (e.g., cycline A, cycline D1, cycline D3, and CDK4) and hampering the cell cycle and cancer cell development.49 In addition, the interaction of let-7 members with the expression of the MYC gene and inhibition of MYC mediated cell growth has been documented in multiple studies.50,51 Interestingly, MYC can also control the expression of let-7 family members including let-7a, −7d, and −7 g by binding to their promoters through a feedback loop framework.52 As far as pancreatic cancer is concerned, our systematic search results indicate that 3 studies have examined and reported let-7a interaction with MYC expression to date. In a 2011 experimental study, Sureban et al demonstrated that the knockdown of the DCAMKL-1 gene in pancreatic cancer cells leads to the downregulation of C-MYC proto-oncogene through pri-let-7a upregulation. They demonstrated that C-MYC is a key downstream target of let-7a miRNA. To confirm this, pancreatic cancer cell line, control, AsPC-1 siSCR and AsPC-1-siDCAMKL-1 cells were analyzed for C-MYC expression by real-time RT-PCR. In comparison to control and AsPC-1-siSCR cells, there was a significant (P < 0.01) 50% decrease in C-MYC mRNA expression in AsPC-1-siDCAMKL-1 cells mediated by overexpression of let-7a.42 Therefore, a drop in c-Myc protein was also seen after DCAMKL-1’s knockdown. As further support for the association of let-7a with the MYC gene, in 2014 the authors performed another study to demonstrate that induction of let-7a expression in pancreatic tumor xenografts by XMD8-92 treatment leads to a reduction of more than 60% in C-MYC mRNA. Recently, the regulatory effect of let-7a on MYC expression has been verified by another group of authors in China. They demonstrated that let-7a inhibition through Lin28B gene upregulation leads to significant overexpression of the MYC gene and prompts cell proliferation, migration, and epithelial–mesenchymal transition (EMT) in PDAC cells. In PDAC tissue samples, the expression of these genes was also exanimated, and the results showed an inverse correlation of expression between let-7a and downstream targets such as C-MYC and RAS oncogenes.44
miR-145
MiR-145 is a completely well-known tumor-suppressor miRNA located in chromosome 9q31. Several studies have revealed that miR-145 is decreased in a variety of solid tumors, such as gastrointestinal, colorectal, and pancreatic cancer.53–55 This microRNA can regulate tumor proliferation via multiple target genes, such as KRAS, C-MYC, and FSCN1.56 Published papers have a number of important evidence to show the relevance of miR-145 ranging from the clinical significance of pancreatic cancer to its therapeutic consequences.57 MiR-145’s direct interaction with the MYC gene is also investigated and reported in various cancers by many studies.58–61 Regarding pancreas cancer, Azmi et al (2017) showed that up-regulation of miR-145 following XPO1 gene inhibition contributes to down-regulation of established target genes, including EGFR, MMP1, MT-MMP, C-MYC, Pak4, and Sox-2.37
miR-34a
Reduced expression of miR-34a in pancreatic cancer cells and tissues has been documented repeatedly, paving the way for this miRNA to be considered as a crucial tumor suppressor gene that is useful in predictive and therapeutic approaches to PDAC.62–65 In response to DNA damage, MiR-34c has been shown to regulate C-MYC inversely and to impede C-MYC -induced DNA synthesis.66 In addition, miR-34a was reported to target C-MYC throughout the oncogene-induced senescence.67,68 In 2018, Gibori et al showed that the administration of a microRNA-mimic to increase miR-34a plus siRNA to silence PLK1 oncogene to orthotopically inoculated PDAC-bearing mice results in an elevated antitumor effect due to inhibition of MYC oncogene, a common target of both miR-34a and PLK1.69
miR-375
A growing chain of evidence has indicated that the expression of miR‐375 is almost always decreased in many types of cancers and functions as a tumor suppressor by inhibiting malignant features of cancer cells.70 There has been little detailed investigation into the relationship of this gene with MYC expression, but some studies have indirectly pointed to the presence of such association in their findings.71 In 2018, Jeppsson et al reported that neuropeptide Y (NPY) enhances the cell proliferation of PDAC cells through miR-375 downregulation. They also transfected the cells with miR-375 inhibitor and assessed various markers of cellular proliferation and cell cycle arrest such as C-MYC, cyclin D1, and p21 by Western blotting. They observed that the addition of NPY resulted in enhanced C-MYC and Cyclin D1 mediated by downregulation of miR-375.39
miR-494
In general, miR‐494 tends to have various roles in multiple tumor forms. MiR‐494 has been shown to prompt factor-related apoptosis of tumor necrosis in non-small cell lung cancer, known to be an oncogenic miRNA that inhibits the transfer of G1/S via liver tumorigenesis.72 On the other hand, miR‐494 is down-regulated in human cholangiocarcinoma, and its re-expression contributes to the inhibition of cancer progression, suggesting an anti-oncogenic function for miR‐494.73 In pancreatic cancer, the level of miR-494 expression in tumor tissues is documented to be significantly down-regulated and the association of this miRNA’s low expression with poor patient overall survival was also confirmed.74 Some studies also demonstrated this miRNA’s anti-tumorigenic effect by targeting the MYC gene in different cancers.75,76 Liu et al (2018) illustrated that miR-494’s expression in PC cell lines and tissues is significantly reduced, and its overexpression can significantly suppress the proliferation of PC cells in vitro and in vivo. Thus, miR-494 induction significantly inhibits PC cell invasion. Furthermore, they verified that both C-MYC and SIRT1 genes are targets of miR-494 by dual luciferase assay, and then confirmed an inverse association in PC samples between miR-494 and C-MYC/SIRT1.
miR-148a
Among the various cancers, Mir-148a consisting of gastric, colorectal, pancreatic, liver, etc. is downregulated. In some other cancers such as glioma and osteosarcoma, the upregulation of this miRNA can also be observed.77 With respect to pancreatic cancer, Sun et al assessed the possible antitumor ability of miR‐148a and its effects on pancreatic cancer metastasis. They found that the expression of miRNA‐148a and the maternally expressed predictive biomarker gene‐3 (MEG‐3) was obviously lower than that in adjacent non-tumorous tissues in human pancreatic cancer tissues. Using Western blot analysis, they also found that miR‐148a mimic transduction significantly reduces the levels of expression in pancreatic cancer cells of C-MYC, cycline D1 and β‐catenin.78
Bioinformatics Evaluations
In order to obtain more information about the associations of miRNAs with pancreatic cancer tumorigenesis considered in this study, we used the online MiRnet tool to predict the target genes for all 6 miRNAs and design an interaction network miRNA-Targets.79 A network of 6 miRNAs, 2118 nodes and 2441 edges (Figure 3A) was created. First, a module consisting of genes that were regulated by all or 5 of those called miRNAs was acquired by selecting the target genes with a Betweenness degree > 5. Not surprisingly, the MYC gene was the final module’s central gene, indicating that all the 6 miRNAs can target this gene. Furthermore, there are three other well-known oncogenes (IGF1R, BCL2, and BCL2L11) which can be targeted simultaneously by miRNAs (Figure 3B). Table 3 shows the degree and intersection scores for all genes and miRNAs listed above.
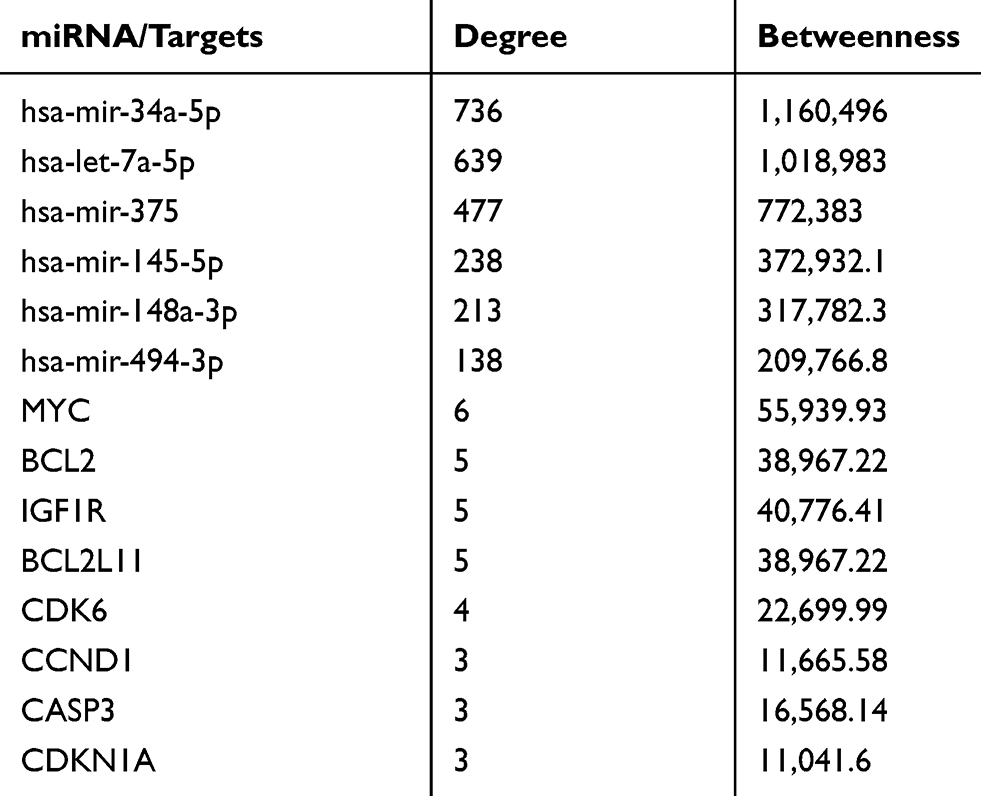 |
Table 3 Degree and Betweenness Scores of miRNAs and Their Targets |
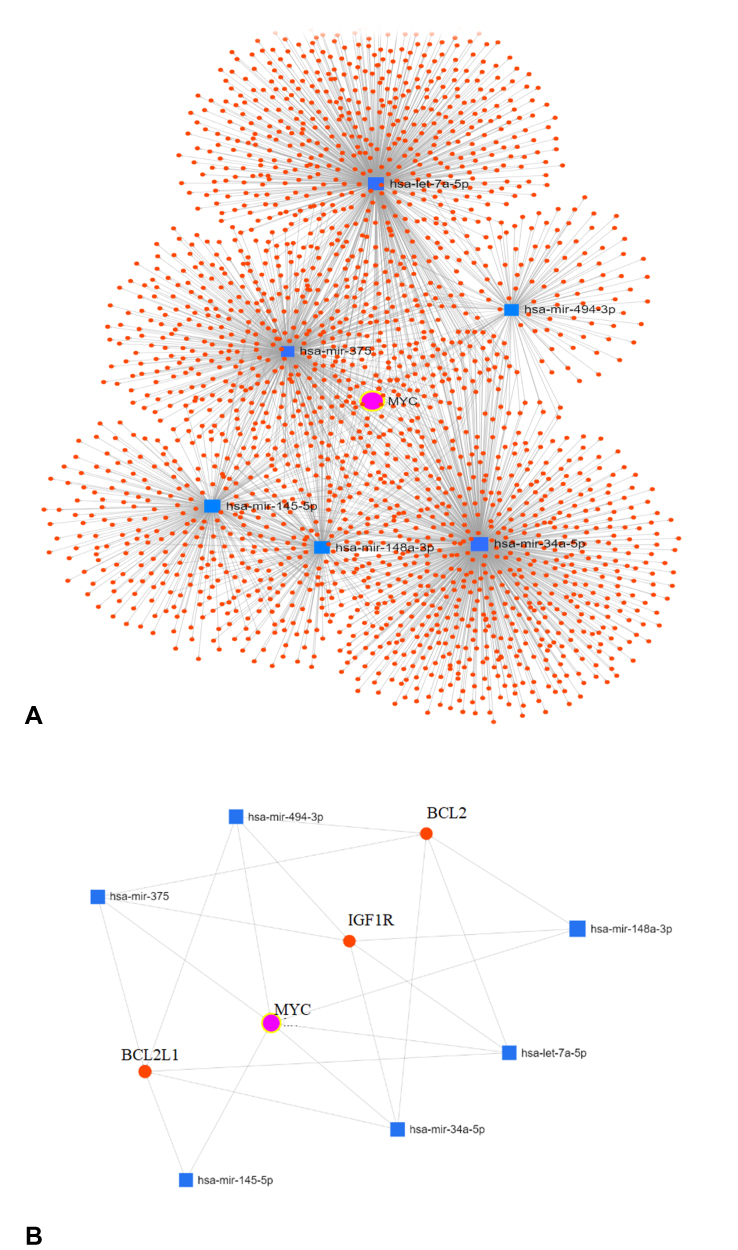 |
Figure 3 (A) The complete MiRNA/Target genes interaction network showing MYC is targeted by all 6 miRNAs as the center of the network. (B) A module showing that some other critical genes also may be targeted by the considered miRNAs. |
Discussion
Cancer is characterized by altering the cell characteristics where it develops, resulting in uncontrolled proliferation, invasion, and metastasis. Most of the primary-stage cancers are curable, but early diagnosis and treatment of cancer is the main challenge for pancreatic cancer.80 The pancreatic cancer therapy methods are based on a variety of factors such as stage and tumor size and include a combination of at least two or more methods of surgery, chemotherapy, radiotherapy, hormone therapy, and target therapy.81 Because the conventional treatments have not achieved significant success in inhibiting tumor progression and increasing patients’ survival time, more sophisticated therapeutic strategies should be sought to suppress tumor cell progression pathways such as those with oncogenic functions or to boost the immune system against tumors.82 Nevertheless, none of the existing antitumor therapies will specifically target a variety of tumor-involved molecular pathways. Recent advances in molecular biology have revealed the pivotal role of miRNAs in tumor initiation, growth, and invasion in the field of pancreatic cancer.83 MiRNAs’ effect on gene expression is important and it is shown that miRNAs can control more than 60% of human protein coding genes.84 MiRNAs’ downstream functions are extensively dependent on cellular content, which is correlated with their target mRNAs’ differential expression. Based on the type of target genes, miRNAs can function in a specific cell-type system either as oncomiRs or as tumor-suppressive. One of the most important oncogenes of pancreatic cancer is the MYC gene. Studies have shown that this oncogene’s overexpression resulting from gene amplification, abnormal transcriptional activation, or disruption in regulatory systems can lead to the activation of many tumorigenic processes such as cell proliferation and invasion. MiRNAs are essential molecules confirmed by targeting their mRNA transcripts in controlling the expression of the MYC gene. Therefore, restoring the expression of MYC-repressing miRNAs tends to be an effective way to repel MYC-driven cancers.85 Several miRNAs have been proposed to target this oncogene via bioinformatics tools, but only a few have been experimentally validated for pancreatic cancer cells and models. In this study, we extracted all the experimental studies that showed miRNAs could target MYC gene expression using a systematic search strategy. As a result, six miRNAs and eight final studies were found, while three of them confirmed Let-7a in PC cells as a direct regulator of MYC expression. Figure 4 schematically represents the process that some miRNAs can target and regulate MYC gene expression leading to inhibition of this gene’s tumorigenesis function in PDAC. The results of our bioinformatics evaluations showed that besides the MYC gene, these miRNAs may target some other oncogenes such as IGF1R, BCL2L11, and BCL2. Targeting insulin-like growth factor 1 receptor (IGF1R) has been confirmed as a potential therapeutic strategy for cancer.86 It is also shown that IGF1R knockdown significantly impeded the proliferation and development of pancreatic cancer cells, increased apoptosis, and inhibited the growth of pancreatic tumors.87 In addition, dysregulation in the expression of the BCL2 gene family that is a prevalent phenomenon in cancer and induces resistance to normal apoptosis inducers inhibits the apoptotic death of some cells. Some studies have documented that in pancreatic cancer cell lines BCL2 correlates with metastatic potential (84). Such findings suggest a possibility that the anti-tumorigenic activity of these miRNAs may be focused on targeting multiple oncogenes as well as MYC on pancreatic cancer cells. On this basis, it would be highly appropriate to consider other essential genes targeted by the desired miRNAs for studies in the field of evaluation of the therapeutic potential of miRNAs targeting the MYC gene. As mentioned above, several other miRNAs appear to be capable of targeting this gene. Consequently, multiplex strategies using a combination of multiple miRNAs targeting the same pathway or gene sunsets may have a robust effect on inducing apoptosis or inhibiting the proliferation of cells in PC cells. Nonetheless, some challenges were met in using miRNA mimics to regulate gene expression. For example, elevated stability and elevated delivery rate of miRNAs to the tumor cells concerned are important points that require more development. In addition, some studies have shown that some of MYC’s own upstream miRNAs as well as some other miRNAs with tumor suppressive effects can also be down-regulated. Therefore, to find more miRNAs with MYC-repressive effects, further studies should be conducted either by bioinformatics or experimental methods. This can help to find the interaction network miRNA-MYC to develop more successful therapeutic strategies for PC using the synergistic effects of these miRNAs.
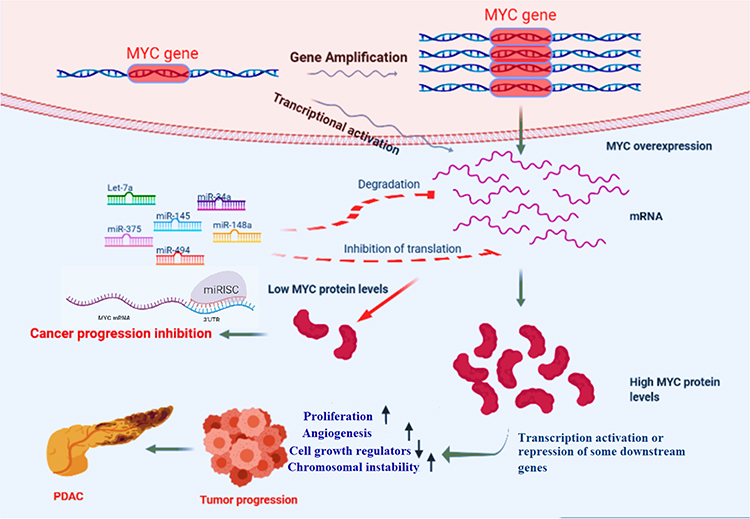 |
Figure 4 A schematic picture of how miRNAs can inhibit pancreatic cancer progression by targeting MYC. Notes: Let-7a, miR-375, miR-34a, miR-145, miR-148a, and miR-494 as experimentally validated miRNAs targeting MYC mRNAs post-transcriptionally can suppress pancreatic cancer tumor progression by inhibiting MYC’s downstream molecular pathways. |
Author Contributions
All the authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agreed to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Miller KD, Goding Sauer A, Ortiz AP, et al. Cancer statistics for hispanics/latinos, 2018. CA. 2018;68(6):425–445.
2. Kindler HL. A glimmer of hope for pancreatic cancer. N Engl J Med. 2018;379(25):2463–2464.
3. Knudsen ES, O’Reilly EM, Brody JR, Witkiewicz AK. Genetic diversity of pancreatic ductal adenocarcinoma and opportunities for precision medicine. Gastroenterology. 2016;150(1):48–63.
4. Witkiewicz AK, McMillan EA, Balaji U, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744.
5. Schleger C, Verbeke C, Hildenbrand R, Zentgraf H, Bleyl U. c-MYC activation in primary and metastatic ductal adenocarcinoma of the pancreas: incidence, mechanisms, and clinical significance. Mod Pathol. 2002;15(4):462.
6. Wirth M, Mahboobi S, Krämer OH, Schneider G. Concepts to target MYC in pancreatic cancer. Mol Cancer Ther. 2016;15(8):1792–1798.
7. Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8(12):976–990.
8. Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45(10):1127.
9. Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Perspect Med. 2014;4(6):a014241.
10. Dang CV. MYC on the path to cancer. Cell. 2012;149(1):22–35.
11. Skoudy A, Hernández-Muñoz I, Navarro PJ. Pancreatic ductal adenocarcinoma and transcription factors: role of c-Myc. J Gastroinstest Cancer. 2011;42(2):76–84.
12. Huang M, Weiss WA. Neuroblastoma and MYCN. Cold Spring Perspect Med. 2013;3(10):a014415.
13. Roussel MF, Robinson GW. Role of MYC in Medulloblastoma. Cold Spring Perspect Med. 2013;3(11):a014308.
14. Hessmann E, Schneider G, Ellenrieder V, Siveke JJO. MYC in pancreatic cancer: novel mechanistic insights and their translation into therapeutic strategies. Oncogene. 2016;35(13):1609.
15. Roussel MF, Cleveland JL, Shurtleff SA, Sherr CJ. Myc rescue of a mutant CSF-1 receptor impaired in mitogenic signalling. Nature. 1991;353(6342):361–363.
16. Baudino TA, McKay C, Pendeville-Samain H, et al. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Develop. 2002;16(19):2530–2543.
17. Li Q, Dang CV. c-Myc overexpression uncouples DNA replication from mitosis. Mol Cell Biol. 1999;19(8):5339–5351.
18. Walz S, Lorenzin F, Morton J, et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature. 2014;511(7510):483.
19. Waddell N, Pajic M, Patch A-M, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501.
20. Sandgren EP, Quaife CJ, Paulovich AG, Palmiter RD, Brinster RL. Pancreatic tumor pathogenesis reflects the causative genetic lesion. Proc Natl Acad Sci. 1991;88(1):93–97.
21. Lin W-C, Rajbhandari N, Liu C, et al. Dormant cancer cells contribute to residual disease in a model of reversible pancreatic cancer. Cancer Res. 2013;73(6):1821–1830.
22. Hayashi K, Jutabha P, Endou H, Anzai N. c-Myc is crucial for the expression of LAT1 in MIA Paca-2 human pancreatic cancer cells. Oncol Rep. 2012;28(3):862–866.
23. Diersch S, Wirth M, Schneeweis C, et al. Kras G12D induces EGFR-MYC cross signaling in murine primary pancreatic ductal epithelial cells. Oncogene. 2016;35(29):3880.
24. Hayes TK, Neel NF, Hu C, et al. Long-term ERK inhibition in KRAS-mutant pancreatic cancer is associated with MYC degradation and senescence-like growth suppression. Cancer Cell. 2016;29(1):75–89.
25. Stellas D, Szabolcs M, Koul S, et al. Therapeutic effects of an anti-Myc drug on mouse pancreatic cancer. JNCI. 2014;106(12).
26. Soucek L, Jucker R, Panacchia L, Ricordy R, Tatò F, Nasi S. Omomyc, a potential Myc dominant negative, enhances Myc-induced apoptosis. Cancer Res. 2002;62(12):3507–3510.
27. Soucek L, Whitfield J, Martins CP, et al. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455(7213):679–683.
28. Zadran S, Remacle F, Levine R. miRNA and mRNA cancer signatures determined by analysis of expression levels in large cohorts of patients. Proc Natl Acad Sci. 2013;110(47):19160–19165.
29. Suárez Y, Sessa WC. MicroRNAs as novel regulators of angiogenesis. Circul Res. 2009;104(4):442–454.
30. Bhardwaj A, Singh S, Singh AP. MicroRNA-based cancer therapeutics: big hope from small RNAs. Mol Cell Pharmacol. 2010;2(5):213–219.
31. Ivkovic TC, Voss G, Cornella H, Ceder Y. microRNAs as cancer therapeutics: a step closer to clinical application. Cancer Lett. 2017;407:113–122.
32. Vervoorts J, Lüscher-Firzlaff J, Lüscher BJ. The ins and outs of MYC regulation by posttranslational mechanisms. J Biol Chem. 2006;281(46):34725–34729.
33. Jackstadt R, Hermeking H. MicroRNAs as regulators and mediators of c-MYC function. Biochim Biophys Acta. 2015;1849(5):544–553.
34. Tazawa H, Kagawa S, Fujiwara T. MicroRNAs as potential target gene in cancer gene therapy of gastrointestinal tumors. Expert Opin Biol Ther. 2011;11(2):145–155.
35. Yamakuchi M, Ferlito M, Lowenstein C. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci. 2008;105(36):13421–13426.
36. Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Int J Surg. 2010;8(5):336–341. doi:10.1016/j.ijsu.2010.02.007
37. Azmi AS, Li Y, Muqbil I, et al. Exportin 1 (XPO1) inhibition leads to restoration of tumor suppressor miR-145 and consequent suppression of pancreatic cancer cell proliferation and migration. Oncotarget. 2017;8(47):82144.
38. Gibori H, Eliyahu S, Krivitsky A, et al. Amphiphilic nanocarrier-induced modulation of PLK1 and miR-34a leads to improved therapeutic response in pancreatic cancer. Nat Commun. 2018;9(1):16.
39. Jeppsson S, Srinivasan S, Chandrasekharan B. Neuropeptide Y (NPY) promotes inflammation-induced tumorigenesis by enhancing epithelial cell proliferation. Am J Physio Gastrointest Liver Physiol. 2017;312(2):G103–G111. doi:10.1152/ajpgi.00410.2015
40. Liu Y, Li X, Zhu S, et al. Ectopic expression of miR-494 inhibited the proliferation, invasion and chemoresistance of pancreatic cancer by regulating SIRT1 and c-Myc. Gene Ther. 2015;22(9):729–738. doi:10.1038/gt.2015.39
41. Sun Y, Wu J, Wu SH, et al. Expression profile of microRNAs in c-Myc induced mouse mammary tumors. Breast Cancer Res Treat. 2009;118(1):185–196. doi:10.1007/s10549-008-0171-6
42. Sureban SM, May R, Lightfoot SA, et al. DCAMKL-1 regulates epithelial-mesenchymal transition in human pancreatic cells through a miR-200a-dependent mechanism. Cancer Res. 2011;71(6):2328–2338. doi:10.1158/0008-5472.CAN-10-2738
43. Sureban SM, May R, Weygant N, et al. XMD8-92 inhibits pancreatic tumor xenograft growth via a DCLK1-dependent mechanism. Cancer Lett. 2014;351(1):151–161. doi:10.1016/j.canlet.2014.05.011
44. Wang Y, Li J, Guo S, et al. Lin28B facilitates the progression and metastasis of pancreatic ductal adenocarcinoma. Oncotarget. 2017;8(36):60414–60428. doi:10.18632/oncotarget.19578
45. Ruzzo A, Graziano F, Vincenzi B, et al. High let-7a microRNA levels in KRAS-mutated colorectal carcinomas may rescue anti-EGFR therapy effects in patients with chemotherapy-refractory metastatic disease. Oncologist. 2012;17(6):823–829.
46. Torrisani J, Bournet B, Du Rieu MC, et al. let-7 MicroRNA transfer in pancreatic cancer-derived cells inhibits in vitro cell proliferation but fails to alter tumor progression. Human Gene Ther. 2009;20(8):831–844.
47. Johnson SM, Grosshans H, Shingara J, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120(5):635–647.
48. Shell S, Park S-M, Radjabi AR, et al. Let-7 expression defines two differentiation stages of cancer. Proc Natl Acad Sci. 2007;104(27):11400–11405.
49. Johnson CD, Esquela-Kerscher A, Stefani G, et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007;67(16):7713–7722.
50. Gunzburg MJ, Sivakumaran A, Pendini NR, et al. Cooperative interplay of let-7 mimic and HuR with MYC RNA. Cell Cycle. 2015;14(17):2729–2733.
51. Kim HH, Kuwano Y, Srikantan S, et al. HuR recruits let-7/RISC to repress c-Myc expression. Genes Develop. 2009;23(15):1743–1748.
52. Wang Z, Lin S, Li JJ, et al. MYC protein inhibits transcription of the microRNA cluster MC-let-7a-1∼ let-7d via noncanonical E-box. J Biol Chem. 2011;286(46):39703–39714.
53. Lei C, Du F, Sun L, et al. miR-143 and miR-145 inhibit gastric cancer cell migration and metastasis by suppressing MYO6. Cell Death Dis. 2017;8(10):e3101.
54. Setua S, Khan S, Doxtater K, et al. miR-145: revival of a dragon in pancreatic cancer. J Nat Sci. 2017;3(3).
55. Xu Q, Liu L-Z, Qian X, et al. MiR-145 directly targets p70S6K1 in cancer cells to inhibit tumor growth and angiogenesis. Nucleic Acids Res. 2011;40(2):761–774.
56. Sachdeva M, Mo YY. miR-145-mediated suppression of cell growth, invasion and metastasis. Am J Transl Res. 2010;2(2):170–180.
57. Setua S, Khan S, Yallapu MM, et al. Restitution of tumor suppressor microRNA-145 using magnetic nanoformulation for pancreatic cancer therapy. J Gastrointest Surg. 2017;21(1):94–105.
58. Shao Y, Qu Y, Dang S, Yao B, Ji M. MiR-145 inhibits oral squamous cell carcinoma (OSCC) cell growth by targeting c-Myc and Cdk6. Cancer Cell Int. 2013;13(1):51.
59. Wang F, Xia J, Wang N, Zong HJO. miR-145 inhibits proliferation and invasion of esophageal squamous cell carcinoma in part by targeting c-Myc. Onkologie. 2013;36(12):754–758.
60. Zhang W, Wang Q, Yu M, Wu N, Wang H. MicroRNA-145 function as a cell growth repressor by directly targeting c-Myc in human ovarian cancer. Tech Cancer Res Treat. 2014;13(2):161–168.
61. Derouet MF, Dakpo E, Wu L, et al. miR-145 expression enhances integrin expression in SK-GT-4 cell line by down-regulating c-Myc expression. Oncotarget. 2018;9(20):15198.
62. Nalls D, Tang S-N, Rodova M, Srivastava RK, Shankar S. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PloS One. 2011;6(8):e24099.
63. Ji Q, Hao X, Zhang M, et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PloS One. 2009;4(8):e6816.
64. Vychytilova-Faltejskova P, Kiss I, Klusova S, et al. MiR-21, miR-34a, miR-198 and miR-217 as diagnostic and prognostic biomarkers for chronic pancreatitis and pancreatic ductal adenocarcinoma. Diagn Pathol. 2015;10(1):38.
65. Tang Y, Tang Y, Cheng Y-S. miR-34a inhibits pancreatic cancer progression through Snail1-mediated epithelial–mesenchymal transition and the Notch signaling pathway. Sci Rep. 2017;7:38232.
66. Cannell IG, Kong YW, Johnston SJ, et al. p38 MAPK/MK2-mediated induction of miR-34c following DNA damage prevents Myc-dependent DNA replication. Proc Natl Acad Sci. 2010;107(12):5375–5380.
67. Christoffersen N, Shalgi R, Frankel L, et al. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Proc Natl Acad Sci. 2010;17(2):236.
68. Yamamura S, Saini S, Majid S, et al. MicroRNA-34a modulates c-Myc transcriptional complexes to suppress malignancy in human prostate cancer cells. PloS One. 2012;7(1):e29722.
69. Gibori H, Eliyahu S, Krivitsky A, et al. Amphiphilic nanocarrier-induced modulation of PLK1 and miR-34a leads to improved therapeutic response in pancreatic cancer. Nat Commun. 2018;9(1):16. doi:10.1038/s41467-017-02283-9
70. Yan JW, Lin JS, He XX. The emerging role of miR‐375 in cancer. Int J Cancer. 2014;135(5):1011–1018.
71. Jung HM, Patel RS, Phillips BL, et al. Tumor suppressor miR-375 regulates MYC expression via repression of CIP2A coding sequence through multiple miRNA–mRNA interactions. Mol Biol Cell. 2013;24(11):1638–1648.
72. Lim L, Balakrishnan A, Huskey N, et al. MicroRNA‐494 within an oncogenic microRNA megacluster regulates G1/S transition in liver tumorigenesis through suppression of mutated in colorectal cancer. Hepatology. 2014;59(1):202–215.
73. Olaru AV, Ghiaur G, Yamanaka S, et al. MicroRNA down‐regulated in human cholangiocarcinoma control cell cycle through multiple targets involved in the G1/S checkpoint. Hepatology. 2011;54(6):2089–2098.
74. Ma Y, Li G, Hu J, Liu X, Shi B. Correlation of miR-494 expression with tumor progression and patient survival in pancreatic cancer. Genet Mol Res. 2015;14(4):18153–18159.
75. He W, Li Y, Chen X, et al. miR‐494 acts as an anti‐oncogene in gastric carcinoma by targeting c‐myc. J Gastroenterol Hepatol. 2014;29(7):1427–1434.
76. Yuan J, Wang K, Xi M. MiR-494 inhibits epithelial ovarian cancer growth by targeting c-Myc. Med Sci Monit. 2016;22:617.
77. Li Y, Deng X, Zeng X, Peng X. The role of mir-148a in cancer. J Cancer. 2016;7(10):1233.
78. Sun YP, Zhu QD, Zhou MT, et al. Restoration of miRNA-148a in pancreatic cancer reduces invasion and metastasis by inhibiting the Wnt/-catenin signaling pathway via downregulating maternally expressed gene-3. Exp Ther Med. 2019;17(1):639–648. doi:10.3892/etm.2018.7026
79. Fan Y, Siklenka K, Arora SK, Ribeiro P, Kimmins S, Xia J. miRNet-dissecting miRNA-target interactions and functional associations through network-based visual analysis. Nucleic Acis Res. 2016;44(W1):W135–W141.
80. Mayer IA, Arteaga CL. The PI3K/AKT pathway as a target for cancer treatment. Annual Rev Med. 2016;67:11–28.
81. Khorana AA, Mangu PB, Berlin J, et al. Potentially curable pancreatic cancer: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2016;34(21):2541–2556.
82. Golan T, Khvalevsky EZ, Hubert A, et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget. 2015;6(27):24560.
83. Li Y, Sarkar FH. MicroRNA targeted therapeutic approach for pancreatic cancer. Int J Biol Sci. 2016;12(3):326.
84. Friedman RC, Farh KK-H, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19(1):92–105.
85. Morton JP, Sansom OJ. MYC-y mice: from tumour initiation to therapeutic targeting of endogenous MYC. Mol Oncol. 2013;7(2):248–258.
86. Miljković M, Girotra M, Abraham R, Erlich R. Novel medical therapies of recurrent and metastatic gastroenteropancreatic neuroendocrine tumors. Digest Dis Sci. 2012;57(1):9–18.
87. Tian X, Hao K, Qin C, et al. Insulin-like growth factor 1 receptor promotes the growth and chemoresistance of pancreatic cancer. Digest Dis Sci. 2013;58(9):2705–2712.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.