")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
MicroRNA modulation combined with sunitinib as a novel therapeutic strategy for pancreatic cancer
Authors Passadouro M, de Lima M, Faneca H
Received 20 March 2014
Accepted for publication 3 May 2014
Published 3 July 2014 Volume 2014:9(1) Pages 3203—3217
DOI https://doi.org/10.2147/IJN.S64456
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Marta Passadouro,1,2 Maria C Pedroso de Lima,1,2 Henrique Faneca1
1Center for Neuroscience and Cell Biology, 2Department of Life Sciences, Faculty of Science and Technology, University of Coimbra, Coimbra, Portugal
Abstract: Pancreatic ductal adenocarcinoma (PDAC) is a highly aggressive and mortal cancer, characterized by a set of known mutations, invasive features, and aberrant microRNA expression that have been associated with hallmark malignant properties of PDAC. The lack of effective PDAC treatment options prompted us to investigate whether microRNAs would constitute promising therapeutic targets toward the generation of a gene therapy approach with clinical significance for this disease. In this work, we show that the developed human serum albumin–1-palmitoyl-2-oleoyl-sn-glycero-3-ethylphosphocholine:cholesterol/anti-microRNA oligonucleotides (+/–) (4/1) nanosystem exhibits the ability to efficiently deliver anti-microRNA oligonucleotides targeting the overexpressed microRNAs miR-21, miR-221, miR-222, and miR-10 in PDCA cells, promoting an almost complete abolishment of microRNA expression. Silencing of these microRNAs resulted in a significant increase in the levels of their targets. Moreover, the combination of microRNA silencing, namely miR-21, with low amounts of the chemotherapeutic drug sunitinib resulted in a strong and synergistic antitumor effect, showing that this combined strategy could be of great importance for therapeutic application in PDAC.
Keywords: pancreatic cancer gene therapy, anti-microRNAs oligonucleotides, delivery nanosystems, albumin-associated lipoplexes
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most predominant type of pancreatic cancer, accounting for more than 90% of new pancreatic cancer cases.1 This disease still remains a therapeutic challenge since only minor significant advances have been achieved, and always with a modest clinical impact.2 Despite its moderate incidence when compared to other carcinomas, PDAC has one of the highest mortality rates, and very low survival improvements have been made over the past 30 years.3 This fact is mainly due to asymptomatic features leading to a late diagnosis in an advanced state of the disease, where early and aggressive metastization to distant organs has already occurred. The overall median survival is 2–8 months, and only 1%–4% of all patients with pancreatic carcinoma survives for 5 years.4 Therefore, there is a pressing need for developing new and efficient therapeutic strategies for pancreatic cancer.
Accumulated evidence has shown that microRNAs are key regulators in cancer, managing a variety of biological processes relevant for tumor development such as proliferation, angiogenesis, and metastization, and therefore they constitute highly promising targets for antitumor therapies.5 These endogenous small noncoding RNA molecules of approximately 22 nucleotides display a critical role as epigenetic regulators of gene expression, acting posttranscriptionally through binding to their target messenger RNA (mRNA), resulting in translation inhibition. Microarray technology has revealed differential microRNA expression patterns depending on the tissue and cell type, and even on the developmental stages of a tumor. In the latter case, the cellular phenotype originated by the disruption of microRNA regulation is not yet well established, demanding a deeper and meticulous investigation.6
At early stages of cancer progression, profound alterations occur in microRNA levels, and oncomiR become overexpressed, whereas tumor-suppressor microRNAs become downregulated, leading to tumor growth and/or repression of apoptosis. A large number of studies have shown that microRNAs, such as the aberrantly expressed microRNAs miR-21, miR-221, miR-222, and miR-10b, act as leading mediators in cancer due to their ability to support tumoral development and cancer cell resistance to chemotherapy.7–9 MiR-21 was shown to regulate the function of several tumor suppressor genes, including PTEN, a phosphatase and tensin homolog gene that negatively regulates the PIK3/Akt survival pathway.10–13 miR-221 and miR-222 are known to target the cyclin-dependent kinase inhibitor p27Kip1, which exerts its antiproliferative action at the G1 phase of the cell cycle, its function being frequently inactivated in many lethal human epithelial cancers.14,15 A few studies have identified miR-10b as a tumor promoter that determines the extent of the expression levels of the homeobox D10 (HoxD10) gene and consequently the RHOC prometastatic gene as a downstream signaling target, both of these genes being involved in metastatic processes in several types of cancers.16,17
The increase of tumor suppressor gene expression has been a successful approach in antitumor strategies, particularly by promoting cell chemosensitivity to a broad range of therapeutic drugs used in cancer treatment.18 For instance, downregulation of miR-21 was reported to directly reinforce susceptibility of breast cancer cells to chemotherapy.10 Modulation of gene expression involves delivery of oligonucleotides against the overexpressed microRNAs. In this regard, cationic liposome/DNA complexes (“lipoplexes”) have been extensively studied, aiming at developing appropriate nanosystems for nucleic acid delivery.19,20 Much effort has been devoted to the synthesis of new cationic lipids, selection of different helper lipids, and association of proteins or fusogenic peptides aiming at enhancing lipoplex biological activity.21–24 Coating cationic liposomes with the most abundant plasma protein, albumin, alleviates some of the undesired interactions between cationic liposome/DNA complexes and serum components, and facilitates intracellular gene delivery by inducing lipoplex binding and uptake into target cells and by promoting endosome membrane destabilization under acidic conditions.25,26 Our previous observations indicated that association of albumin to lipoplexes, prepared with 1-palmitoyl-2-oleoyl-sn-glycero-3-ethylphosphocholine (EPOPC):cholesterol (Chol) cationic liposomes at the 4/1 lipid/DNA (+/-) charge ratio, strongly increases their transfection activity with reporter and therapeutic genes in several types of cells, both in vitro and in vivo, showing the high gene delivery efficiency of this nanosystem.27,28 Nevertheless, the human serum associated (HSA)-EPOPC:Chol/DNA (+/-) (4/1) lipoplex nanoformulation was never tested as an oligonucleotide delivery system.
Although gemcitabine constitutes the current front line therapy for pancreatic cancer, with a better outcome in unresectable tumor cases, new drugs are becoming the focus of attention for the treatment of progressive pancreatic neuroendocrine tumors, namely sunitinib malate, which has been recently approved for this purpose in clinical trials.29,30 Sunitinib is a competitive inhibitor of the catalytic activity of a strictly related receptor tyrosine kinase (RTK) group, including vascular endothelial growth factor receptors and platelet-derived growth factor receptors, and due to its multitargeted profile, the activity of sunitinib is likely mediated by multiple distinct antitumor mechanisms.31 This drug acts by blocking the activity of those RTKs in major pathways related with tumor growth, proliferation, and metastasis dispersal, thus exhibiting a potent antitumor and antiangiogenic effect.32 For patients with advanced stage of pancreatic adenocarcinoma who have been first submitted to gemcitabine-based treatments with no significant results, there are no reliable second line therapies, and sunitinib has already been pointed out as a promising drug for treating such patients.33
Considering the instrumental role of microRNAs in tumorigenesis and the success of combining several drugs targeting major effectors of the tumorigenic process as the most promising treatment for this disease, we evaluated the potential of a new therapeutic strategy based on the combination of low amounts of chemotherapeutic drugs and oligonucleotides against different microRNAs, delivered by the developed albumin-associated nanosystem, aiming at achieving a significant and synergistic antitumor effect in PDAC.
Materials and methods
Cell lines and culture conditions
The Hs766T cell line was obtained from American Type Culture Collection (Manassas, VA, USA) and used as an in vitro tumoral model for human pancreatic carcinoma. The cells were maintained in adherent culture using Dulbecco’s Modified Eagle’s Medium (DMEM) (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) and 100 μM each of penicillin and streptomycin from Sigma-Aldrich (St Louis, MO, USA). HPNE, an immortalized normal pancreatic epithelium cell line, was kindly provided by Dr Ming-Sound Tsao from the Ontario Cancer Institute (Toronto, ON, Canada). HPNE cells were grown in keratinocyte serum-free medium, supplemented with epidermal growth factor, bovine pituitary extract, and 1× antibiotic-antimycotic (all reagents from Thermo Fisher Scientific). Both Hs766T and HPNE cells were grown at 37°C, under 5% CO2, in humidified atmosphere.
Antisense inhibitors and drugs
Anti-microRNA oligonucleotides (AMOs) against miR-221, miR-222, miR-21, and miR-10b and scrambled oligonucleotides (control), as well as 5′-fluorescein-labeled oligonucleotides for confocal microscopy and digoxigenin (DIG)-labeled oligonucleotides for miR-21 detection, were all purchased from Exiqon (Vedbaek, Denmark) as miRCURY locked nucleic acids (LNATM).
The chemotherapeutic drugs docetaxel and gemcitabine were purchased from Sigma-Aldrich, and stock solutions were prepared in distilled water and subsequently stored at −20°C and at room temperature, respectively. Sunitinib malate (Sutent®) was kindly offered by Pfizer, Inc. (New York, NY, USA), and the stock solutions were prepared in dimethyl sulfoxide (Sigma-Aldrich) and stored at −20°C.
Preparation of cationic liposomes and lipoplexes
Small unilamellar cationic liposomes were prepared with lipids (Avanti Polar Lipids, Alabaster, AL, USA) dissolved in CHCl3. The cationic lipid EPOPC and Chol were mixed at the 1:1 molar ratio and dried under a nitrogen flux. The dried lipid film was then rehydrated with deionized water to a final lipid concentration of 4 mM. The obtained multilamellar liposomes were then submitted to sonication for 3 minutes and extruded 21 times through two staked polycarbonate filters of 50 nm pore diameter using a Lipofast device (Avestin Inc., Ottawa, ON, Canada) to obtain small unilamellar liposomes. Finally, the liposome suspension was diluted three times with deionized water and filter-sterilized using a 0.22 μm pore-diameter filter (Schleicher and Schuell BioScience, Dassel, Germany). The suspension was stored at 4°C until use. For intracellular distribution studies, EPOPC:Chol liposomes were labeled with 0.1% carboxyfluorescein-dioleoylphosphatidylethanolamine. Lipoplex preparation was performed by adding the components in the following order: HEPES-buffered saline solution (100 mM NaCl; 20 mM Hepes, pH 7.4), liposome suspension in the appropriate amount to achieve the 4/1 (+/−) lipid/LNA charge ratio, and HSA solution at a ratio of 32 μg of HSA/μg of AMOs. This mixture was incubated at room temperature for 15 minutes, and the necessary amount of LNAs was gently added and submitted to a further 15-minute incubation period.
Mean diameter and zeta potential
Nanosystems were characterized with respect to their mean diameter and zeta potential using a Zetasizer Nano ZS (Malvern Instruments Ltd, Worcestershire, UK), which measures these parameters by a phase analysis light scattering method. The analysis was performed at 25°C in HEPES-buffered saline solution, and lipoplexes were prepared immediately before analysis.
Transfection assays
Transfection assays were performed in Hs766T cells using HSA-EPOPC:Chol/AMOs (+/−) 4/1 lipoplexes containing AMOs or scrambled oligonucleotides. For RNA expression analysis, 1.5×105 cells/well were seeded in 12-well culture plates 24 hours before transfection, aimed at achieving 80% of confluence. For target protein analysis, 3×105 cells/well were seeded in 6-well culture plates 24 hours before transfection. For cell viability assays, 0.35×105 cells/well were seeded in 48-well culture plates 24 hours prior to transfection. Before transfection, cell medium was replaced by DMEM-high glucose (HG) medium without serum or antibiotics, and then 0.15 mL of lipoplexes per 1 mL of DMEM were gently added to each well and incubated with cells for 4 hours (5% CO2 at 37°C). After this period of incubation, cell medium was replaced by DMEM-HG with serum and antibiotics, and cells were further incubated for 48 hours and 72 hours for RNA and protein analysis, respectively, and for 48 hours and 72 hours for cell viability assays.
Intracellular distribution of lipoplexes and oligonucleotides
In order to evaluate intracellular distribution of the nanosystems, Hs766T cells seeded in 12-well culture plates (previously covered with coverslips) 24 hours before transfection were incubated with lipoplexes prepared from carboxyfluorescein-labeled EPOPC:Chol liposomes for 4 hours in DMEM-HG (without serum or antibiotics). The transfection medium was then removed, and cells were carefully washed twice with a phosphate-buffered saline solution (PBS) and incubated for 30 minutes with 200 nM of Lysotrack Red DND-99, purchased from Molecular Probes (Thermo Fisher Scientific), which labels acidic compartments of living cells. Cells were washed three times with PBS and fixed with 4% of paraformaldehyde solution for 15 minutes at room temperature. Nuclei labeling was accomplished through 5-minute incubation at room temperature with the fluorescent DNA binding dye Hoechst 33342 (1 μg/mL) (Thermo Fisher Scientific). Cells were then mounted in Mowiol 40–88 medium from Sigma-Aldrich, and images were taken in a confocal microscope (LSM-510 META; Carl Zeiss Meditec AG, Jena, Germany) using a 40× objective. For evaluation of cytoplasmic delivery of AMOs, lipoplexes were prepared with 100 nM of 5′-fluorescein-labeled AMOs, and cells were submitted to the previously described protocol in order to acquire confocal microscopy images.
Extraction of total RNA and complementary DNA synthesis
Total RNA was isolated from transfected cells and purified using the miRCURY RNA Isolation Kit – Cell and Plant (Exiqon), following the manufacturer’s protocol. Briefly, after cell lysis, the total RNA was adsorbed to a silica matrix, washed with the recommended buffers, eluted with elution buffer by centrifugation, and quantified in a Nanodrop UV-Vis spectrophotometer (Thermo Fisher Scientific).
For quantification of microRNA expression levels, one first-strand complementary DNA (cDNA) synthesis reaction was performed using the Universal cDNA Synthesis Kit (Exiqon), providing a template for all microRNA real-time assays, by performing a 60-minute incubation at 42°C, followed by a heat inactivation step of the reverse transcriptase for 5 minutes at 95°C. Finally, cDNA was diluted 1:60 with RNase free water and stored at 4°C. For determination of target mRNA expression levels, cDNA synthesis was performed using the One Strand cDNA Synthesis Kit purchase from BioRad (Hercules, CA, USA). cDNA was then incubated for 5 minutes at 25°C, 30 minutes at 42°C, followed by a heat inactivation step of the reverse transcriptase for 5 minutes at 85°C. Finally, cDNA was diluted 1:3 with RNase-free water and stored at 4°C.
Quantitative real-time PCR
For quantification of microRNA expression levels, the resulting cDNA was submitted to real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) using the specific primer set for each microRNA under analysis, specifically miR-221, miR-222, miR-21, miR-10b, and the reference RNA (U6 small nuclear RNA), in combination with miRCURY LNA Universal RT microRNA PCR system from Exiqon. A master mix was designed for each primer set, according to the kit manufacturer’s recommendations for the real-time PCR setup of individual assays. For each reaction, performed in duplicate, 6 μL of master mix were added to 4 μL of template cDNA. The reactions were monitored using a real-time instrument ABI Prism 7300 qPCR System from Applied BioSystems (Thermo Fisher Scientific). The PCR conditions were 10 minutes at 95°C for polymerase activation and 40 cycles of amplification with 10 seconds at 95°C and 1 minute at 60°C; ramp-rate 1.6°C/second. Threshold values for threshold cycle determination (Ct) were generated automatically by the StepOne Software v.2.2.2 from Applied Biosystems (Thermo Fisher Scientific, Waltham, MA, USA).
For quantification of target mRNA expression levels, the resulting cDNA was subjected to real-time qRT-PCR using the specific primer set for each target mRNA under analysis, obtained from Qiagen (Venlo, the Netherlands), and iQ SYBR Green Supermix Kit from BioRad. Each reaction was performed in duplicate, by adding 10 μL of master mix to 2.5 μL of template cDNA. The reaction conditions consisted of enzyme activation at 95°C for 10 minutes, followed by 40 cycles at 95°C for 15 seconds (denaturation), 30 seconds at 55°C (annealing), and 35 seconds at 72°C (elongation).
For both microRNA and mRNA quantification, a melting curve protocol was started immediately after amplification and consisted of 1 minute heating at 55°C, followed by 80 steps of 10 seconds with a 2°C increase at each step. Threshold values for Ct determination were generated automatically by the StepOne Software v.2.2.2. Relative microRNA and mRNA levels were determined following the ΔΔCt method in comparison with control cells.
Western blot analysis
Seventy-two hours after transfection, Hs766T cells were washed twice with a PBS and solubilized in RIPA buffer (25 mM Tris-HCl, pH 7.7; 150 nM NaCl; 1% NP-40; 1% sodium deoxycholate; 0.1% sodium dodecyl sulfate) containing a protease inhibitor cocktail from Sigma-Aldrich, 2 mM dithiothreitol, and 0.1 mM phenylmethylsulfonyl fluoride. The whole-cell suspension was subjected to sonication for 3 seconds and centrifuged at 14,000 rpm for 8 minutes at 4°C. The supernatant was collected and stored at −20°C until use. Protein concentration was determined using the Bio-Rad Dc protein assay. Heat-denatured protein samples (40 μg per lane) were resuspended in loading buffer (20% glycerol, 10% sodium dodecyl sulfate, 0.1% bromophenol blue), loaded onto a 10% polyacrylamide gel, and resolved by electrophoretic separation. After electrophoresis, proteins were transferred to a polyvinylidene fluoride membrane (Millipore Corporation, Billerica, MA, USA). The membrane was blocked for nonspecific binding for 60 minutes in a Tris-buffered saline solution (TBS) containing 1% of Tween 20 (TBS-T) and 5% of bovine serum albumin (BSA), followed by incubation overnight at 4°C with primary antibodies: rabbit monoclonal antibody against p27Kip1 protein (Cell Signaling Technology, Danvers, MA, USA), rabbit monoclonal antibody against PTEN protein (Cell Signaling Technology), rabbit monoclonal antibody against HoxD10 protein (Abcam, Cambridge, UK), and mouse monoclonal antibody against RHOC protein (Abcam). The primary antibodies were diluted at 1:2,000 in TBS-5% milk or TBS-5% BSA. The membrane was washed three times with TBS-T for 10 minutes and then incubated for 1 hour at room temperature with goat anti-rabbit antibody (GE Healthcare UK Ltd, Little Chalfont, UK) at a dilution of 1:10,000 as a secondary antibody for p27Kip1, PTEN, and HoxD10, and with a goat anti-mouse antibody (GE Healthcare) at a dilution of 1:10,000 as a secondary antibody for RHOC. The membrane was washed thoroughly in a TBS-T solution, and the bound antibody was detected using the enhanced chemifluorescence detection reagent (ECF; GE Healthcare), according to the manufacturer’s recommendations. Images were obtained using a VersaDoc Imaging System Model 3000 from BioRad, and detection was performed at 570 nm. The analysis of band intensity was made using the Quantity One software also from BioRad.
Fluorescence in situ hybridization
Fluorescence in situ hybridization was performed as described by Lu and Tsourkas34 with some modifications. Briefly, Hs766T cells were seeded onto multichambered cover glass slides (Lab-Tek, Rochester, NY, USA) appropriate for confocal microscopy. Forty-eight hours after transfection with lipoplexes containing anti-miR-21 or scrambled (control) oligonucleotides, cells were washed with PBS, fixed with 4% paraformaldehyde for 30 minutes at room temperature, and permeabilized at 4°C in 70% ethanol for 4 hours. Cells were then incubated with fresh acetylation solution (0.1 M triethanolamine and 0.5% [volume/volume {v/v}] acetic anhydride) for 30 minutes at room temperature, rinsed twice in TBS, and prehybridized in the absence of LNA probe in the hybridization buffer (50% formamide, 5× SSC [0.75 M NaCl, 0.075 M sodium citrate], 5× Denhardt’s solution [1% Ficoll {type 400}, 1% polyvinylpyrrolidone, and 1% BSA], 250 μg/mL yeast transfer RNA, 500 μg/mL salmon sperm DNA, 2% [weight/volume {w/v}] blocking reagent [Roche, Basel, Switzerland], 0.1% CHAPs, 0.5% Tween 20) for 2 hours at 52°C. The hybridization step was carried out overnight, at the same temperature, using the DIG-labeled LNA probe for miR-21 and a scrambled probe. Three stringency washes were performed also at 52°C to completely remove the nonhybridized probe. Endogenous peroxidase activity was inactivated by incubation in 3% hydrogen peroxide in TBS-T for 30 minutes, followed by three washes with TBS-T. The slides were then placed in blocking solution (10% heat-inactivated goat serum and 0.5% blocking agent in TBS-T) for 1 hour at room temperature and incubated for the same period of time with a hydrogen peroxide-conjugated anti-DIG antibody (Roche) purchased from Sigma-Aldrich. To amplify the antibody signal, slides were further incubated with a tyramide signal amplification plus cyanine 3 solution (PerkinElmer Inc., Waltham, MA, USA) for 10 minutes in the dark, in accordance with the manufacturer’s protocol. Cells were finally stained with the fluorescent DNA-binding dye Hoechst 33342 (1 μg/mL) (Thermo Fisher Scientific) for 5 minutes in the dark, washed with cold PBS, and mounted in Mowiol 40–88 medium. Confocal images were acquired using a point scanning confocal microscope Zeiss LSM 510 Meta, using a 60× oil objective.
Cell viability assays
Cellular viability and proliferation were evaluated by a modified alamar blue assay, under different experimental conditions.35 This assay measures the redox capacity of tumoral cells and allows the determination of cell viability without cell detachment. To evaluate the effect of the combined strategies involving AMOs and chemotherapeutic drugs, cells were seeded onto 48-well culture plates and transfected as mentioned in the section “Transfection assays”. Twenty-four hours after transfection, cells were treated with different amounts of drugs for a period of 24 hours, and cell viability was then measured. Briefly, 300 μL of DMEM-HG medium containing 10% (v/v) alamar blue dye (prepared from a 0.1 mg/mL stock solution of alamar blue) was added to each well, and cells were incubated at 37°C for 1 hour in a 5% CO2 humidified atmosphere. One-hundred and fifty microliters of supernatant was collected from each well, transferred to 96-well plates, and absorbance was measured at 570 nm and 600 nm in a SPECTRAmax PLUS384 spectrophotometer (Molecular Devices, Union City, CA, USA). Cell viability (as a percentage of untreated control cells) was calculated according to the formula,
| (1) |
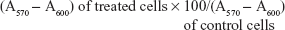
were A is absorbance.
Statistical analysis
Data are presented as the mean ± standard deviation. The data were analyzed using the GraphPath Prism, version 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA). Statistical analyses were performed by analysis of variance (ANOVA) using Dunnett’s multiple comparison test or Student’s t-test. P-value <0.05 was considered statistically significant.
Results and discussion
Efficient oligonucleotide delivery mediated by HSA-EPOPC:Chol/AMOs (+/−) (4/1) lipoplexes
Cationic liposomes brought new insight into bionanotechnology by facilitating the introduction of nucleic acids into target cells, thus allowing a controlled modification of their genetic expression profile and, consequently, a specific effect.23,36,37 However, most of the nanosystems based on cationic liposomes have been applied for DNA delivery, but whether these carriers have the ability to promote efficient and controlled release of oligonucleotides into tumor cells has yet to be demonstrated. Therefore, our initial studies addressed the potential of a previously developed gene delivery formulation, HSA-EPOPC:Chol/DNA (+/−) (4/1), to efficiently release AMOs targeting overexpressed microRNAs involved in cancer, towards the generation of a new therapeutic approach.
The analysis of the physicochemical properties of our HSA-EPOPC:Chol/AMOs (+/−) (4/1) formulation reveals a neutral zeta potential (0.4±1.5 mV), which is most probably due to the presence of HSA that masks the positive charge from cationic liposomes and a mean diameter of approximately 450 nm. This neutral zeta potential indicates that the interaction of lipoplexes with the negatively charged cellular membrane is not due to electrostatic interactions, but rather to the interaction of the associated HSA with cytoplasmic membrane receptors.26 The cellular internalization of these lipoplexes, prepared from carboxyfluorescein-labeled liposomes, was evaluated in a PDAC cell line (Hs766T cells) by confocal microscopy. As illustrated in Figure 1A, an intense green fluorescence, corresponding to lipoplexes, was observed throughout the cytoplasm of almost all cells, demonstrating efficient cellular internalization of this nanosystem. Moreover, the results presented in Figure 1A show that lipoplexes (green fluorescence) were not colocalized with the lysosomal compartments (red fluorescence), suggesting their successful release from the endolysosomal pathway to the cytoplasm, consequently avoiding nucleic acid degradation inside the lysosomes.
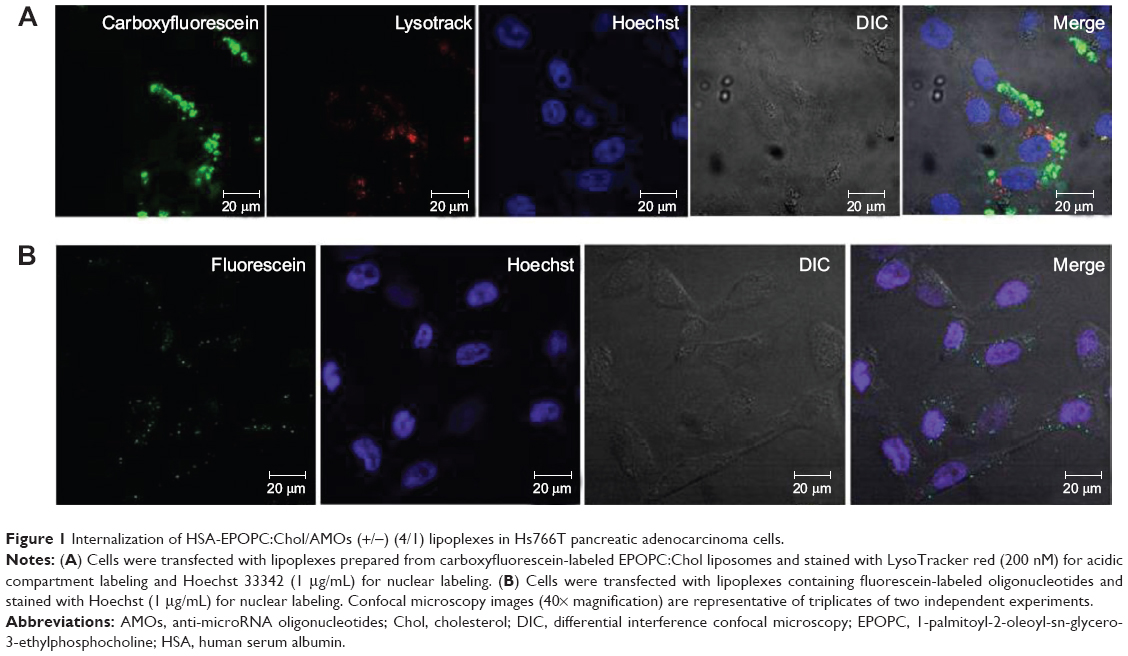 | Figure 1 Internalization of HSA-EPOPC:Chol/AMOs (+/−) (4/1) lipoplexes in Hs766T pancreatic adenocarcinoma cells. |
In order to evaluate the efficacy of the HSA-EPOPC:Chol/AMOs (+/−) (4/1) nanosystem to mediate the intracellular delivery of AMOS, Hs766T cells were transfected with lipoplexes prepared with fluorescein-labeled oligonucleotides. As shown in Figure 1B, 4 hours after transfection, fluorescent particles (green dots) were homogeneously distributed throughout the cytoplasm of almost all cells. This observation suggests that the HSA-EPOPC:Chol/AMOs (+/−) (4/1) lipoplex formulation has the ability to efficiently complex AMOs, promote binding and internalization into tumor target cells, and deliver their content into the cell cytoplasm, showing that this nanosystem presents great potential to be used in antitumor strategies involving the delivery of AMOs.
Robust microRNA inhibition after transfection with HSA-EPOPC:Chol/AMOs (+/−) (4/1) lipoplexes
Each type of cancer can be characterized by a distinct microRNA signature, and emerging evidence indicates that several microRNAs, such as miR-221/miR-222, miR-21, and miR-10b, display a significant role in managing tumor survival and aggressiveness.38
MiR-221 and miR-222 are known to target the tumor suppressor gene coding for the cyclin-dependent kinase inhibitor p27Kip1, and their role was established in PDCA as key inhibitors of cell cycle arrest, apoptosis, and sensitization of cells to gemcitabine.7,15,39 Upregulation of these two microRNAs is often related with poor patient survival rate.40,41 MiR-21 has been identified as an upregulated microRNA in almost all cancer types, including PDAC, and among other important microRNAs involved in tumoral regulation, miR-21 stood out as the one with most significant expression in PDCA associated to metastatic status or proliferation index.42,43 Studies performed by Nakata et al indicated that miR-10b is also an upregulated microRNA in pancreatic cell lines, with up to 10-fold increased levels as compared to normal cells.17 Moreover, these authors showed that transfection with miR-10b was associated with invasiveness of pancreatic cancer cell lines, making miR-10b another important microRNA to be manipulated towards the development of new PDAC therapies.
Although this group of four upregulated microRNAs have been pointed out as key regulators of tumorigenesis, involved in processes such as proliferation, angiogenesis, and metastization, there are other important microRNAs implicated in these major biological processes that also constitute vital targets in oncogenesis.
In order to determine the expression profile of miR-221, miR-222, miR-21, and miR-10b in an in vitro metastatic model of pancreatic adenocarcinoma, the levels of these microRNAs were measured in Hs766T cells. As illustrated in Figure 2, these four microRNAs are differentially expressed in this cell line. The microRNA cluster miR-221/miR-222 is more prominently overexpressed (14-fold and 8 fold-increase, respectively) than miR-21 and miR-10b, both exhibiting a 3-fold-increase in their expression levels when compared with those obtained in an in vitro model of normal pancreatic epithelium (HPNE cell line).
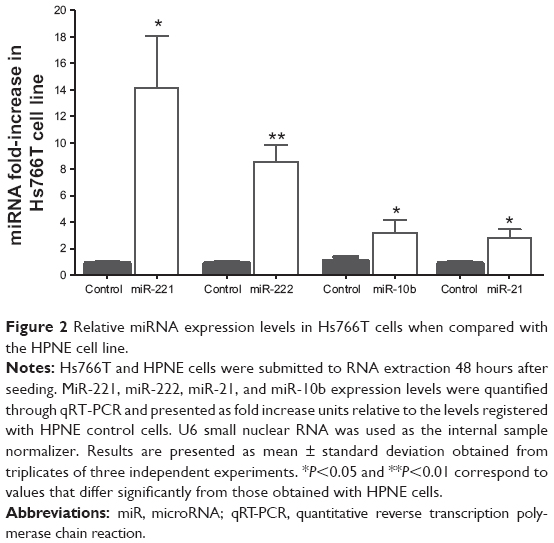 | Figure 2 Relative miRNA expression levels in Hs766T cells when compared with the HPNE cell line. |
Following the demonstration of the feasibility of the HSA-EPOPC:Chol-based nanosystem to mediate efficient delivery of AMOs, we evaluated the effect of intracellularly delivered anti-microRNAs to promote microRNA silencing. For this purpose, Hs766T cells were transfected with lipoplexes containing antisense oligonucleotides targeting miR-221, miR-222, miR-21, or miR-10b (scrambled oligonucleotides were used as a control), and the expression levels were analyzed for each microRNA after 48 hours. As shown in Figure 3A, a significant reduction in the levels of all tested microRNAs was obtained when cells were transfected with 80 nM of AMOs. In the case of miR-21, nearly to 99% of microRNA silencing was achieved. A similar inhibition pattern was observed for miR-10b, with a 94% decrease in its levels, and a maximum inhibition of 79% and 89% was attained for the expression levels of miR-221 and miR-222, respectively. Concentrations of AMOs higher than 80 nM did not show further significant improvement in reducing the levels of microRNAs, and lower concentrations resulted in a lesser efficacy of microRNAs inhibition (data not shown), revealing that 80 nM was the optimal AMOs concentration to promote microRNA silencing.
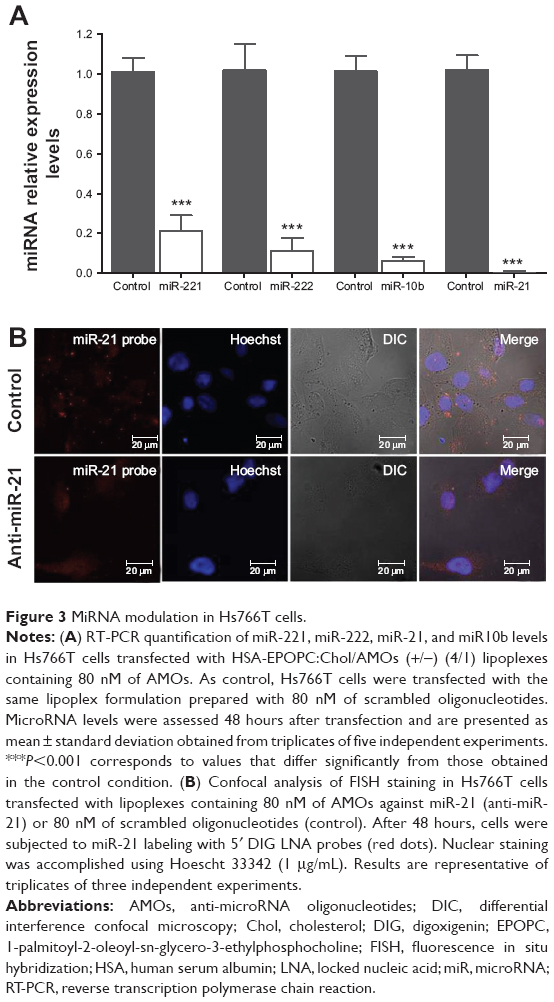 | Figure 3 MiRNA modulation in Hs766T cells. |
The efficient miR-21 silencing in Hs766T cells was also evident from fluorescence in situ hybridization experiments. Figure 3B displays typical images obtained from these essays, showing a huge decrease in miR-21 staining (red dots) in the cell cytoplasm following transfection with our nanosystem containing anti-miR-21 oligonucleotides when compared to that observed in cells transfected with lipoplexes prepared with scrambled oligonucleotides. These results are in agreement with those showing the high intracellular delivery of AMOs promoted by HSA-EPOPC:Chol/AMOS (+/−) (4/1) lipoplex formulation (Figure 1B), thus demonstrating the efficacy of the developed nanosystem to mediate microRNA silencing.
MicroRNA targets are differentially modulated in PDAC
In previous reports, p27kip1 protein was shown to play an important role in regulating cell cycle arrest, being described as a potential target in prostate cancer treatment,40 and both miR-221 and miR-222 were considered as potent regulators of cell cycle through p27kip1 protein.39
On the other hand, Meng et al11 and Liu et al44 pointed to miR-21 as responsible for directly modulating the expression of the PTEN gene in hepatocarcinoma, showing that the decrease of miR-21 levels results in the decline of hepatocarcinoma cell proliferation, acceleration of apoptosis, and cell invasiveness decay. PTEN is a tumor suppressor factor, directly acting as a central negative regulator in playing a dual phosphatase activity in the PI3K signaling pathways, which in turn control major biological processes such as cellular growth, proliferation, and protein synthesis. Several reports have suggested PTEN is an important mediator of carcinogenesis in pancreas.45,46 However, tumor samples collected from pancreatic cancer patients exhibit only an estimated 1% of PTEN mutations, which points towards the hypothesis of posttranscriptional regulation of the expression levels of the PTEN gene, most likely involving microRNAs as the most important mechanism in this process.47
MiR-10b was also identified as a microRNA with altered expression patterns in several cancers, being associated to suppression of HoxD10 protein synthesis and consequently allowing the expression of the RHOC gene, as a downstream signaling target, which is known to be involved in metastatic processes by inducing cell migration.16,48–50
The recognized oncomiR activity of these four microRNAs and their demonstrated deregulation in PDAC make them promising targets for new therapeutic strategies involving AMO delivery. In this regard, we further evaluated the effect of microRNA silencing on the mRNA and protein levels of the molecular targets p27kip1, PTEN, HoxD10, and RHOC, aiming to analyze the potential of the developed nanosystem in a therapeutic context and clarify the mechanisms involved in an antitumor response.
Forty-eight hours after transfection of Hs766T cells with lipoplexes containing anti-miR oligonucleotides, mRNA levels were quantified by qRT-PCR. Our results show that, despite successful microRNA inhibition mediated by the developed nanosystem (Figure 3A), the AMOs targeting miR-221 and miR-222 were not able to significantly increase the p27kip1 mRNA levels (Figure 4A). The lack of total inhibition of miR-221 and miR-222 levels observed after cell transfection might be responsible for this effect. In fact, the remaining miR-221 and miR-222 in cell cytoplasm could be sufficient to induce the posttranscriptional inhibition of the p27kip1 gene. On the other hand, it is also possible that miR-221 and miR-222 are not able to induce cleavage of p27kip1 mRNA, but rather its translational repression by a less efficient mechanism, justifying the absence of a significant increase in the p27kip1 mRNA levels after treatment with anti-miR-221 or anti-miR-222 oligonucleotides.51 As shown in Figure 4B, miR-21 silencing resulted in an increase of approximately 40% in PTEN mRNA levels after cell treatment with AMOs, which is most probably due to the almost total miR-21 silencing induced by the anti-miR-21 oligonucleotides (Figure 3). Regarding miR-10b inhibition, two targets were addressed: HoxD10, as a direct target, and RHOC, as a downstream target but still the main regulator of an important pathway related with cell migration.17 Transfection of Hs766T cells with 80 nM of anti-miR-10b oligonucleotides promoted a 1.9-fold increase in the mRNA levels of HoxD10 gene and 27% decrease in the mRNA levels of RHOC when compared to that observed with cells treated with scrambled oligonucleotides (Figure 4C). This result meets the expected outcome since HoxD10 exerts its regulatory role in the transcription levels of its downstream target, RHOC, negatively setting the expression levels of this gene.50
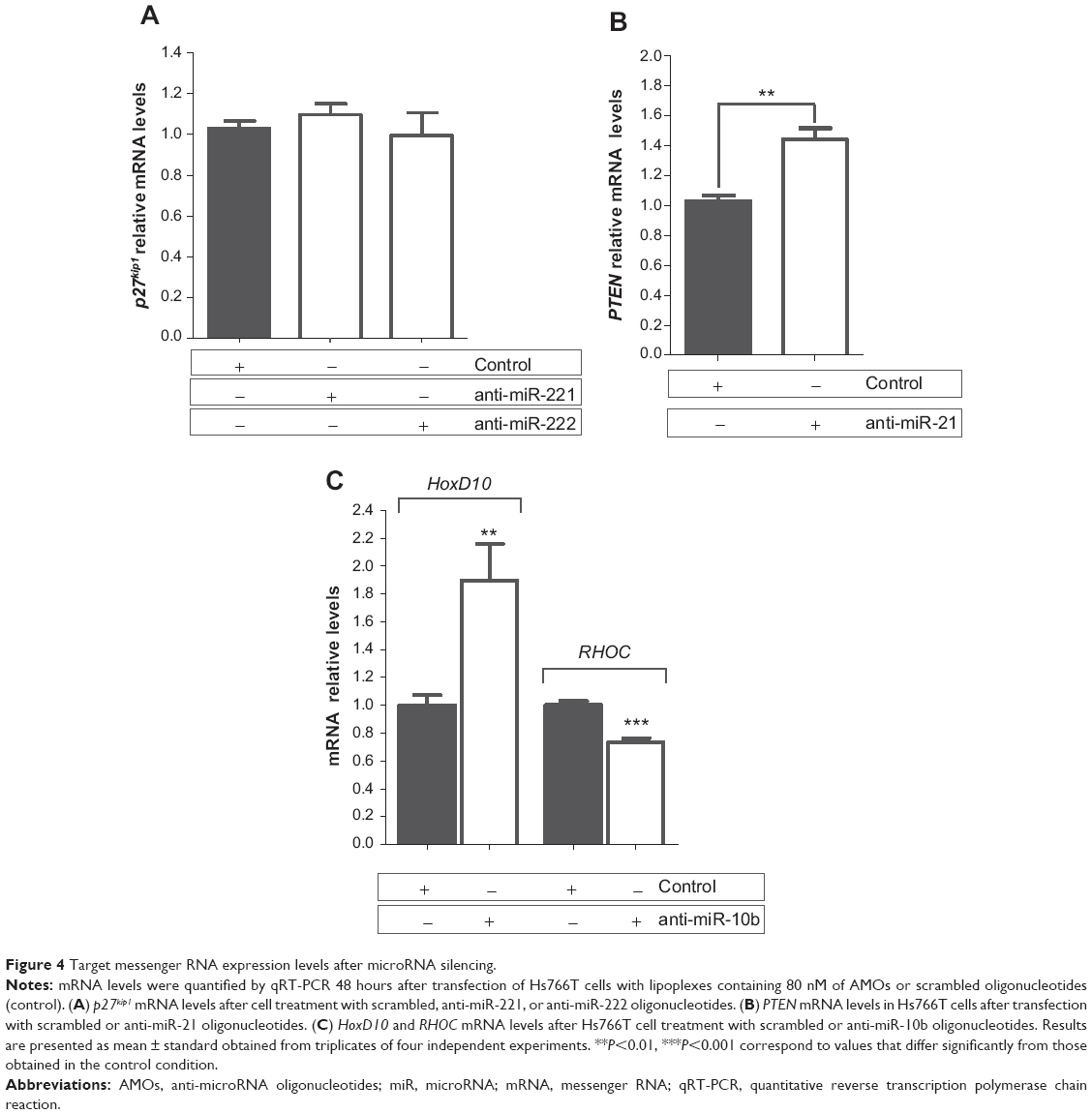 | Figure 4 Target messenger RNA expression levels after microRNA silencing. |
Although no significant changes in p27kip1 mRNA levels were observed after cell treatment with AMOs targeting miR-221 and miR-222, western blot analysis showed an approximately 1.1-fold and 1.6-fold increase of p27kip1 protein levels following treatment with anti-miR-221 and anti-miR-222 oligonucleotides, respectively (Figure 5A and B). The difference found for the increase of p27kip1 expression could be due either to an insufficient miR-221 inhibition (Figure 3), thus avoiding significant increase of the protein levels, or to a predominant role of miR-222 in the posttranscription regulation of the p27kip1 gene. The combination of both AMOs, simultaneously targeting miR-221 and miR-222, did not result in a significant increase of the p27kip1 mRNA and protein levels when compared to that observed with anti-miR-222 alone (data not shown).
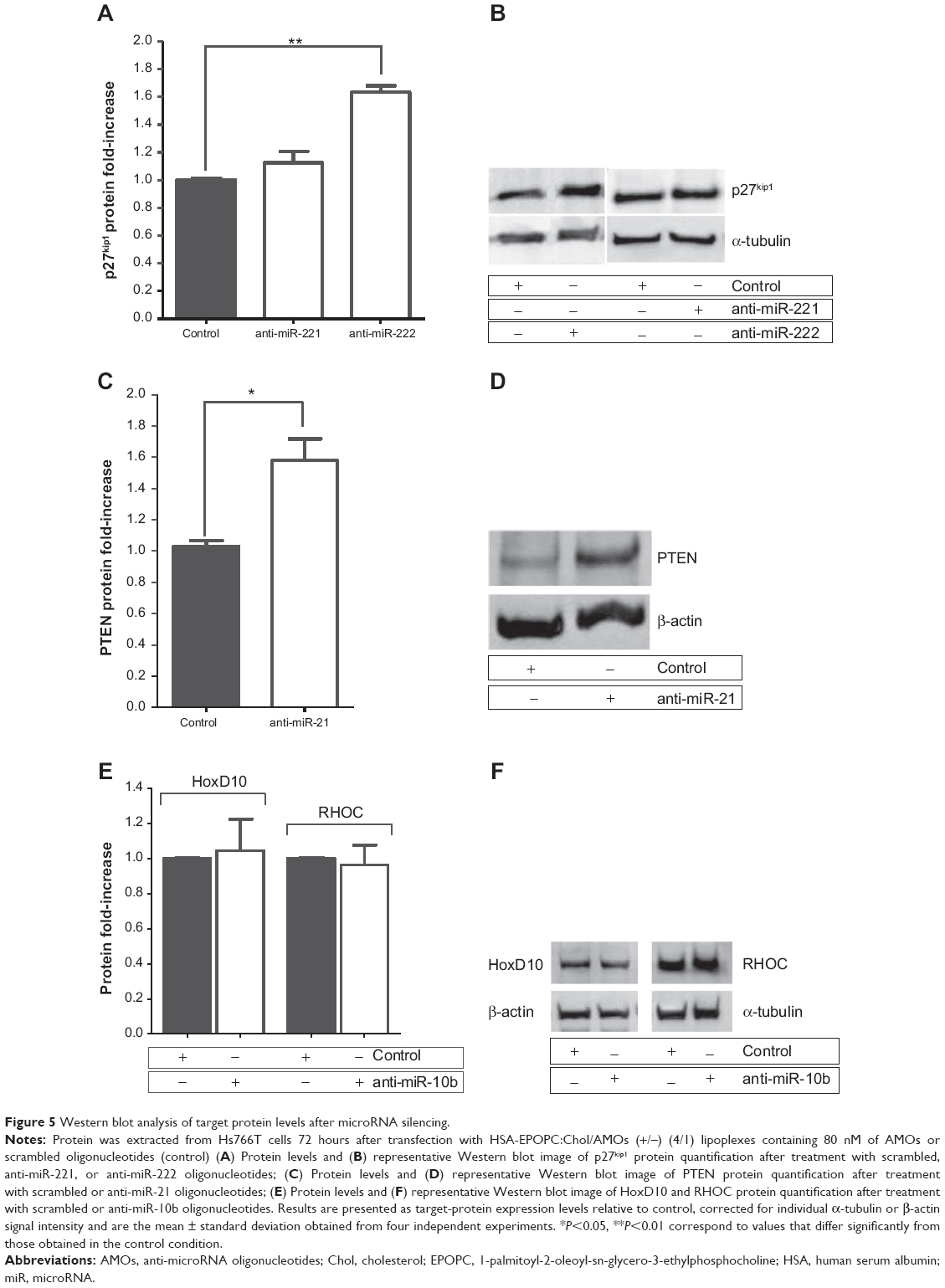 | Figure 5 Western blot analysis of target protein levels after microRNA silencing. |
Regarding PTEN protein expression levels, it was observed that the almost total miR-21 silencing (Figure 3) promoted by transfection of Hs766T cells with lipoplexes containing 80 nM of AMOs against this microRNA resulted in a substantial enhancement in the PTEN levels. In fact, in these conditions, an increase of 60% in the expression levels of this protein was obtained when compared to that observed with Hs766T cells treated with the same amount of scrambled oligonucleotides, showing that our strategy successfully modulates PTEN protein levels.
In the case of HoxD10 and RHOC, although mRNA levels were modulated towards an antitumoral profile, the analysis performed by western blot showed no significant alterations in the protein levels after miR-10b silencing. Despite the effective silencing of miR-10b induced by AMOs treatment, the inability to successfully change HoxD10 and RHOC protein levels led us to consider that both effectors might have a multiplicity of regulators in PDAC that promote their translational repression, which probably demands a more broad strategy to modulate their expression levels.
Overall, the obtained results show that transfection of PDAC cells with the HSA-EPOPC:Chol/AMOs (+/−) (4/1) nanosystem targeting miR-21, miR-221, or miR-222 promotes a significant posttranscriptional modulation of important tumor suppressor genes, such as PTEN and p27kip1, respectively.
Combination of oligonucleotides against miR-21 with sunitinib results in a synergistic antitumor effect in PDAC
Gemcitabine has long been the only standard treatment for pancreatic cancer, but increasing resistance over time impelled researchers to seek other drugs in order to improve patient survival. Docetaxel has been used in combination with gemcitabine as front line therapy to reduce the size of the tumor and overcome its metastatic phase. Nevertheless, neither of these drugs or their combination were revealed to be an effective treatment for pancreatic cancer.52 On the other hand, sunitinib malate, a potent RTK inhibitor, has been demonstrated to be a successful drug in neuroendocrine pancreatic cancer clinical trials, and new data points towards a more meaningful treatment for this disease.53 The possibility of combining the inhibition of microRNAs, thus sensitizing cancer cells by decreasing some of their main regulators of tumorigenesis, with chemotherapeutic drugs that have shown high potential in clinical trials emerged as a promising antitumor strategy. Indeed, combining two or more therapeutic approaches with different mechanisms can exert a synergistic effect over cancer progression and tumor resistance to chemotherapeutic drugs.54
In this regard, we investigated whether a two-step, sequential treatment involving the modulation of aberrantly expressed microRNAs to sensitize tumoral cells to the action of drugs, and the subsequent treatment with chemotherapeutic agents could result in a significant and synergistic antitumor effect. For this purpose, we evaluated the in vitro antitumor activity mediated by HSA-EPOPC:Chol/AMOs (+/−) (4/1) lipoplexes, containing oligonucleotides against miR-221, miR-222, or miR-21 (since these AMOs presented the most promising results in terms of tumor suppressor gene modulation in our PDAC model) in combination with small amounts of chemotherapeutic drugs, docetaxel, gemcitabine, or sunitinib malate. From the results obtained using different doses for each chemotherapeutic agent (data not shown), only the lower concentration resulting in a small but still significant effect on tumor cell viability was chosen to be applied in the combined strategies (1 μM docetaxel, 5 μM gemcitabine, and 15 μM sunitinib). These low amounts of drugs were used to avoid the adverse effects that are usually associated with the higher clinical doses of chemotherapeutic agents. As illustrated in Figure 6, treatment of Hs766T cells with 1 μM of docetaxel, 5 μM of gemcitabine, or 15 μM of sunitinib malate resulted in a decrease of approximately 16%, 10%, and 18% in cell viability, respectively.
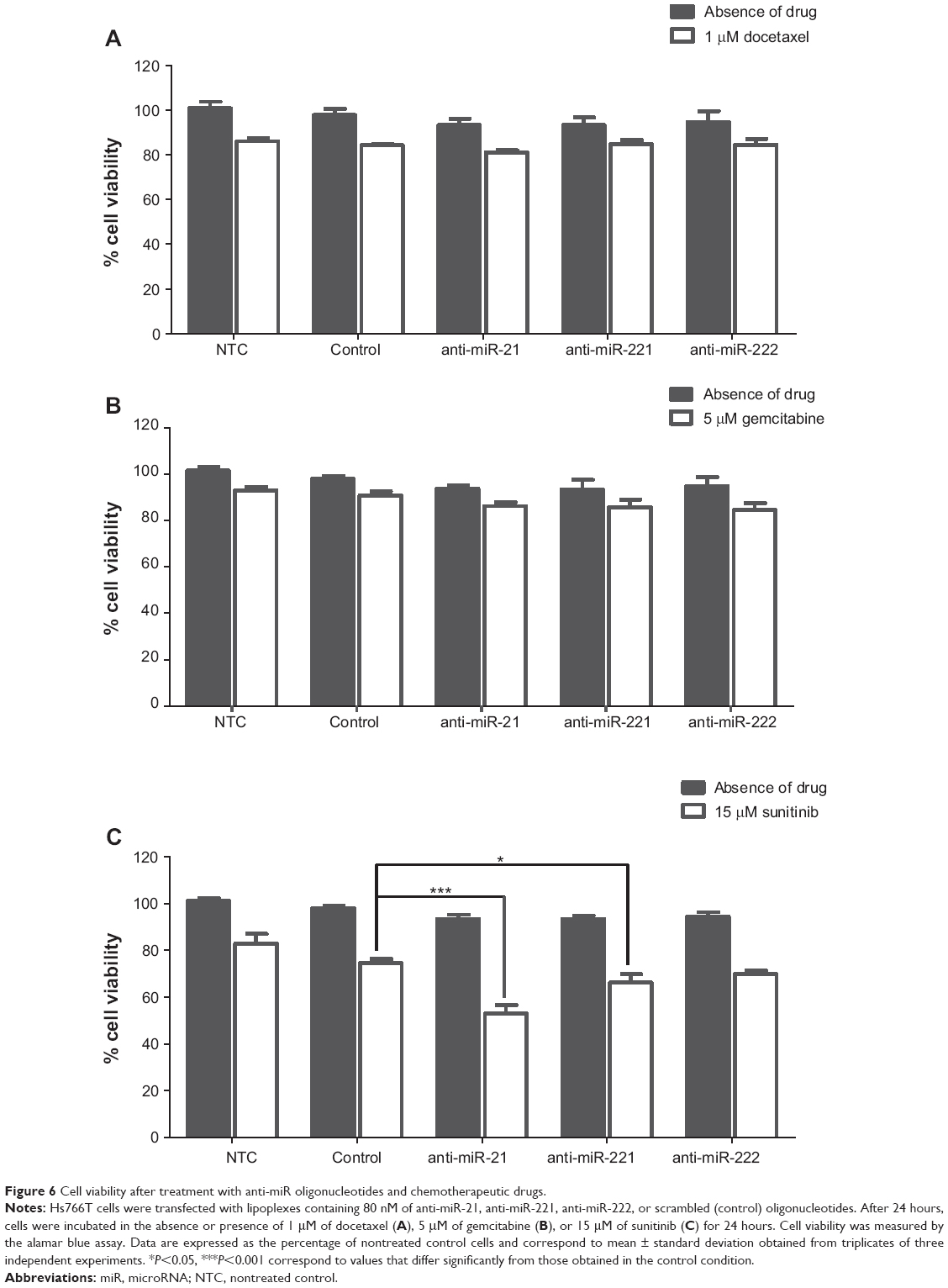 | Figure 6 Cell viability after treatment with anti-miR oligonucleotides and chemotherapeutic drugs. |
The impact of microRNA inhibition per se on cell viability was even smaller than that observed with the low amounts of chemotherapeutic drugs alone, this being verified for any of the three studied microRNAs, miR-21, miR-221, or miR222 (Figure 6). Although target protein expression levels were substantially increased upon microRNA silencing (Figure 5), a reduction of only approximately 5% in the viability of Hs766T cells was obtained when compared to that observed in the control condition (cells treated with scrambled oligonucleotides). Nevertheless, in agreement with other authors, this is an expected result since this kind of approach represents a fine-tuning of molecular signaling rather than a single molecular effector with major impact on cell metabolism/viability.55,56
Combination of AMOs with chemotherapeutic drugs did not result in any significant therapeutic effect in the case of docetaxel or gemcitabine, as no further considerable reduction in cell viability was achieved (Figure 6A and B). However, when Hs766T cells were sequentially treated with AMOs (against miR-21, miR-221, or miR-222) and sunitinib, a substantial reduction in cell viability was observed as compared to the extent of cell death (21%) registered with scrambled oligonucleotides and sunitinib (Figure 6C). Importantly, cell treatment involving the combination of oligonucleotides anti-miR-21 with sunitinib resulted in a cell viability decrease of approximately 45%, showing that this combined strategy promoted a significant and synergistic antitumor effect, which was much higher than that observed with any of the two strategies by themselves (Figure 6C). PTEN, a direct miR-21 target, is an important cell cycle regulator, and therefore its upregulation (Figure 5C and D) strongly affects apoptosis signaling pathways, inducing cell sensitization to sunitinib. Moreover, miR-21 has been pointed out to have great impact in almost all types of cancers since it targets many important protein mediators involved in tumorigenesis, which could also contribute to the high and synergistic antitumor effect of this combined strategy.57–59 On the other hand, miR-221 or miR-222 inhibition followed by treatment with sunitinib promoted a smaller, but still considerable, increase in the antitumor effect, inducing a decrease of 32% in cell viability (Figure 6C). A similar result was obtained with a combined treatment involving simultaneous transfection with anti-miR-221 and anti-miR-222 oligonucleotides (data not shown). Although a different modulation of p27kip1 expression levels was obtained with anti-miR-221 and anti-miR-222 oligonucleotides (Figure 5A and B), a similar reduction in cell viability was observed, showing that these microRNAs may also target other molecular regulators with a crucial role in carcinogenesis that have not been addressed in this study.60
The results obtained with these combined approaches, involving the PDAC cell treatment with AMOs (against miR-21, miR-221, or miR-222) and sunitinib (Figure 6C) are even more remarkable considering the fact that the same combined strategies involving the drugs gemcitabine or docetaxel (Figure 6A and B), which represent the therapeutic front line for PDAC, promoted a much lower antitumor activity.
Conclusion
Overall, our results clearly show that the HSA-EPOPC:Chol/AMOs (+/−) (4/1) nanosystem has the ability to efficiently deliver antisense oligonucletides into PDAC cells, inducing an almost total inhibition of microRNAs (miR-21, miR-10b, miR-221, and miR-222) aberrantly expressed in this cancer model. Moreover, our data constitute evidence that the strong reduction in the levels of these microRNAs resulted in a significant modulation of their targets, this being particularly evident for miR-21 and miR-221/miR-222, where their inhibition promoted a significant increase in the levels of their protein targets, PTEN and p27Kip1 protein, respectively. With this work we demonstrate for the first time the notable synergistic antitumor effect in PDAC cells promoted by the combination of microRNA inhibition with low amounts of the chemotherapeutic drug sunitinib, which is much higher than that obtained with any of the two strategies by themselves. This combined strategy could be of great importance for clinical application in this disease due to the high therapeutic activity and the reduced side effects associated with the use of low drug concentrations.
Acknowledgments
This work was financed by the grants PTDC/SAU-BMA/114482/2009 and PEst-C/SAU/LA0001/2013–2014 from the Portuguese Foundation for Science and Technology and the European Community Fund through the COMPETE program. Marta Passadouro is a recipient of a fellowship from the Portuguese Foundation for Science and Technology (SFRH/BD/46903/2008). The authors would like to thank Ana Luísa Cardoso and Luísa Cortes for their help in obtaining the confocal microscopy images.
Disclosure
The authors report no conflicts of interest in this work.
References
Saif MW. Pancreatic neoplasm in 2011: an update. JOP. 2011;12(4): 316–321. | ||
Hidalgo M, Von Hoff DD. Translational therapeutic opportunities in ductal adenocarcinoma of the pancreas. Clin Cancer Res. 2012;18(16): 4249–4256. | ||
Siegel R, Naishadham D, Jemal A. Cancer Statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29. | ||
Di Marco M, Di Cicilia R, Macchini M, et al. Metastatic pancreatic cancer: is gemcitabine still the best standard treatment? (Review). Oncol Rep. 2010;23(5):1183–1192. | ||
Mardin WA, Mees ST. MicroRNAs: novel diagnostic and therapeutic tools for pancreatic ductal adenocarcinoma? Ann Surg Oncol. 2009; 16(11):3183–3189. | ||
Zhang Y, Li M, Wang H, et al. Profiling of 95 microRNAs in pancreatic cancer cell lines and surgical specimens by real-time PCR analysis. World J Surg. 2009;33(4):698–709. | ||
Szafranska AE, Davison TS, John J, et al. MicroRNA expression alterations are linked to tumorigenesis and non-neoplastic processes in pancreatic ductal adenocarcinoma. Oncogene. 2007;26(30):4442–4452. | ||
Volinia S, Calin GA, Liu CG, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103(7):2257–2261. | ||
Bloomston M, Frankel WL, Petrocca F, et al. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA. 2007;297(17):1901–1908. | ||
Wang ZX, Lu BB, Wang H, Cheng ZX, Yin YM. MicroRNA-21 modulates chemosensitivity of breast cancer cells to doxorubicin by targeting PTEN. Arch Med Res. 2011;42(4):281–290. | ||
Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133(2): 647–658. | ||
Giovannetti E, Funel N, Peters GJ, et al. MicroRNA-21 in pancreatic cancer: correlation with clinical outcome and pharmacologic aspects underlying its role in the modulation of gemcitabine activity. Cancer Res. 2010;70(11):4528–4538. | ||
Han M, Liu M, Wang Y, et al. Antagonism of miR-21 reverses epithelial-mesenchymal transition and cancer stem cell phenotype through AKT/ERK1/2 inactivation by targeting PTEN. PLoS One. 2012;7(6): e39520. | ||
Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8(4):253–267. | ||
Vervoorts J, Lüscher B. Post-translational regulation of the tumor suppressor p27(KIP1). Cell Mol Life Sci. 2008;65(20):3255–3264. | ||
Nakayama I, Shibazaki M, Yashima-Abo A, et al. Loss of HOXD10 expression induced by upregulation of miR-10b accelerates the migration and invasion activities of ovarian cancer cells. Int J Oncol. 2013;43(1):63–71. | ||
Nakata K, Ohuchida K, Mizumoto K, et al. MicroRNA-10b is overexpressed in pancreatic cancer, promotes its invasiveness, and correlates with a poor prognosis. Surgery. 2011;150(5):916–922. | ||
Lai D, Visser-Grieve S, Yang X. Tumour suppressor genes in chemotherapeutic drug response. Biosci Rep. 2012;32(4):361–374. | ||
Pedroso de Lima MC, Neves S, Filipe A, Düzgüneş N, Simões S. Cationic liposomes for gene delivery: from biophysics to biological applications. Curr Med Chem. 2003;10(14):1221–1231. | ||
Simões S, Fonseca C, Faneca H, Düzgünes N, Pedroso de Lima MC. Protein-associated lipoplexes: novel strategies to enhance gene delivery mediated by lipid-based particles, S.T.P. Pharma Sci. 2002;12(1): 339–344. | ||
Hattori Y, Nakamura T, Ohno H, Fujii N, Maitani Y. siRNA delivery into tumor cells by lipid-based nanoparticles composed of hydroxyethylated cholesteryl triamine. Int J Pharm. 2013;443(1–2):221–229. | ||
Yu B, Hsu SH, Zhou C, et al. Lipid nanoparticles for hepatic delivery of small interfering RNA. Biomaterials. 2012;33(25):5924–5934. | ||
Simões S, Filipe A, Faneca H, et al. Cationic liposomes for gene delivery. Expert Opin Drug Deliv. 2005;2(2):237–254. | ||
Pedroso de Lima MC, Simões S, Pires P, Faneca H, Düzgüneş N. Cationic lipid-DNA complexes in gene delivery: from biophysics to biological applications. Adv Drug Deliv Rev. 2001;47(2–3):277–294. | ||
Simões S, Slepushkin V, Pires P, Gaspar R, Pedroso de Lima MC, Düzgüneş N. Human serum albumin enhances DNA transfection by lipoplexes and confers resistance to inhibition by serum. Biochim Biophys Acta. 2000;1463(2):459–469. | ||
Faneca H, Simões S, Pedroso de Lima MC. Association of albumin or protamine to lipoplexes: enhancement of transfection and resistance to serum. J Gene Med. 2004;6(6):681–692. | ||
Faneca H, Faustino A, Pedroso de Lima MC. Synergistic antitumoral effect of vinblastine and HSV-Tk/GCV gene therapy mediated by albumin-associated cationic liposomes. J Control Release. 2008;126(2):175–184. | ||
Faneca H, Cabrita AS, Simões S, Pedroso de Lima MC. Evaluation of the antitumoral effect mediated by IL-12 and HSV-tk genes when delivered by a novel lipid-based system. Biochim Biophys Acta. 2007; 1768(5):1093–1102. | ||
Zhu CP, Shi J, Chen YX, Xie WF, Lin Y. Gemcitabine in the chemoradiotherapy for locally advanced pancreatic cancer: a meta-analysis. Radiother Oncol. 2011;99(2):108–113. | ||
Raymond E, Hammel P, Dreyer C, et al. Sunitinib in pancreatic neuroendocrine tumors. Target Oncol. 2012;7(2):117–125. | ||
European Medicines Agency. Assessment Report for Sutent (sunitinib). Procedure No EMA/H/C/000687/II/0021. London: European Medicines Agency; 2010. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/000687/WC500100433.pdf. Accessed June 26, 2014. | ||
Raymond E, Faivre S, Hammel P, Ruszniewski P. Sunitinib paves the way for targeted therapies in neuroendocrine tumors. Target Oncol. 2009;4(4):253–254. | ||
O’Reilly EM, Niedzwiecki D, Hall M, et al; Cancer and Leukemia Group B. A Cancer and Leukemia Group B phase II study of sunitinib malate in patients with previously treated metastatic pancreatic adenocarcinoma (CALGB 80603). Oncologist. 2010;15(12):1310–1319. | ||
Lu J, Tsourkas A. Quantification of miRNA Abundance in Single Cells Using Locked Nucleic Acid-FISH and Enzyme-Labeled Fluorescence. In: Shah K, editor. Molecular Imaging. New York, NY: Springer; 2011:77–88. | ||
Konopka K, Pretzer E, Felgner PL, Düzgüneş N. Human immunodeficiency virus type-1 (HIV-1) infection increases the sensitivity of macrophages and THP-1 cells to cytotoxicity by cationic liposomes. Biochim Biophys Acta. 1996;1312(3):186–196. | ||
Balazs DA, Godbey W. Liposomes for use in gene delivery. J Drug Deliv. 2011;2011:326497. | ||
Faneca H, Cardoso ALC, Trabulo S, Duarte S, Pedroso de Lima MC. Cationic liposome-based systems for nucleic acid delivery: from the formulation development to therapeutic applications. In: Coelho JF, editor. Drug delivery systems: Advanced technologies potentially applicable in personalised treatments, Advances in Predictive, Preventive and Personalized Medicine. 2013;4:153–184. | ||
Liu C. The role of microRNAs in tumors. Arch Pharm Res. 2013;36(10): 1169–1177. | ||
Park JK, Lee EJ, Esau C, Schmittgen TD. Antisense inhibition of microRNA-21 or -221 arrests cell cycle, induces apoptosis, and sensitizes the effects of gemcitabine in pancreatic adenocarcinoma. Pancreas. 2009;38(7):e190–e199. | ||
Galardi S, Mercatelli N, Giorda E, et al. miR-221 and miR-222 expression affects the proliferation potential of human prostate carcinoma cell lines by targeting p27Kip1. J Biol Chem. 2007;282(32):23716–23724. | ||
Gillies JK, Lorimer IA. Regulation of p27Kip1 by miRNA 221/222 in glioblastoma. Cell Cycle. 2007;6(16):2005–2009. | ||
Dillhoff M, Liu J, Frankel W, Croce C, Bloomston M. MicroRNA-21 is overexpressed in pancreatic cancer and a potential predictor of survival. J Gastrointest Surg. 2008;12(12):2171–2176. | ||
Zhu S, Wu H, Wu F, Nie D, Sheng S, Mo YY. MicroRNA-21 targets tumor suppressor genes in invasion and metastasis. Cell Res. 2008; 18(3):350–359. | ||
Liu C, Yu J, Yu S, et al. MicroRNA-21 acts as an oncomir through multiple targets in human hepatocellular carcinoma. J Hepatol. 2010;53(1): 98–107. | ||
Stanger BZ, Stiles B, Lauwers GY, et al. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell. 2005;8(3):185–195. | ||
Maitra A, Hruban RH. A new mouse model of pancreatic cancer: PTEN gets its Akt together. Cancer Cell. 2005;8(3):171–172. | ||
Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–150. | ||
Sasayama T, Nishihara M, Kondoh T, Hosoda K, Kohmura E. MicroRNA-10b is overexpressed in malignant glioma and associated with tumor invasive factors, uPAR and RHOC. Int J Cancer. 2009;125(6): 1407–1413. | ||
Baffa R, Fassan M, Volinia S, et al. MicroRNA expression profiling of human metastatic cancers identifies cancer gene targets. J Pathol. 2009;219(2):214–221. | ||
Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007; 449(7163):682–688. | ||
Gu S, Kay MA. How do miRNAs mediate translational repression? Silence. 2010;1(1):11. | ||
Saif MW, Syrigos K, Penney R, Kaley K. Docetaxel second-line therapy in patients with advanced pancreatic cancer: a retrospective study. Anticancer Res. 2010;30(7):2905–2909. | ||
Hubner RA, Valle JW. Sunitinib for advanced pancreatic neuroendocrine tumors. Expert Rev Anticancer Ther. 2011;11(12):1817–1827. | ||
Sun TM, Du JZ, Yao YD, et al. Simultaneous delivery of siRNA and paclitaxel via a “two-in-one” micelleplex promotes synergistic tumor suppression. ACS Nano. 2011;5(2):1483–1494. | ||
Sevignani C, Calin GA, Siracusa LD, Croce CM. Mammalian microRNAs: a small world for fine-tuning gene expression. Mamm Genome. 2006;17(3):189–202. | ||
Ying SY, Chang DC, Lin SL. The microRNA (miRNA): overview of the RNA genes that modulate gene function. Mol Biotechnol. 2008;38(3): 257–268. | ||
Eto K, Iwatsuki M, Watanabe M, et al. The microRNA-21/PTEN pathway regulates the sensitivity of HER2-positive gastric cancer cells to trastuzumab. Ann Surg Oncol. 2014;21(1):343–350. | ||
Costa PM, Cardoso AL, Nóbrega C, et al. MicroRNA-21 silencing enhances the cytotoxic effect of the antiangiogenic drug sunitinib in glioblastoma. Hum Mol Genet. 2013;22(5):904–918. | ||
Huang Y, Yang YB, Zhang XH, Yu XL, Wang ZB, Cheng XC. MicroRNA-21 gene and cancer. Med Oncol. 2013;30(1):376. | ||
Sarkar S, Dubaybo H, Ali S, et al. Down-regulation of miR-221 inhibits proliferation of pancreatic cancer cells through up-regulation of PTEN, p27(kip1), p57(kip2), and PUMA. Am J Cancer Res. 2013;3(5):465–477. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.