")
Back to Journals » Cancer Management and Research » Volume 11
microRNA-944 inhibits the malignancy of hepatocellular carcinoma by directly targeting IGF-1R and deactivating the PI3K/Akt signaling pathway
Received 28 December 2018
Accepted for publication 25 February 2019
Published 29 March 2019 Volume 2019:11 Pages 2531—2543
DOI https://doi.org/10.2147/CMAR.S199818
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Alexandra R. Fernandes
This paper has been retracted.
Lili Lv, 1 Xiaodong Wang, 2 Tonghui Ma 3
1Department of Oncology and Hematology, The Second Hospital of Jilin University, Changchun, Jilin 130000, People’s Republic of China; 2Department of Digestive Endoscopy, The Second Hospital of Jilin University, Changchun, Jilin 130000, People’s Republic of China; 3Jilin Provincial Key Laboratory on Molecular and Chemical Genetics, The Second Hospital of Jilin University, Changchun, Jilin 130000, People’s Republic of China
Purpose: Recent studies have identified microRNA-944 (miR-944) as a cancer-related miRNA, but its expression and precise functions in hepatocellular carcinoma (HCC) remain unknown.
Patients and methods: miR-944 expression in HCC tissues and cell lines were detected by RT-qPCR. A series of functional assays were utilized to examine the influence of miR-944 on the malignant phenotypes of HCC cells in vitro and in vivo. More importantly, the associated mechanisms underlying the activity of miR-944 in HCC cells were investigated using bioinformatics, luciferase reporter assays, RT-qPCR, and western blot analysis.
Results: In this study, we report for the first time, a significant downregulation of miR-944 in HCC tissues and cell lines and the correlation between its downregulation and malignant clinical parameters, including Edmondson-Steiner grade, TNM stage, and venous infiltration. Low miR-944 expression predicted poorer overall survival rate and disease-free survival rate in patients with HCC. Functionally, exogenous miR-944 expression attenuated cell proliferation, clone formation, metastasis, and epithelial-mesenchymal transition and increased apoptosis in HCC, whereas miR-944 knockdown produced the opposite results. In addition, ectopic miR-944 expression hindered HCC tumor growth in vivo. Mechanistically, insulin-like growth factor 1 receptor (IGF-1R) was demonstrated to be the direct target gene of miR-944 in HCC cells. Furthermore, the expression level of miR-944 was inversely correlated with IGF-1R expression in HCC tissues. Rescue experiments showed that IGF-1R was at least partially responsible for the effects of miR-944 on the malignant phenotypes of HCC cells. In addition, the PI3K/Akt pathway was notably deactivated, both in vitro and in vivo, upon miR-944 upregulation.
Conclusion: This study provides the first evidence that miR-944 directly targets IGF-1R and inhibits the aggressiveness of HCC, in vitro and in vivo, by decreasing PI3K/Akt signaling. Hence, targeting miR-944 may open a new avenue for the treatment of patients with HCC.
Keywords: hepatocellular carcinoma, microRNA-944, insulin-like growth factor 1 receptor, PI3K/Akt pathway, epithelial-mesenchymal transition
Introduction
Hepatocellular carcinoma (HCC) is one of the most common primary liver malignancies and ranks as the third leading cause of cancer-associated deaths globally.1 The morbidity of HCC is continuing to grow and causes a heavy healthcare-related economic burden in developing countries, including China.2 In recent years, there have been remarkable developments in therapeutic strategies to combat this disease, including surgical resection, radiofrequency ablation, liver transplantation, and radiochemotherapy, but the clinical outcomes of patients with HCC are still dissatisfying due to frequent intrahepatic spread and extrahepatic metastasis.3 The other major reason for the poor prognosis of patients with HCC is that the etiology and exact mechanisms underlying hepatocarcinogenesis are not fully understood.4,5 Therefore, uncovering the complex molecular mechanisms responsible for the pathogenesis of HCC is urgently needed to enable the discovery of novel biomarkers for diagnosis and prognosis and to identify promising therapeutic targets for patients with HCC.
microRNAs (miRNAs) are single-stranded, non-coding, short RNA molecules measuring approximately 17–23 nucleotides in length.6 miRNAs are able to post-transcriptionally modulate gene expression by promoting mRNA degradation and inducing translational inhibition by directly binding to complementary sites within the 3′-untranslated regions (3′-UTRs) of their target genes.7 More than 1,500 mature miRNAs have been identified in the human genome and these miRNAs regulate approximately one third of all human protein-coding genes.8 Increasing evidence indicates that a variety of miRNAs are aberrantly expressed in HCC. For example, miR-34a,9 miR-506,10 and miR-64511 are downregulated in HCC, whereas miR-197,12 miR-552,13 and miR-65014 are expressed at high levels in HCC. The dysregulated miRNAs exert their functions as oncogenes or tumor suppressors and play crucial roles in the initiation and progression of HCC.15–17 Therefore, therapies that target miRNAs may be attractive strategies for HCC treatment.
Recently, miR-944 has been reported to serve both tumor-suppressing18,19 and tumor-promoting20–22 roles, depending on the type of human malignancy. These conflicting observations indicate that the role of miR-944 in tumorigenesis is tissue-specific. However, details of the expression, clinical significance, and precise functions of miR-944 in HCC are not yet known. Hence, we attempted to determine the expression pattern and clinical value of miR-944 in HCC. The detailed roles of miR-944 in the malignant development of HCC and the associated mechanisms were also examined.
Material and methods
Patients and tissue specimens
HCC and adjacent normal tissues (ANTs) were obtained from 61 patients with HCC who underwent surgical resection at The Second Hospital of Jilin University. None of these patients received radiofrequency ablation, radiotherapy, or chemotherapy before surgery. Tumor stage was classified according to the TNM classification system advocated by the International Union against Cancer and was followed up until June 2018.
Tissues were collected from the 61 patients, frozen in liquid nitrogen, and then stored at −80 °C for further analyses. The present study was approved by the Ethics Committee of The Second Hospital of Jilin University and was performed in accordance with the ethical standards of the Declaration of Helsinki. In addition, written informed consent was provided by all patients.
Cell culture
The immortalized human hepatic cell line L02, and a panel of HCC cell lines (Hep3B, Bel-7402, SMMC-7721, Huh7, and SK-HEP-1) were purchased from the Shanghai Cell Bank of the Chinese Academy of Science (Shanghai, China). All cell lines were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin (all from Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 37 °C in an incubator containing 5% CO2.
Cell transfection assays
miR-944 mimics, a miR-944 inhibitor, negative control miRNA mimics (miR-NC), and a negative control miRNA inhibitor (NC inhibitor) were provided by Shanghai GenePharma Co., Ltd. (Shanghai, China). A small interfering RNA (siRNA) used to knockdown IGF-1R expression (si-IGF-1R) and a control siRNA (si-ctrl) were chemically synthesized by Guangzhou RiboBio Co., Ltd. (Guangzhou, China).
The pcDNA3.1-IGF-1R (pc-IGF-1R) plasmid was employed to increase the expression of IGF-1R and an empty pcDNA3.1 plasmid was used as a control. Plasmids were chemically synthesized by the Chinese Academy of Sciences (Changchun, China).
For transfection, cells were plated into 6-well plates. Transfection was performed when the cells had grown to approximately 60–70% density. Lipofectamine™ 2000 (Invitrogen; Thermo Fisher Scientific) was used for transfections and assays were performed according to the product specifications. After 8 h of culture, culture medium was replaced with fresh DMEM containing 10% FBS.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
TRIzol reagent (Invitrogen) was used to extract total RNA, following the manufacturer’s recommendations. The concentration of isolated RNA was evaluated using a Nanodrop-ND-1000 (Thermo Fisher Scientific, Inc.). To quantify miRNA expression, cDNA was prepared from total RNA using the miScript Reverse Transcription Kit (Qiagen GmbH, Hilden, Germany). Thereafter, the amplification of cDNA was performed using the miScript SYBR Green PCR Kit (Qiagen GmbH). Alternatively, reverse transcription was performed using the TaqMan RT reagent (Applied Biosystems; Thermo Fisher Scientific, Inc.), followed by qPCR using the SYBR®-Green PCR Master Mix Kit (Applied Biosystems; Thermo Fisher Scientific, Inc.). U6 snRNA and GAPDH were used as controls for quantitating miR-944 and IGF-1R expression, respectively. Each assay was performed in triplicate and relative gene expression was calculated using the 2−ΔΔCq method.
Cell Counting Kit-8 (CCK-8) and clone formation assays
For CCK-8 assays, transfected cells were collected and inoculated into 96-well plates (2,000 cells/well). Each group had three repeated wells. After 0, 1, 2, or 3 d, 10 μL of CCK-8 reagent (Beyotime Institute of Biotechnology, Inc., Shanghai, China) was added to each well. Cells were then incubated at 37 °C for 2 h and the absorbance at 450 nm (A450) was determined using a Spectramax M5 microplate reader (Molecular Devices LLC, Sunnyvale, CA, USA).
For colony formation assays, 1,000 cells were seeded into 6-well plates. After 14 d of culture, cells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. The number of clones (defined as ≥50 cells/clone) was counted using an inverted light microscope (IX83; Olympus Corporation, Tokyo, Japan).
Apoptosis assays
Apoptosis was determined using an Annexin V-Fluorescein Isothiocyanate (FITC) Apoptosis Detection Kit (Biolegend, San Diego, CA, USA). Transfected cells were detached with trypsin (Gibco) and washed twice with ice-cold PBS at 4 °C. After centrifugation, cells were resuspended in 100 µL of 1X binding buffer and double-stained with 5 µL Annexin V-FITC and 5 µL propidium iodide. Following 20 min of incubation in the dark at room temperature, apoptosis rate was measured using a flow cytometer (FACScan™, BD Biosciences, Franklin Lakes, NJ, USA).
Cell migration and invasion assays
After incubation for 48 h, 5×104 transfected cells suspended in FBS-free DMEM were placed into the upper compartments of 8-µm pore size Corning Transwell chambers (Corning Inc., Corning, NY, USA). A total of 600 μL of DMEM supplemented with 10% FBS was added to the lower chambers as a chemoattractant. Following 24 h of incubation at 37 °C, migrated cells were fixed with 4% paraformaldehyde at room temperature for 30 min. A 0.05% crystal violet (Beyotime Institute of Biotechnology) solution was then added to stain the migrated cells at room temperature for 30 min. Finally, the number of migrated cells was counted in five randomly selected fields/membranes under an inverted IX83 light microscope. Cell invasion assays were performed using the same procedures as the cell migration assays, expect that the Corning Transwell chambers were pre-coated with Matrigel (BD Biosciences, San Jose, CA, USA).
Tumor xenograft model
All animal experiments were approved by the Animal Care and Use Committee of The Second Hospital of Jilin University and were performed in accordance with the Animal Protection Law of the People’s Republic of China-2009 for experimental animals. miR-944 mimics- or miR-NC-transfected cells were harvested and injected subcutaneously into the dorsal right flank of 6 week-old BALB/c nude mice (Shanghai SLAC Laboratory Animal Co. Ltd., Shanghai, China). Tumor xenografts were measured every 3 d and their volume was calculated using the equation: volume = length × (width)2/2. On day 28, nude mice were sacrificed and tumor xenografts were resected and weighed. Total RNA and protein was also isolated from tumor xenografts and used for RT-qPCR and western blotting analysis, respectively.
Target prediction and luciferase reporter assays
TargetScan (
A fragment of the IGF-1R 3′-UTR, containing the wild-type (wt) miR-944 target site, was amplified by Shanghai GenePharma Co., Ltd. The binding sequences in the 3′-UTR of IGF-1R were mutated, and the IGF-1R 3′-UTR wt and mutant (mut) fragments were inserted into the pmirGLO luciferase reporter plasmid (Promega, Madison, WI, USA). Cells were plated in triplicate in 24-well plates and allowed to settle for 12 h. The chemically synthesized luciferase reporter plasmids, along with miR-944 mimics or miR-944 inhibitor, were co-transected into cells, according to the Lipofectamine™ 2000 reagent protocol. Luciferase activity was detected 48 h post-transfection using a Dual-Luciferase Reporter Assay System (Promega). Renilla luciferase activity was used for normalization.
Protein extraction and western blotting analysis
Homogenized tissues or cells were lysed with an active protein extraction kit (KGP1050; Nanjing KeyGen Biotech Co., Ltd., Nanjing, China) containing protease inhibitor and protein was quantified using a BCA Protein Assay Kit (Beyotime Institute of Biotechnology). Equal amounts of protein were electrophoresed on 10% sodium dodecyl sulfate-polyacrylamide gels and then transferred onto polyvinylidene fluoride (PVDF) membranes. Membranes were blocked with 5% non-fat powdered milk at room temperature for 2 h and were subsequently incubated overnight at 4 °C with the appropriate primary antibodies. After washing thrice with TBS containing 0.1% Tween-20 (TBST), membranes were incubated for 2 h at room temperature with a horseradish peroxidase-conjugated goat anti-mouse (catalog no. ab205719; Abcam, Cambridge, MA, USA) or goat anti-rabbit (catalog no. ab205718; Abcam) secondary antibody at a 1:5,000 dilution. They were then extensively washed with TBST. Protein bands were detected using an Enhanced Chemiluminescence Detection System (Pierce; Thermo Fisher Scientific, Inc.).
The primary antibodies used for western blotting analysis were as follows: anti-E-cadherin (cat. no. ab1416), anti-N-cadherin (cat. no. ab76011), anti-vimentin (cat. no. ab92547), anti-p-PI3K (cat. no. ab182651), and anti-PI3K (cat. no. ab191606) were purchased from Abcam and anti-IGF-1R (cat. no. sc-81464), anti-p-Akt (cat. no. sc-81433), anti-Akt (cat. no. sc-81464), and anti-GAPDH (cat. no. sc-365062) antibodies were purchased from Santa Cruz Biotechnology, Inc. (Dallas, Texas, USA).
Statistical analysis
Results were expressed as mean ± standard deviation from at least three separate experiments. A Chi-square test was applied to assess the relationship between miR-944 expression and clinicopathological parameters in patients with HCC. A log-rank test was used to examine the association between miR-944 expression and overall survival and disease-free survival of patients with HCC. The correlation between miR-944 and IGF-1R mRNA levels in HCC tissues was determined by Spearman’s correlation analysis. Differences between two or multiple groups were evaluated by two-tailed Student’s t-test or one-way analysis of variance (ANOVA), respectively. The Student-Newman-Keuls test was used as the post hoc test after ANOVA. SPSS 19.0 software (IBM Corporation, Armonk, NY, USA) was used for all statistical analyses, and a P-value <0.05 was defined as statistically significant.
Results
miR-944 is downregulated in HCC and indicates poor prognosis for patients with HCC
To investigate the relationship between miR-944 and HCC, RT-qPCR was employed to measure miR-944 expression in 61 pairs of HCC tissues and ANTs. The data showed that miR-944 expression was significantly decreased in HCC tissues compared with ANTs (Figure 1A, P<0.05). In addition, we determined the expression difference of miR-944 in five HCC cell lines (Hep3B, Bel-7402, SMMC-7721, Huh7, SK-HEP-1) and one immortalized human hepatic cell line (L02). Consistent with the trend in tissue specimens, miR-944 expression was significantly lower in all five HCC cell lines than that in L02 cells (Figure 1B, P<0.05).
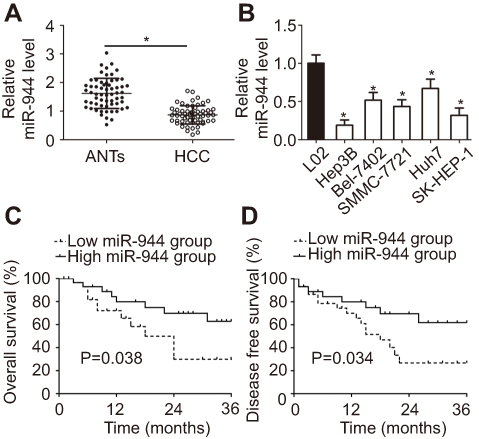 | Figure 1 miR-944 is downregulated in HCC tissues and cell lines. (A) Expression level of miR-944 was determined in 61 pairs of HCC tissues and ANTs. miR-944 expression was significantly lower in HCC tissues than in ANTs. *P<0.05 vs ANTs. (B) The relative expression of miR-944 was detected in the five HCC cell lines, Hep3B, Bel-7402, SMMC-7721, Huh7, and SK-HEP-1. The immortalized human hepatic cell line, L02, was used as a control. *P<0.05 vs L02. (C, D) Patients with HCC in the miR-944 low expression group had poorer overall and disease-free survival rates than those in the high miR-944 expression group. |
We then explored the clinical utility of miR-944 in patients with HCC. The patients with HCC were categorized into two subgroups (low or high miR-944 expression groups) based on the median value of miR-944 levels in HCC tissues. As shown in Table 1, low miR-944 expression was significantly correlated with Edmondson-Steiner grade (P=0.007), TNM stage (P=0.013), and venous infiltration (P=0.001) in patients with HCC, suggesting that tumors with decreased miR-944 levels may be more aggressive. Furthermore, patients with HCC who had lower miR-944 expression had a poorer overall survival rate (Figure 1C, P = 0.038) and disease-free survival rate (Figure 1D, P = 0.034). These observations indicated that miR-944 may be a promising biomarker for HCC diagnosis and prognosis.
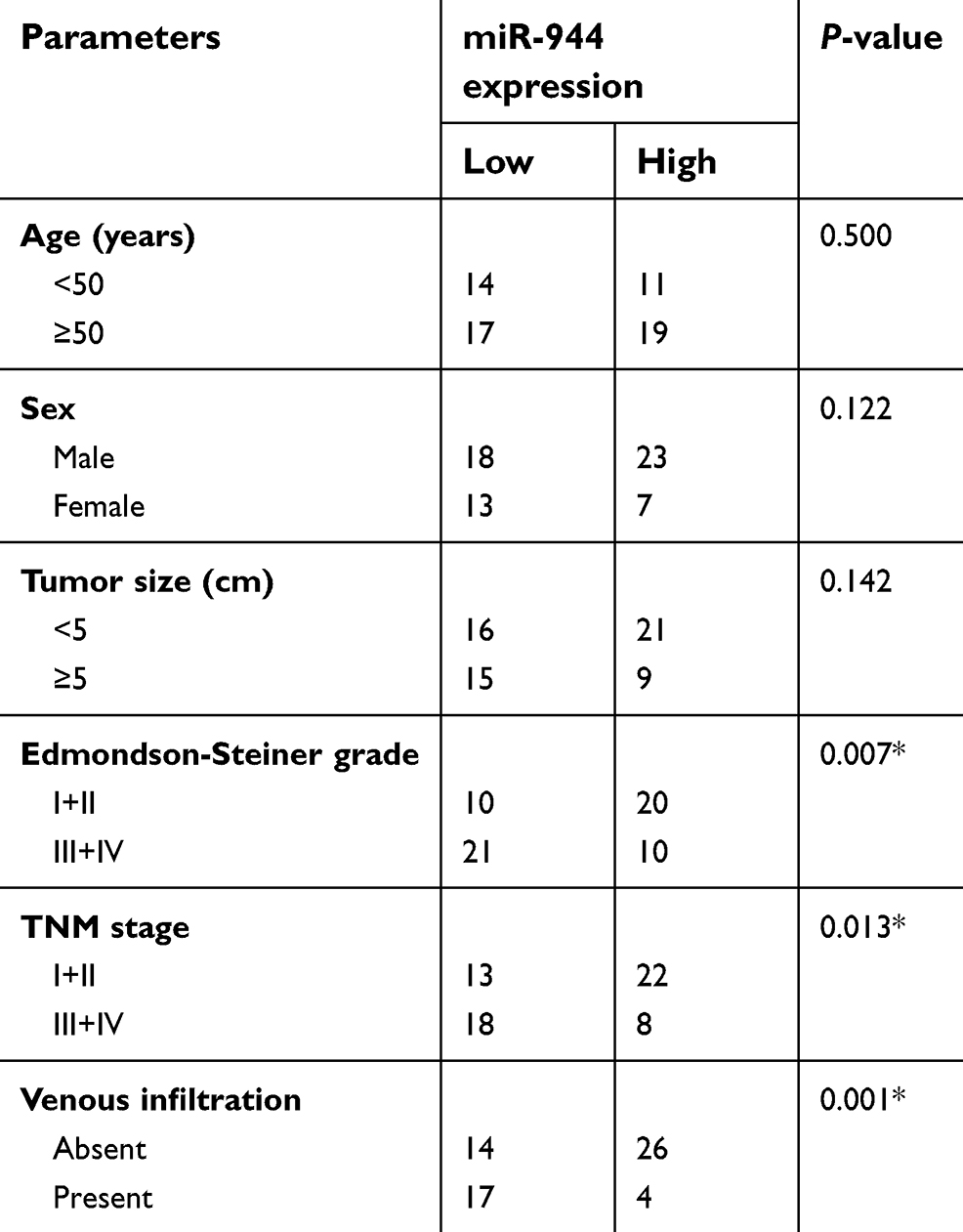 | Table 1 The association between miR-944 and the clinicopathological parameters of patients with HCC |
miR-944 suppresses the growth of HCC cells in vitro
Since miR-944 was significantly downregulated in HCC, we speculated that miR-944 may have a crucial role in the aggressiveness of HCC cells. To evaluate this hypothesis, Hep3B and Huh7 cell lines, which exhibited relatively low and relatively high miR-944 levels, respectively, were chosen for subsequent functional experiments. Hep3B cells were transfected with miR-944 mimics, while a miR-944 inhibitor was introduced into Huh7 cells. The efficiency of transfection was confirmed by RT-qPCR. miR-944 was overexpressed in Hep3B cells and silenced in Huh7 cells (Figure 2A, P<0.05). CCK-8 and clone formation assays demonstrated that the upregulation of miR-944 suppressed the proliferative and clone-formation capacities of Hep3B cells, whereas inhibition of miR-944 promoted these abilities (Figure 2B and C, P<0.05). Furthermore, the influence of miR-944 on the apoptosis of HCC cells was examined. Apoptosis assay results showed that transfection with miR-944 mimics increased the percentage of apoptotic Hep3B cells, whereas transfection with a miR-944 inhibitor decreased the percentage of apoptotic Huh7 cells (Figure 2D, P<0.05). Altogether, miR-944 exerted an inhibitory effect on the growth of HCC cells in vitro.
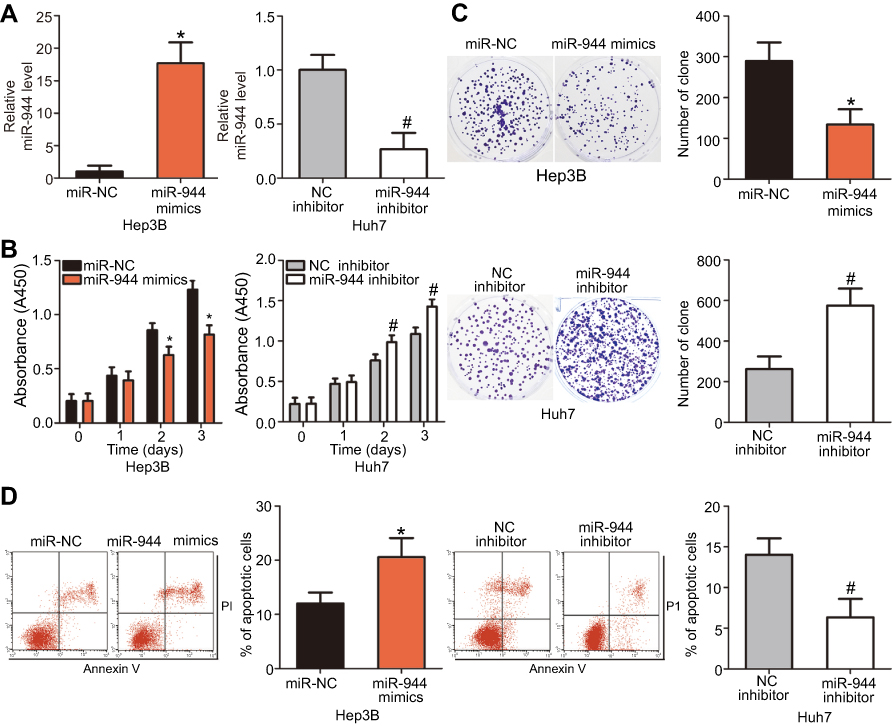 | Figure 2 miR-944 inhibits the proliferation and clone formation and induces apoptosis in HCC cells. (A) The transfection efficiency of miR-944 mimics and a miR-944 inhibitor was assessed by RT-qPCR. miR-944 was significantly upregulated in Hep3B cells, while it was efficiently knocked down in Huh7 cells. *P<0.05 vs miR-NC. #P<0.05 vs NC inhibitor. (B) CCK-8 assays indicated that overexpression of miR-944 attenuated the proliferation of Hep3B cells, while miR-944 silencing promoted Huh7 cell proliferation. *P<0.05 vs miR-NC. #P<0.05 vs NC inhibitor. (C) miR-944 upregulation clearly decreased the clone formation of Hep3B cells, while miR-944 knockdown had the opposite effect on Huh7 cells. *P<0.05 vs miR-NC. #P<0.05 vs NC inhibitor. (D) The percentage of apoptotic Hep3B cells increased after transfection with miR-944 mimics, whereas the apoptosis rate was suppressed in miR-944 inhibitor-transfected Huh7 cells. *P<0.05 vs miR-NC. #P<0.05 vs NC inhibitor. |
miR-944 restricts the metastasis and epithelial-mesenchymal transition (EMT) of HCC cells in vitro
Cell migration and invasion assays were performed to explore the precise roles of miR-944 in the metastasis of HCC cells. Results showed that the migratory and invasive abilities of HCC cells were notably attenuated by exogenous miR-944 expression (Figure 3A, P<0.05), but significantly increased by miR-944 knockdown (Figure 3B, P<0.05). Tumor metastasis is a complex process that is regulated by several cellular processes, including EMT. Hence, western blotting analysis was performed to determine the expression levels of EMT markers in Hep3B and Huh7 cells after transfection with miR-944 mimics or a miR-944 inhibitor, respectively. As indicated in Figure 3C, miR-944 overexpression markedly increased the expression of the epithelial marker, E-cadherin, and suppressed the expression of the mesenchymal markers, N-cadherin and vimentin, in Hep3B cells. In contrast, the protein levels of E-cadherin were downregulated, while N-cadherin and vimentin levels were upregulated in Huh7 cells upon miR-944 silencing (Figure 3C). These results implied that miR-944 decreased metastasis and EMT in HCC cells in vitro.
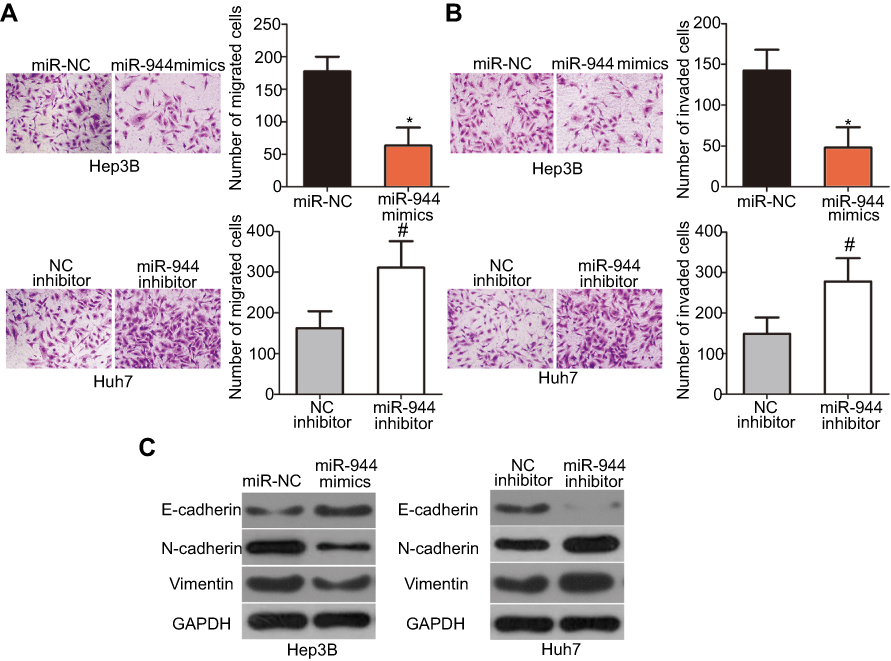 | Figure 3 Inhibitory effects of miR-944 on the metastasis and EMT of HCC cells. (A and B) Cell migration and invasion was determined using cell migration and invasion assays after treatment with miR-944 mimics or a miR-944 inhibitor. The migratory and invasive capacities of Hep3B cells were significantly attenuated by exogenous miR-944 expression, while these abilities increased following miR-944 downregulation on Huh7 cells. *P<0.05 vs miR-NC. #P<0.05 vs NC inhibitor. (C) The protein levels of E-cadherin, N-cadherin, and vimentin were detected by western blotting analysis. |
IGF-1R is a downstream target of miR-944 in HCC cells
To elucidate the potential effectors of miR-944 in the progression of HCC, bioinformatics tools were used to predict the putative target of miR-944. The 3′-UTR of IGF-1R contains two major miR-944 binding sites (Figure 4A). IGF-1R, a well-known oncogene, has been reported to be a regulator of hepatocarcinogenesis.23–29 Therefore, IGF-1R was chosen for further validation. Luciferase reporter assays were performed to test whether miR-944 directly targets the 3′-UTR of IGF-1R in HCC cells. Ectopic miR-944 expression resulted in a significant decrease in the luciferase activity of the plasmid harboring the wt IGF-1R 3′-UTR (1 and 2) in Hep3B cells, whereas silencing miR-944 expression increased the luciferase activity of the wt IGF-1R 3′-UTR (1 and 2) in Huh7 cells (P<0.05). However, the luciferase activity of the mutant IGF-1R 3′-UTR (1 and 2) was unaffected by either miR-944 induction or knockdown (Figure 4B). To further explore whether IGF-1R was a bona fide downstream target of miR-944, RT-qPCR and western blotting were used to determine how changes in miR-944 expression affected IGF-1R expression in HCC cells. The mRNA and protein levels of IGF-1R were suppressed in Hep3B cells after miR-944 overexpression, whereas downregulation of miR-944 led to the opposite results in Huh7 cells (Figure 4C and D, P<0.05). These results suggest that IGF-1R is a direct target gene of miR-944 in HCC cells.
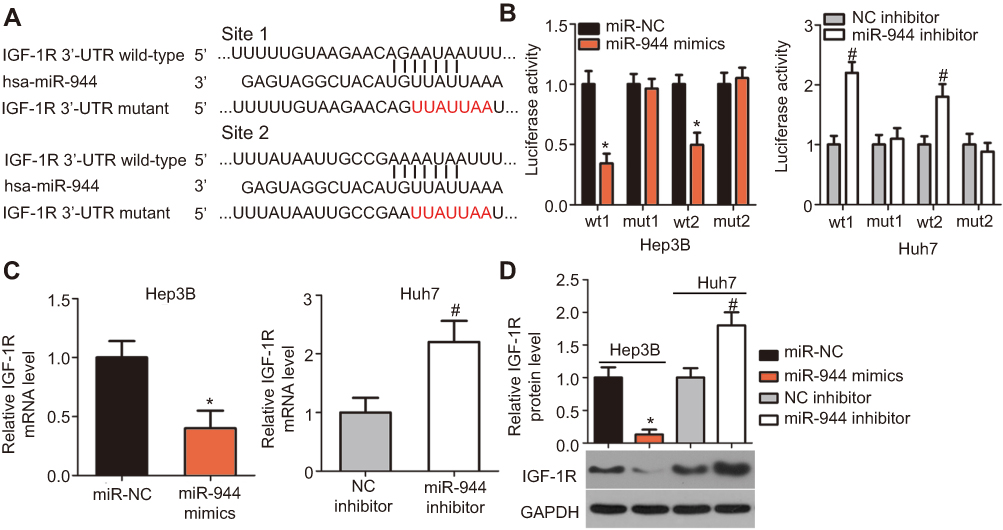 | Figure 4 IGF-1R is a downstream target of miR-944 in HCC cells. (A) The putative miR-944 binding site in the 3′-UTR of IGF-1R was predicted by bioinformatics tools. The mutant binding sequences are also shown. (B) Exogenous miR-944 expression suppressed the luciferase activity of the wt reporter plasmid in Hep3B cells. Silencing of miR-944 expression increased the luciferase activity in Huh7 cells that contained the wt 3′-UTR of IGF-1R. *P<0.05 vs miR-NC. #P<0.05 vs NC inhibitor. (C and D) The mRNA and protein levels of IGF-1R in miR-944-overexpressing Hep3B or miR-944-silenced Huh7 cells were measured by RT-qPCR and western blotting, respectively. *P<0.05 vs miR-NC. #P<0.05 vs NC inhibitor. |
Mir-944 is inversely correlated with IGF-1R expression in HCC tissues
To further investigate the relationship between miR-944 and IGF-1R in HCC, we investigated IGF-1R mRNA and protein expression in HCC tissues and ANTs. IGF-1R mRNA was expressed at high levels in HCC tissues as compared to the ANTs (Figure 5A, P<0.05). As all patients with HCC could be divided into two subgroups, low or high miR-944 expression groups, we next explored the relationship between miR-944 expression and IGF-1R expression in HCC tissues. The mRNA and protein levels of IGF-1R were significantly higher in the low miR-944 expression group compared to the high miR-944 expression group (Figure 5B and C, P<0.05). Furthermore, an inverse association between miR-944 and IGF-1R mRNA expression levels was demonstrated by Spearman’s correlation analysis (Figure 5D; R2= 0.3402, P < 0.0001). These results further suggest that IGF-1R is a downstream target of miR-944 in HCC cells.
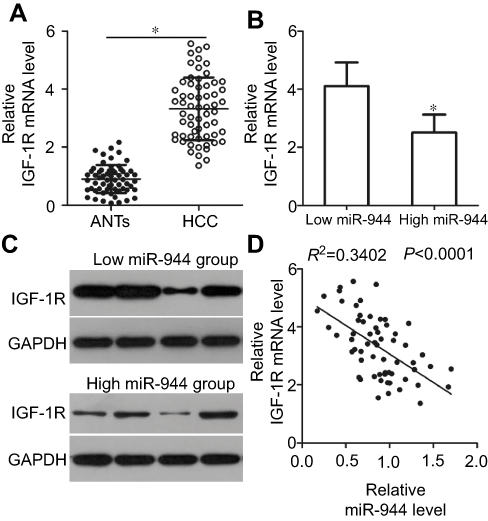 | Figure 5 Correlation analysis between miR-944 and IGF-1R levels in HCC tissues. (A) IGF-1R mRNA levels in HCC tissues and adjacent normal tissues (ANTs) were analyzed by RT-qPCR. IGF-1R mRNA level was higher in HCC tissues as compared to the ANTs. *P<0.05 vs ANTs. (B and C) IGF-1R mRNA and protein levels were significantly lower in the high miR-944 expression group HCC tissues as compared to the low miR-944 expression group tissues. *P<0.05 vs low miR-944 expression group. (D) Correlation analysis identified an inverse relationship between miR-944 and IGF-1R mRNA levels in HCC tissues. R2 = 0.3402, P < 0.0001. |
IGF-1R mediates the functional roles of miR-944 in HCC cells
Rescue experiments were performed to further clarify whether IGF-1R was essential for the effects of miR-944 in HCC cells. IGF-1R expression was restored in miR-944 mimics-transfected Hep3B cells after co-transfection with the IGF-1R-overexpressing plasmid, pcDNA3.1-IGF-1R (pc-IGF-1R), while IGF-1R siRNA (si-IGF-1R) knocked down IGF-1R expression in miR-944 inhibitor-transfected Huh7 cells (Figure 6A, P<0.05). Reintroduction of IGF-1R expression partially counteracted the tumor-suppressing effects of miR-944 mimics on the proliferation (Figure 6B; P < 0.05), clone formation (Figure 6C; P < 0.05), apoptosis (Figure 6D; P < 0.05), migration (Figure 7A; P < 0.05), invasion (Figure 7B; P < 0.05), and EMT (Figure 7C) of Hep3B cells. Meanwhile, the tumor-promoting activity of miR-944 inhibition on the malignant phenotypes of Huh7 cells was attenuated by co-transfection with si-IGF-1R. These results suggest that miR-944 regulates the malignant progression of HCC, at least partially, by regulating IGF-1R expression.
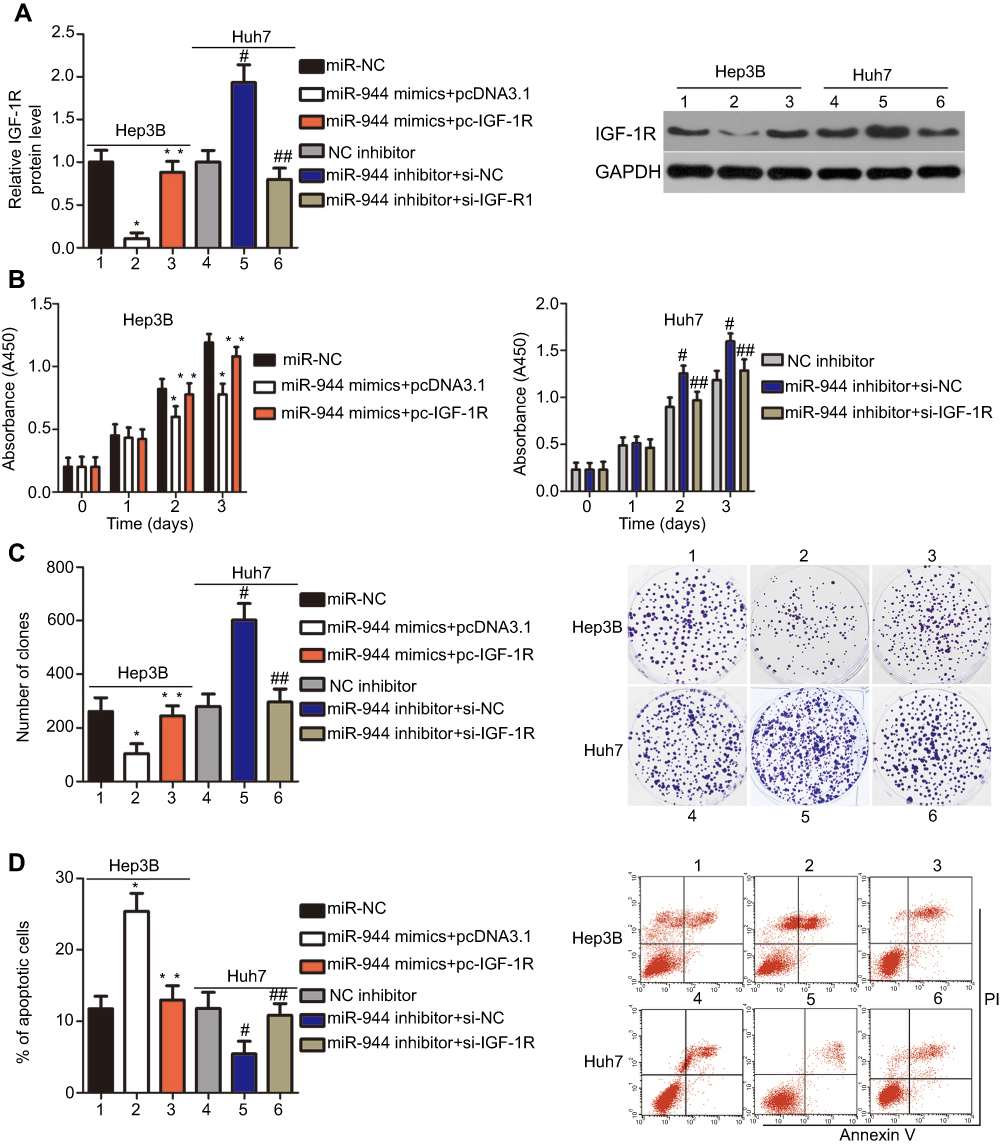 | Figure 6 Modulation of IGF-1R partially reverses the miR-944-mediated proliferation, colony formation, and apoptosis effects in HCC cells. miR-944-overexpressing Hep3B cells were transfected with pcDNA3.1-IGF-1R (pc-IGF-1R) or pcDNA3.1. IGF-1R siRNA (si-IGF-1R) and si-NC were introduced into miR-944-silenced Huh7 cells. The transfected cells were collected after different incubation times and used in the subsequent assays. (A) Western blotting analysis was used to quantify IGF-1R protein levels. *P<0.05 vs miR-NC. **P<0.05 vs miR-944 mimics+pcDNA3.1. #P<0.05 vs NC inhibitor. ##P<0.05 vs miR-944 inhibitor+si-NC. (B–D) The proliferation, colony formation, and apoptosis of the aforementioned cells were determined using the CCK-8, clone formation, and cell apoptosis assays, respectively. *P<0.05 vs miR-NC. **P<0.05 vs miR-944 mimics+pcDNA3.1. #P<0.05 vs NC inhibitor. ##P<0.05 vs miR-944 inhibitor+si-NC. |
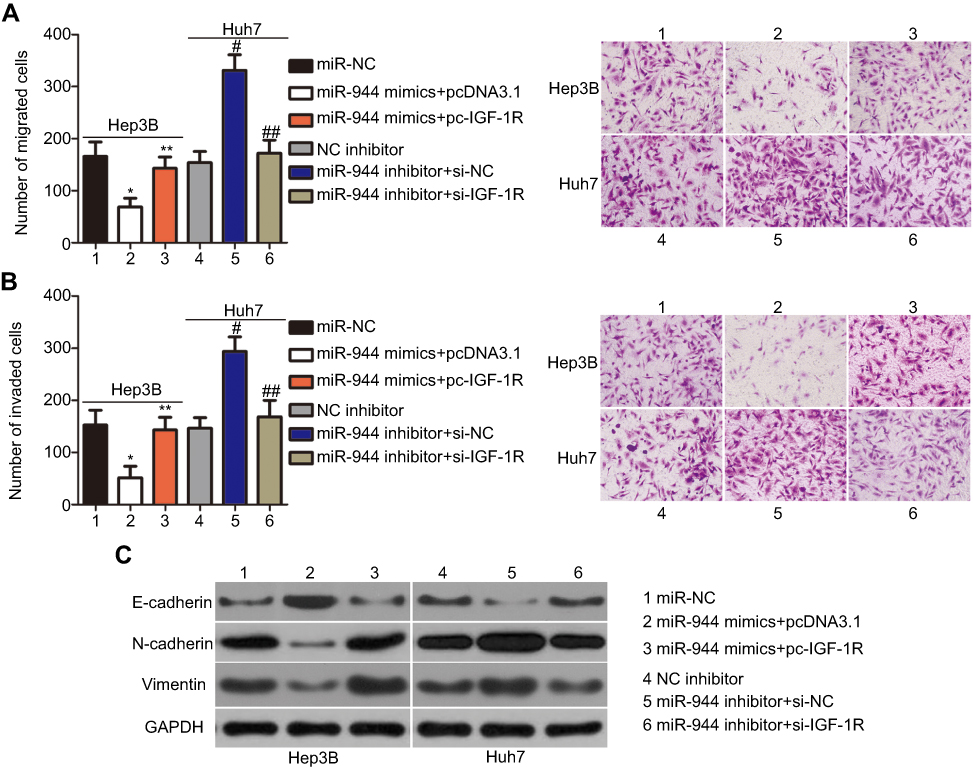 | Figure 7 Modulation of IGF-1R partially rescues the miR-944-mediated metastasis and EMT of HCC cells. (A and B) miR-944 mimics-transfected Hep3B cells were co-transfected with pc-IGF-1R, while si-IGF-1R and si-NC were introduced into Huh7 cells that were transfected with a miR-944 inhibitor. Cell migration and invasion assays were conducted to determine the migration and invasion capacities of the transfected HCC cells. *P<0.05 vs miR-NC. **P<0.05 vs miR-944 mimics+pcDNA3.1. #P<0.05 vs NC inhibitor. ##P<0.05 vs miR-944 inhibitor+si-NC.(C) Western blotting analysis was utilized to measure the protein levels of E-cadherin, N-cadherin, and vimentin in Hep3B and Huh7 cells treated as described. ×200 magnification. |
miR-944 is involved in the regulation of the PI3K/Akt pathway through IGF-1R
We further attempted to identify the molecular mechanism by which the miR-944/IGF-1R axis affected the growth and metastasis of HCC cells. The PI3K/Akt signaling pathway has recently been reported to be regulated by IGF-1R in several human cancers, including HCC.24,30,31 Therefore, we hypothesized that the functional roles of miR-944 in the aggressiveness of HCC cells may be associated with the PI3K/Akt pathway. Western blotting analysis revealed that the enforced expression of miR-944 in Hep3B cells decreased the protein levels of p-PI3K and p-Akt. Conversely, silencing miR-944 expression in Huh7 cells caused an increase in the levels of p-PI3K and p-Akt (Figure 8). More importantly, the recovery of IGF-1R expression rescued the downregulation of p-PI3K and p-Akt induced by miR-944 upregulation in Hep3B cells. Similarly, the increased expression of p-PI3K and p-Akt proteins caused by miR-944 suppression was attenuated in Huh7 cells by si-IGF-1R co-transfection. These results demonstrated that miR-944 was able to regulate HCC progression, at least in part, through the IGF-1R/PI3K/Akt pathway.
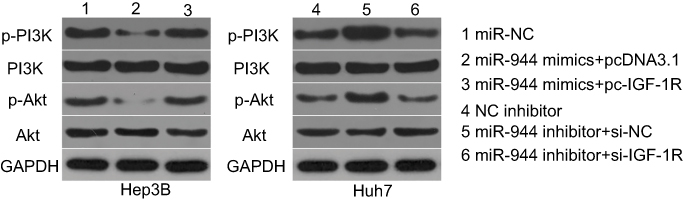 | Figure 8 miR-944 suppresses the activation of the PI3K/Akt pathway in HCC cells. Hep3B cells were co-transfected with miR-944 mimics and pc-IGF-1R or pcDNA3.1, while Huh7 cells were co-transfected with a miR-944 inhibitor and si-IGF-1R or si-NC. Transfected cells were harvested after 72 h of incubation. Western blotting analysis was utilized to measure p-PI3K, PI3K, p-Akt, and Akt protein levels. |
miR-944 suppresses HCC tumor growth in vivo by regulating the IGF-1R/PI3K/Akt pathway
To examine the effect of miR-944 on tumor growth in vivo, Hep3B cells transfected with miR-944 mimics were injected into nude mice. miR-NC-transfected cells were used as controls. Tumor growth (Figure 9A and B, P<0.05) and weight (Figure 9C, P<0.05) were significantly impaired in the miR-944 mimics group. To further investigate whether the inhibitory role in HCC tumor growth in vivo was due to miR-944 overexpression, RT-qPCR analysis was performed to quantify miR-944 expression in tumor xenografts. Significantly higher miR-944 expression was observed in tumor xenografts derived from miR-944 mimics-treated nude mice than in those derived from miR-NC-treated mice (Figure 9D, P<0.05). In addition, the protein levels of IGF-1R, p-PI3K, and p-Akt decreased in the tumor xenografts from the miR-944 mimics group (Figure 9E). These results indicated that miR-944 hindered HCC tumor growth in vivo, at least in part, by inhibiting IGF-1R and the activation of the PI3K/Akt pathway.
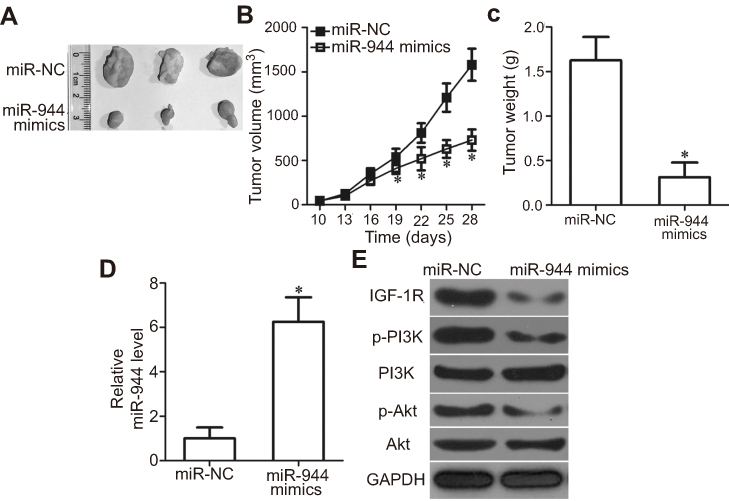 | Figure 9 miR-944 inhibits tumor growth in vivo. (A) Representative images of tumor xenografts from nude mice inoculated with Hep3B cells that were transfected with miR-944 mimics or miR-NC. (B and C) Comparison of tumor volume and weight in tumor xenografts derived from miR-944-mimics- or miR-NC-transfected Hep3B cells. *P<0.05 vs miR-NC. (D) miR-944 expression in tumor xenografts derived from nude mice was measured by RT-qPCR. *P<0.05 vs miR-NC. (E) IGF-1R, p-PI3K, PI3K, p-Akt, and Akt protein levels in tumor xenografts were detected by western blotting. |
Discussion
Dysregulated miRNAs are frequently reported in HCC and this has been shown to be important in regulating the pathogenesis and progression of the disease.15,32,33 More importantly, abnormally expressed miRNAs are suggested to be promising indicators for diagnosis and prognosis, as well as therapeutic targets for patients with HCC.34 Therefore, an in-depth understanding of the relevant mechanisms whereby miRNAs affect HCC is urgent and important. However, studies investigating the expression patterns and precise roles of miR-944 in HCC are still quite limited. Therefore, in this study, we investigated miR-944 expression in HCC and explored the detailed roles of miR-944 in the malignant progression of HCC. The molecular mechanisms related to the tumor suppressor activity of miR-944 in HCC were also examined. To the best of our knowledge, this is the first study demonstrating that miR-944 exerts anticancer effects on HCC cells by specifically targeting IGF-1R and deactivating the PI3K/Akt signaling pathway.
miR-944 is expressed at low levels in colorectal cancer, and low miR-944 expression is significantly correlated with the tumor invasion stage, lymph node metastasis, distant metastasis, and TNM stage. Colorectal cancer patients with decreased miR-944 expression have shorter 5-year overall and progression-free survival times than those with high miR-944 expression.18 miR-944 is also downregulated in gastric35 and non-small cell lung19 cancers. In contrast, miR-944 expression is upregulated in endometrial,20 cervical,21 and breast22 cancers. However, the expression status of miR-944 in HCC remains to be elucidated. Herein, RT-qPCR analysis showed that miR-944 was downregulated in HCC tissues and cell lines and its downregulation was associated with Edmondson-Steiner grade, TNM stage, and venous infiltration in patients with HCC. Notably, patients with HCC who had low miR-944 expression exhibited poorer prognosis than the patients with high miR-944 expression. These observations suggest that miR-944 is a potential biomarker for the diagnosis and prognosis of patients with HCC.
miR-944 plays tumor-suppressive or tumor-promoting activities during carcinogenesis and cancer progression. For example, miR-944 upregulation inhibits the metastasis of colorectal cancer cells by regulating the MACC1/Met/AKT signaling pathway.18 miR-944 directly targets EPHA7 to suppress cell proliferation in non-small cell lung cancer.19 In contrast, the resumption of miR-944 expression increases cell growth and metastasis and improves the cytotoxicity of cisplatin in breast cancer by regulating the BNIP3-MMP-caspase-3 pathway.22 miR-944 also acts as an oncogenic miRNA in endometrial20 and cervical21 cancers. A previous study also reported that miR-944 knockdown restrained the proliferation of Hep3B cells.36 However, the specific roles of miR-944 in HCC are not well established. In this study, we demonstrated that miR-944 overexpression attenuated HCC cell proliferation, clone formation, metastasis, and EMT and increased apoptosis, whereas miR-944 silencing led to the opposite results. Additionally, ectopic miR-944 expression hindered HCC tumor growth in vivo. These findings support the notion that miR-944 may be an attractive target when treating patients with this deadly disease.
IGF-1R, a transmembrane tyrosine kinase receptor of the insulin receptor family, is known to be a direct target gene of miR-944 in HCC cells. It is highly expressed in a variety of human cancers, including bladder cancer,37 endometrial cancer,38 cervical cancer,39 and oral squamous cell carcinoma.40 IGF-1R is also expressed at high levels in HCC tissues and cell lines.23 High IGF-1R expression is significantly correlated with TNM stage in patients with HCC. Survival analysis has shown that patients with HCC who display high IGF-1R expression have poorer overall and disease-free survival than those with low IGF-1R expression. In addition, multivariate analysis has identified high IGF-1R expression as an independent prognostic marker of poor survival in patients with HCC.23 IGF-1R is able to activate multiple components of the PI3K/Akt pathway and it contributes to the aggressive characteristics of HCC, including cell proliferation, apoptosis, migration, invasion, and chemotherapy resistance.24–29 In our current study, miR-944 was shown to be a negative regulator of IGF-1R and could inhibit the malignant phenotypes of HCC in vitro and in vivo by decreasing PI3K/Akt signaling. These results suggest that miR-944 restoration-mediated silencing of IGF-1R expression may be an effective approach to treat patients with HCC.
Conclusion
In summary, we determined the clinical and biological roles of the miR-944/IGF-1R axis in HCC and identified the PI3K/Akt signaling pathway as the downstream effector. The miR-944/IGF-1R axis suppressed the progression and development of HCC and the PI3K/Akt pathway was notably deactivated in this process. These observations suggest that miR-944 is a potential prognostic indicator and may provide alternative therapeutic opportunities for patients with HCC. However, we did not explore whether the morphological evidence for EMT-like characteristics in HCC cells could be affected by miR-944. It was a limitation of the present study that may be resolved in future investigations.
Disclosure
The authors have no conflicts of interest to declare.
References
1. McGlynn KA, Petrick JL, London WT. Global epidemiology of hepatocellular carcinoma: an emphasis on demographic and regional variability. Clin Liver Dis. 2015;19:223–238. doi:10.1016/j.cld.2015.01.001
2. Zhu ZX, Huang JW, Liao MH, Zeng Y. Treatment strategy for hepatocellular carcinoma in China: radiofrequency ablation versus liver resection. Jpn J Clin Oncol. 2016;46:1075–1080. doi:10.1093/jjco/hyw134
3. Greten TF, Wang XW, Korangy F. Current concepts of immune based treatments for patients with HCC: from basic science to novel treatment approaches. Gut. 2015;64:842–848. doi:10.1136/gutjnl-2014-307990
4. Tunissiolli NM, Castanhole-Nunes MMU, Biselli-Chicote PM, et al. Hepatocellular carcinoma: a comprehensive review of biomarkers, clinical aspects, and therapy. Asian Pac J Cancer Prev. 2017;18:863–872. doi:10.22034/APJCP.2017.18.4.863
5. Fujiwara N, Friedman SL, Goossens N, Hoshida Y. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. J Hepatol. 2018;68:526–549. doi:10.1016/j.jhep.2017.09.016
6. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297.
7. Wijnhoven BP, Michael MZ, Watson DI. MicroRNAs and cancer. Br J Surg. 2007;94:23–30. doi:10.1002/bjs.5673
8. Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–D144. doi:10.1093/nar/gkj112
9. Zhou Y, Liu K, Liu Y, Tan L. MicroRNA-34a inhibit hepatocellular carcinoma progression by repressing hexokinase-1. J Cell Biochem. 2018;120(5):7147–7153.
10. Wang Z, Si M, Yang N, et al. MicroRNA-506 suppresses invasiveness and metastasis of human hepatocellular carcinoma cells by targeting IL8. Am J Cancer Res. 2018;8:1586–1594.
11. Tao J, Liu Z, Wang Y, et al. MicroRNA-645 represses hepatocellular carcinoma progression by inhibiting SOX30-mediated p53 transcriptional activation. Int J Biol Macromol. 2019;121:214–222. doi:10.1016/j.ijbiomac.2018.10.032
12. Hu Z, Wang P, Lin J, et al. MicroRNA-197 promotes metastasis of hepatocellular carcinoma by activating Wnt/beta-catenin signaling. Cell Physiol Biochem. 2018;51:470–486. doi:10.1159/000495242
13. Li C, Wang Z, Chen S, Zhang J, Qu K, Liu C. MicroRNA-552 promotes hepatocellular carcinoma progression by downregulating WIF1. Int J Mol Med. 2018;42:3309–3317. doi:10.3892/ijmm.2018.3882
14. Han LL, Yin XR, Zhang SQ. miR-650 promotes the metastasis and epithelial-mesenchymal transition of hepatocellular carcinoma by directly inhibiting LATS2 expression. Cell Physiol Biochem. 2018;51:1179–1192. doi:10.1159/000495495
15. Zhu Z, Zhang X, Wang G, Zheng H. Role of microRNAs in hepatocellular carcinoma. Hepat Mon. 2014;14:e18672. doi:10.5812/hepatmon
16. Vasuri F, Visani M, Acquaviva G, et al. Role of microRNAs in the main molecular pathways of hepatocellular carcinoma. World J Gastroenterol. 2018;24:2647–2660. doi:10.3748/wjg.v24.i25.2647
17. Wang Z, Wu Z, Huang P. The function of miRNAs in hepatocarcinogenesis induced by hepatitis B virus X protein (Review). Oncol Rep. 2017;38:652–664. doi:10.3892/or.2017.5716
18. Wen L, Li Y, Jiang Z, Zhang Y, Yang B, Han F. miR-944 inhibits cell migration and invasion by targeting MACC1 in colorectal cancer. Oncol Rep. 2017;37:3415–3422. doi:10.3892/or.2017.5611
19. Liu M, Zhou K, Cao Y. MicroRNA-944 affects cell growth by targeting EPHA7 in non-small cell lung cancer. Int J Mol Sci. 2016;17:1493. doi:10.3390/ijms17101493
20. He Z, Xu H, Meng Y, Kuang Y. miR-944 acts as a prognostic marker and promotes the tumor progression in endometrial cancer. Biomed Pharmacother. 2017;88:902–910. doi:10.1016/j.biopha.2017.01.117
21. Xie H, Lee L, Scicluna P, et al. Novel functions and targets of miR-944 in human cervical cancer cells. Int J Cancer. 2015;136:E230–E241. doi:10.1002/ijc.29160
22. He H, Tian W, Chen H, Jiang K. MiR-944 functions as a novel oncogene and regulates the chemoresistance in breast cancer. Tumour Biol. 2016;37:1599–1607. doi:10.1007/s13277-015-3844-x
23. Lin SB, Zhou L, Liang ZY, Zhou WX, Jin Y. Expression of GRK2 and IGF1R in hepatocellular carcinoma: clinicopathological and prognostic significance. J Clin Pathol. 2017. doi:10.1136/jclinpath-2016-203998
24. Yulyana Y, Ho IA, Sia KC, et al. Paracrine factors of human fetal MSCs inhibit liver cancer growth through reduced activation of IGF-1R/PI3K/Akt signaling. Mol Ther. 2015;23:746–756. doi:10.1038/mt.2015.13
25. Liu W, Kang L, Han J, et al. miR-342-3p suppresses hepatocellular carcinoma proliferation through inhibition of IGF-1R-mediated Warburg effect. Onco Targets Ther. 2018;11:1643–1653. doi:10.2147/OTT.S161586
26. Han X, Wang X, Zhao B, et al. MicroRNA-187 inhibits tumor growth and metastasis via targeting of IGF-1R in hepatocellular carcinoma. Mol Med Rep. 2017;16:2241–2246. doi:10.3892/mmr.2017.6788
27. Xu Y, Huang J, Ma L, et al. MicroRNA-122 confers sorafenib resistance to hepatocellular carcinoma cells by targeting IGF-1R to regulate RAS/RAF/ERK signaling pathways. Cancer Lett. 2016;371:171–181. doi:10.1016/j.canlet.2015.11.034
28. Wu J, Zhu AX. Targeting insulin-like growth factor axis in hepatocellular carcinoma. J Hematol Oncol. 2011;4:30. doi:10.1186/1756-8722-4-30
29. Yao WF, Liu JW, Sheng GL, Huang DS. Blockade of IGF-IR exerts anticancer effects in hepatocellular carcinoma. Mol Med Rep. 2011;4:719–722. doi:10.3892/mmr.2011.486
30. Song Y, Zhao Y, Ding X, Wang X. microRNA-532 suppresses the PI3K/Akt signaling pathway to inhibit colorectal cancer progression by directly targeting IGF-1R. Am J Cancer Res. 2018;8:435–449.
31. Zhang Y, Goodfellow R, Li Y, et al. NEDD4 ubiquitin ligase is a putative oncogene in endometrial cancer that activates IGF-1R/PI3K/Akt signaling. Gynecol Oncol. 2015;139:127–133. doi:10.1016/j.ygyno.2015.07.098
32. Lin L, Yin X, Hu X, Wang Q, Zheng L. The impact of hepatitis B virus x protein and microRNAs in hepatocellular carcinoma: a comprehensive analysis. Tumour Biol. 2014;35:11695–11700. doi:10.1007/s13277-014-2658-6
33. Akkiz H. The emerging role of microRNAs in hepatocellular carcinoma. Euroasian J Hepatogastroenterol. 2014;4:45–50. doi:10.5005/jp-journals-10018-1095
34. Yang N, Ekanem NR, Sakyi CA, Ray SD. Hepatocellular carcinoma and microRNA: new perspectives on therapeutics and diagnostics. Adv Drug Deliv Rev. 2015;81:62–74. doi:10.1016/j.addr.2014.10.029
35. Pan T, Chen W, Yuan X, Shen J, Qin C, Wang L. miR-944 inhibits metastasis of gastric cancer by preventing the epithelial-mesenchymal transition via MACC1/Met/AKT signaling. FEBS Open Bio. 2017;7:905–914. doi:10.1002/2211-5463.12215
36. Zheng H, Zou AE, Saad MA, et al. Alcohol-dysregulated microRNAs in hepatitis B virus-related hepatocellular carcinoma. PLoS One. 2017;12:e0178547. doi:10.1371/journal.pone.0178547
37. Sun HZ, Wu SF, Tu ZH. Blockage of IGF-1R signaling sensitizes urinary bladder cancer cells to mitomycin-mediated cytotoxicity. Cell Res. 2001;11:107–115. doi:10.1038/sj.cr.7290075
38. Pengchong H, Tao H. Expression of IGF-1R, VEGF-C and D2-40 and their correlation with lymph node metastasis in endometrial adenocarcinoma. Eur J Gynaecol Oncol. 2011;32:660–664.
39. Huang YF, Shen MR, Hsu KF, Cheng YM, Chou CY. Clinical implications of insulin-like growth factor 1 system in early-stage cervical cancer. Br J Cancer. 2008;99:1096–1102. doi:10.1038/sj.bjc.6604661
40. Gao L, Wang X, Wang X, et al. IGF-1R, a target of let-7b, mediates crosstalk between IRS-2/Akt and MAPK pathways to promote proliferation of oral squamous cell carcinoma. Oncotarget. 2014;5:2562–2574. doi:10.18632/oncotarget.1812
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.