")
Back to Journals » OncoTargets and Therapy » Volume 12
microRNA-628 inhibits the proliferation of acute myeloid leukemia cells by directly targeting IGF-1R
Authors Chen L, Jiang X, Chen H, Han Q, Liu C, Sun M
Received 25 October 2018
Accepted for publication 18 December 2018
Published 29 January 2019 Volume 2019:12 Pages 907—919
DOI https://doi.org/10.2147/OTT.S192137
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjeev K. Srivastava
This paper has been retracted
Lu Chen, Xin Jiang, Haoyue Chen, Qiaoyan Han, Chunhua Liu, Miao Sun
Department of Hematology, Jingjiang People’s Hospital, The Seventh Affiliated Hospital of Yangzhou University, Jiangsu 214500, P.R. China
Background: A variety of microRNAs (miRNAs) are aberrantly expressed in acute myeloid leukemia (AML), and these dysregulated miRNAs perform crucial roles in tumorigenesis and progression of AML. miR-628-3p (miR-628), one of the miRNAs dysregulated in multiple types of human cancers, exerts antitumor roles in different cancer types. However, no specific study has explored the expression pattern and role of miR-628 in AML.
Materials and methods: In this study, RT-qPCR was performed to detect miR-628 expression in AML tissues and cell lines. CCK-8 assay, flow cytometry analysis and xenograft tumor experiment was carried out to determine the functions of miR-628 in AML cells. The possible mechanism underlying the activity of miR-628 in AML cells was also explored using a series of experiments.
Results: Our results revealed the downregulated expression of miR-628 in patients with AML and AML cell lines. Ectopic expression of miR-628 resulted in the inhibition of AML cell proliferation and induction of cell cycle arrest and apoptosis in vitro and attenuation of tumor growth in vivo. Insulin-like growth factor 1 receptor (IGF-1R) was identified as a direct target gene of miR-628 in AML cells. IGF-1R expression was upregulated in patients with AML and upregulation of IGF-1R expression inversely correlated with miR-628 level. Furthermore, IGF-1R knockdown imitated the tumor suppressive effect of miR-628 in AML cells. Restoration of IGF-1R expression abrogated the effects of miR-628 on the proliferation, cycle status, and apoptosis rate of AML cells. miR-628 inhibited the activation of phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/protein kinase B (Akt) pathway in AML cells both in vitro and in vivo through the inhibition of IGF-1R expression.
Conclusion: Our results demonstrate that miR-628 exhibits antitumor effects in AML through the direct targeting of IGF-1R and regulation of PI3K/Akt pathway, suggestive of its potential role as a therapeutic target in patients with this aggressive hematological malignant tumor.
Keywords: acute myeloid leukemia, microRNA-628, proliferation, cell cycle, apoptosis, insulin-like growth factor 1 receptor, PI3K/Akt pathway
Introduction
Acute myeloid leukemia (AML), a very aggressive leukemic subtype, is a bone marrow malignancy characterized with abnormal growth of bone marrow stromal cells.1 It accounts for about 15%–20% of cases of acute leukemia in children.2 Genetic abnormalities that interfere with the growth, metastasis, and differentiation of normal hematopoietic progenitor cells are involved in AML pathogenesis;3,4 however, the detailed mechanisms underlying the occurrence and development of AML are incompletely understood. At present, the treatment strategies for patients with AML include chemotherapy, targeted therapy, and hematopoietic stem cell transplantation.5 The significant development in treatment strategies has greatly improved the therapeutic outcomes of patients with AML over last several decades. Unfortunately, a large number of patients with AML still relapse and die of the disease.6,7 In this regard, elucidation of the molecular processes and mechanisms responsible for AML development and progression is of great importance to develop reliable therapeutic approaches for patients with this disease. microRNAs (miRNAs) have great importance in the progression and development of AML.8 miRNAs belong to a group of noncoding and short RNA molecules composed of 17–23 nucleotides.9 miRNAs function as gene regulators by directly binding to the 3′-untranslated regions (3′-UTRs) of their target genes and inducing degradation and/or translational suppression of messenger RNAs (mRNAs).10 To date, over 1,881 human miRNAs have been identified in miRBase (Release 21; http://www.mirbase.org/) that are thought to modulate the expression of about 30% human protein-coding genes.11 The expression patterns of miRNAs are altered in almost all human malignancies, suggesting that the changes in miRNA expression may have crucial effect on carcinogenesis and cancer progression.12–14 Several miRNAs that serve as tumor suppressors or oncogenes have been reported to be dysregulated in AML.15–17 Therefore, miRNAs may serve as the potential targets for AML treatment.
The expression of miR-628-3 p (miR-628) is dysregulated in multiple types of human cancers, wherein it performs antitumor roles.18–21 However, no specific study has explored the expression pattern and role of miR-628 in AML. In this study, we measured the expression level of miR-628 in AML and examined the functions of miR-628 in AML. To reveal the possible underlying mechanism, bioinformatics analysis was performed to search for the potential target of miR-628. The analysis indicated that insulin-like growth factor 1 receptor (IGF-1R) was a potential target of miR-628. Herein, a series of experiments were performed to illustrate whether IGF-1R was a direct target gene of miR-628 in AML cells. The results of this study indicate that the miR-628/IGF-1R axis closely correlates with the malignant progression of AML.
Materials and methods
Human samples
Bone marrow specimens were obtained from 39 patients with AML and 23 healthy controls from Jingjiang People’s Hospital, The Seventh Affiliated Hospital of Yangzhou University, between March 2015 and September 2017. The clinical and molecular characteristics of AML patients are provided in the Table 1. None of the patients had received chemotherapy, radiotherapy, targeted therapy, or hematopoietic stem cell transplantation before bone marrow aspiration. Patients who had been treated with abovementioned therapeutic techniques were excluded from this study. This study was approved by the Ethics Committee Hospital of Jingjiang People’s Hospital (ethic number: 20150312) and was performed in accordance with the Declaration of Helsinki and the guidelines of the Ethics Committee of Jingjiang People’s Hospital, The Seventh Affiliated Hospital of Yangzhou University. Written informed consent was also provided by all participants.
 | Table 1 Clinical and molecular characteristics of acute myeloid leukemia patients |
Cell lines
A total of three human AML cell lines (HL-60, Kasumi-1, and THP-1) and a normal bone marrow cell line (HS-5) were purchased from the American Type Culture Collection (Manassas, VA, USA). DMEM containing 10% heat-inactivated FBS (both from Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), 100 U/mL of penicillin, and 100 mg/mL of streptomycin (Sigma-Aldrich, St Louis, MO, USA; Merck KGaA, Darmstadt, Germany) was used to culture all AML cell lines. All cultures were maintained at 37°C in a humidified incubator containing 5% CO2.
Transfection of mimics, small-interfering RNA (siRNA), and plasmid
The miR-628 mimics and miRNA mimics negative control (miR-NC) were chemically synthesized by Shanghai Genechem Co., Ltd. (Shanghai, P.R. China). To knockdown IGF-1R expression, an siRNA against IGF-1R (IGF-1R siRNA) and a negative control siRNA (NC siRNA) were purchased from Shanghai GenePharma Co., Ltd. (Shanghai, P.R. China). IGF-1R expression plasmid pcDNA3.1-IGF-1R (pc-IGF-1R) and empty pcDNA3.1 plasmid were obtained from GeneCopoeia, Inc. (Rockville, MD, USA). Cells were seeded into six-well plates at a density of 5×105 cells/well. The miRNA mimics, siRNA, or plasmid was transfected into cells using Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer’s protocols. Cells were incubated at 37°C with 5% CO2. Transfected cells were collected after incubation for different time points and used in the subsequent experiments.
Reverse-transcription quantitative polymerase chain reaction (RT-qPCR)
Mononuclear cells were isolated from the bone marrow samples using Ficoll-Paque Plus (GE Healthcare, Chicago, IL, USA), in accordance with the manufacturer’s protocols. TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc.) was used to extract total RNA from mononuclear cells and cultured cell lines, and the RNA was reverse transcribed into complementary DNA (cDNA) using TaqMan MicroRNA RT kit (Applied Biosystems; Thermo Fisher Scientific, Inc.). miR-628 expression was determined using TaqMan MicroRNA Assay kit (Applied Biosystems; Thermo Fisher Scientific, Inc.). To quantify IGF-1R mRNA expression, cDNA was synthesized from total RNA using a PrimeScript RT Reagent kit, and the synthesized cDNA was subjected to qPCR using a SYBR Premix Ex Taq kit (both from Takara Biotechnology Co., Ltd., Dalian, P.R. China). U6 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as the housekeeping genes to normalize the expression levels of miR-628 and IGF-1R mRNA, respectively. The 2−ΔΔCq method was used to analyze the relative gene expression.22
Cell counting kit-8 (CCK-8) assay
The regulatory role of miR-628 on the proliferation of AML cells was evaluated using the CCK-8 assay. In detail, the transfected cells in 200 μL of culture medium were seeded in 96-well plates at a density of 3×103 cells/well. Cellular proliferation was determined every 24 hours for 3 days. A total of 10 μL of CCK-8 assay solution (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) was added into each well at each time point. Following 2 hours of incubation at 37°C with 5% CO2, the optical density was detected at 450 nm wavelength using an ELx808 absorbance reader (BioTek Instruments, Inc., Winooski, VT, USA).
Flow cytometry analysis of cell cycle and apoptosis
After 48 hours of transfection, the cells were harvested, washed twice with ice-cold PBS (Gibco; Thermo Fisher Scientific, Inc.), and fixed with 70% ethanol at 4°C for 1 hour. Cells were incubated with 50 μL of RNase 1 at room temperature for 10 minutes to degrade RNA. Cells were centrifugated at 157× g at 4°C for 5 minutes, followed by the addition of 25 μL of propidium iodide solution and 425 μL of cell staining buffer (both from BioLegend, San Diego, CA, USA). Cell cycle status was evaluated using a flow cytometer (FACScan; BD Biosciences, Franklin Lakes, NJ, USA).
Cell apoptosis was assessed after 48 hours of transfection using an Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit (BioLegend). Briefly, the transfected cells were washed with ice-cold PBS, centrifugated, and resuspended in 100 μL of binding buffer. The transfected cells were double-stained with 5 μL of Annexin V-FITC and 5 μL of propidium iodide for 30 minutes at room temperature in the dark. A flow cytometer was used to measure the number of apoptotic cells.
Xenograft tumor experiment
BALB/c nude mice (4–6 weeks old) were purchased from the Shanghai Laboratory Animal Center (Shanghai, P.R. China). miR-628 mimics or miR-NC was transfected into HL-60 cells. After 24 hours of incubation, the transfected cells were collected and subcutaneously administered into the hind flanks of nude mice. The width and length of tumor xenografts were detected every 2 days using a Vernier caliper. The tumor volumes were analyzed using the equation: tumor volume (mm3) = width2 (mm2) × length (mm)/2. The tumor xenografts were excised from the mice sacrificed after 4 weeks of cell implantation and weighed. Xenograft tumor experiment was performed in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the Ethics Review Committee of Jingjiang People’s Hospital, The Seventh Affiliated Hospital of Yangzhou University, and was conducted in accordance with the Declaration of Helsinki.
Bioinformatics prediction
Three different miRNA target prediction softwares, including TargetScan (http://www.targetscan.org/), miRDB (http://www.mirdb.org/), and miRanda (http://www.microrna.org), were used to predict the putative targets of miR-628.
Luciferase reporter assay
The luciferase reporter plasmids harboring the wild-type (WT) and mutated (MUT) 3′-UTR of IGF-1R gene were designed and obtained from Shanghai GenePharma Co., Ltd.; these were referred as pmiR-IGF-1R-3′-UTR WT and pmiR-IGF-1R-3′-UTR MUT, respectively. For the reporter assay, cells were plated in 24-well plates at a concentration of 1×105 cells/well. miR-628 mimics or miR-NC was cotransfected with pmiR-IGF-1R-3′-UTR WT or pmiR-IGF-1R-3′-UTR MUT into cells using Lipofectamine 2000, following the manufacturer’s protocols. After 48 hours, the transfected cells were harvested and the luciferase activity was evaluated using a Dual-Luciferase Reporter Assay System (Promega Corp., Madison, WI, USA), according to the manufacturer’s protocols. Firefly luciferase activity was normalized to that of Renilla luciferase activity.
Western blot analysis
Total cellular protein was isolated from the cultured cells or bone marrow specimens using a radioimmunoprecipitation assay lysis buffer (Sigma-Aldrich). A bicinchoninic acid protein assay reagent kit (Pierce; Thermo Fisher Scientific, Inc.) was used to detect the concentration of total protein. Equal amounts of proteins were loaded, separated by 10% SDS-PAGE, and transferred onto polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA, USA). After blocking with 5% nonfat milk in Tris-buffered saline containing 0.1% Tween-20, the membranes were incubated overnight at 4°C with the following primary antibodies: rabbit anti-human monoclonal IGF-1R antibody (ab182408; 1:1,000 dilution; Abcam, Cambridge, UK), rabbit anti-human monoclonal antibody to phosphorylated phosphatidylinositol-4,5-bisphosphate 3-kinase (p-PI3K; ab182651; 1:1,000 dilution; Abcam), rabbit anti-human monoclonal PI3K antibody (ab191606; 1:1,000 dilution; Abcam), mouse anti-human monoclonal antibody to phosphorylated protein kinase B (p-Akt; sc-81433; 1:1,000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse anti-human monoclonal Akt antibody (sc-56878; 1:1,000 dilution; Santa Cruz Biotechnology), and rabbit anti-human monoclonal GAPDH antibody (ab181603; 1:1,000 dilution; Santa Cruz Biotechnology). Horseradish peroxidase-conjugated goat antirabbit secondary antibody (ab6721 and ab6789; Abcam) was used (1:5,000 dilution) for 2 hours at room temperature, and the protein signals were detected using an enhanced chemiluminescence reagent (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Quantity One software version 4.62 (Bio-Rad Laboratories, Inc.) was used for the quantification of protein expression.
Statistical analysis
All data were expressed as the mean ± standard error from at least three independent experiments. The difference between two groups was analyzed using Student’s t-test. One-way ANOVA, followed by the Student–Newman–Keuls post hoc test, was used to compare the differences between multiple groups. Spearman’s correlation analysis was carried out to determine the correlation between miR-628 and IGF-1R mRNA levels in patients with AML. All statistical analyses were performed using SPSS 21.0 software (IBM Corporation, Armonk, NY, USA). P<0.05 indicated statistically significant difference.
Results
miR-628 expression is downregulated in AML
To investigate the biological role of miR-629 in AML, we first detected the expression level of miR-628 in the bone marrow specimens derived from 29 patients with AML and 23 healthy controls. The results of RT-qPCR analysis revealed the notable downregulation in the expression level of miR-628 in patients with AML compared with healthy controls (Figure 1A, P<0.05). We also performed RT-qPCR analysis to determine the expression status of miR-628 in three human AML cell lines (HL-60, Kasumi-1, and THP-1) and a normal bone marrow cell line (HS-5). The expression level of miR-628 was significantly lower in all AML cell lines than in HS-5 cells (Figure 1B, P<0.05). These results suggest that miR-628 downregulation may be related to the development of AML.
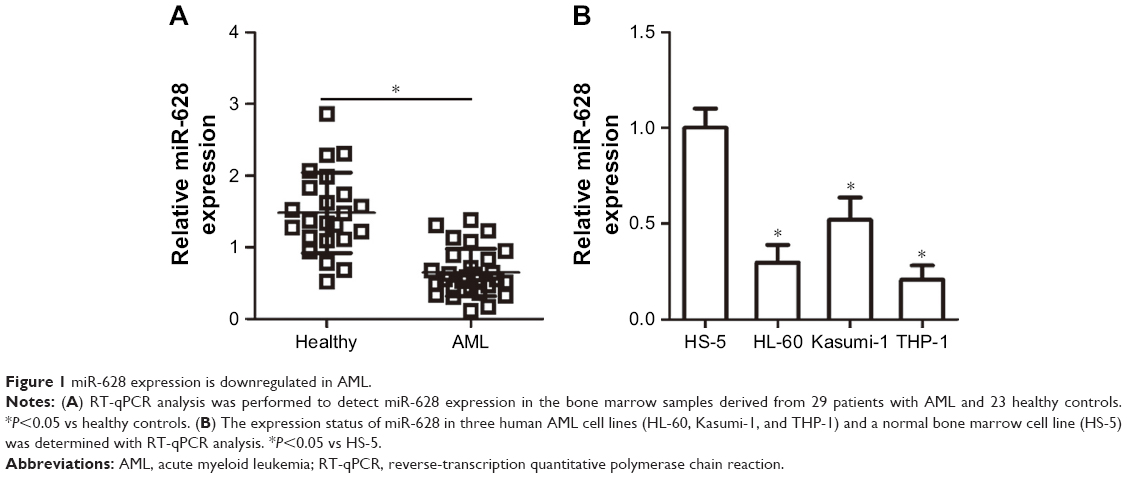 | Figure 1 miR-628 expression is downregulated in AML. |
Ectopic miR-628 expression inhibits AML growth in vitro
HL-60 and THP-1 cell lines exhibited a relatively lower miR-628 expression among the three AML cell lines; therefore, we chose the two cell lines as models to investigate the effects of miR-628 on AML progression. miR-628 was exogenously expressed by transfecting miR-628 mimics into HL-60 and THP-1 cells, and the expression was confirmed by RT-qPCR analysis (Figure 2A, P<0.05). CCK-8 assay was used to evaluate the impact of miR-628 overexpression on AML cell proliferation. We observed that miR-628 overexpression significantly inhibited the proliferation of HL-60 and THP-1 cells compared with the cells transfected with miR-NC (Figure 2B, P<0.05). Considering the inhibitory effect of miR-628 expression on AML cell proliferation, we investigated whether miR-628 expression affected AML cell cycle and apoptosis. Flow cytometry analysis was performed to determine cell cycle status and apoptosis of HL-60 and THP-1 cells transfected with miR-628 mimics or miR-NC. The results showed that the ectopic expression of miR-628 in HL-60 and THP-1 cells resulted in a decrease in the percentage of cells in S phase and an increase in the percentage of cells in G0/G1 phase (Figure 2C, P<0.05). Furthermore, the apoptotic rate of HL-60 and THP-1 cells significantly increased following transfection with miR-628 mimics (Figure 2D, P<0.05). These results suggest that miR-628 inhibits AML cell growth in vitro via induction of apoptosis and cell cycle arrest.
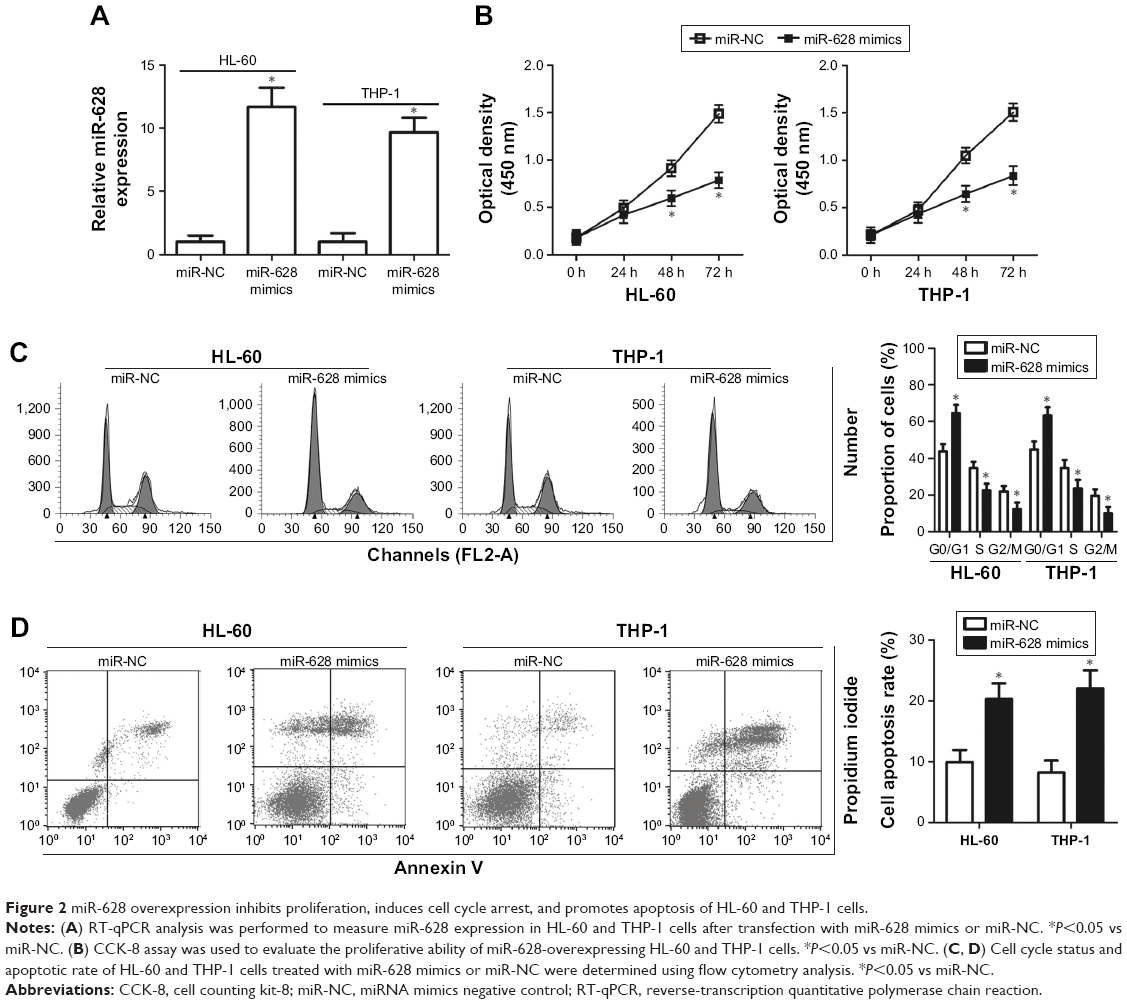 | Figure 2 miR-628 overexpression inhibits proliferation, induces cell cycle arrest, and promotes apoptosis of HL-60 and THP-1 cells. |
IGF-1R is a direct target gene of miR-628 in AML cells
miRNA modulates gene expression by binding to the 3′-UTR of their target genes, subsequently leading to mRNAs degradation and/or translation suppression. To clarify the mechanisms underlying the roles of miR-628, three public miRNA databases, TargetScan, miRDB, and miRanda, were scanned to predict the potential target of miR-628. As indicated in Figure 3A, the 3′-UTR of IGF-1R contains a highly conserved binding site for miR-628. IGF-1R was of particular interest because of its involvement in AML progression.23–26 Luciferase reporter assay was performed to confirm this hypothesis. Luciferase reporter plasmid carrying the WT or mutant 3′-UTR-binding site of IGF-1R was chemically synthesized and cotransfected with miR-628 mimics or miR-NC into HL-60 and THP-1 cells. The results showed that the resumption of miR-628 expression decreased the luciferase activity in HL-60 and THP-1 cells transfected with the WT IGF-1R 3′-UTR (P<0.05); however, the luciferase activity of the plasmid harboring the mutant IGF-1R 3′-UTR was unaffected (Figure 3B), suggesting that miR-628 may recognize and bind to the 3′-UTR of IGF-1R.
 | Figure 3 miR-628 directly targets IGF-1R in AML cells. |
We detected IGF-1R expression level in the bone marrow specimens derived from 29 patients with AML and 23 healthy controls using RT-qPCR analysis and found that IGF-1R mRNA expression was significantly upregulated in patients with AML (Figure 3C, P<0.05). Furthermore, an inverse correlation was observed between miR-628 and IGF-1R mRNA levels in patients with AML (Figure 3D; r=−0.5393, P=0.0025). We examined whether miR-628 affects the endogenous IGF-1R expression in AML cells. RT-qPCR and Western blot analysis showed that the enforced miR-628 expression resulted in the reduction of IGF-1R expression in HL-60 and THP-1 cells at both mRNA (Figure 3E, P<0.05) and protein (Figure 3F, P<0.05) levels. These observations demonstrate that IGF-1R is a direct target of miR-628 in AML cells.
IGF-1R expression inhibition imitates the miR-628-induced phenotype in AML cells
Considering IGF-1R as a direct target gene of miR-628, we investigated the functional role of IGF-1R in AML cells by introducing an siRNA targeting IGF-1R (IGF-1R siRNA) into HL-60 and THP-1 cells. Western blot analysis confirmed the downregulated expression of IGF-1R in IGF-1R siRNA-transfected HL-60 and THP-1 cells compared with the cells transfected with NC siRNA (Figure 4A, P<0.05). CCK-8 assay indicated that IGF-1R knockdown significantly reduced the proliferation of HL-60 and THP-1 cells (Figure 4B, P<0.05). In addition, the cell cycle status and apoptosis rate of HL-60 and THP-1 cells treated with IGF-1R siRNA or NC siRNA were determined through flow cytometry analysis. Transfection of IGF-1R siRNA in HL-60 and THP-1 cells induced a significant decrease in the percentage of cells in S phase and increased the number of cells in G0/G1 phase (Figure 4C, P<0.05). As shown in Figure 4D, the percentage of apoptotic HL-60 and THP-1 cells was higher in the cells subjected to IGF-1R silencing than in the control cells treated with NC siRNA (P<0.05). Together, these data demonstrate that IGF-1R expression inhibition could mimic the inhibitory roles of miR-628 in AML cells, suggesting that IGF-1R serves as a downstream target of miR-628.
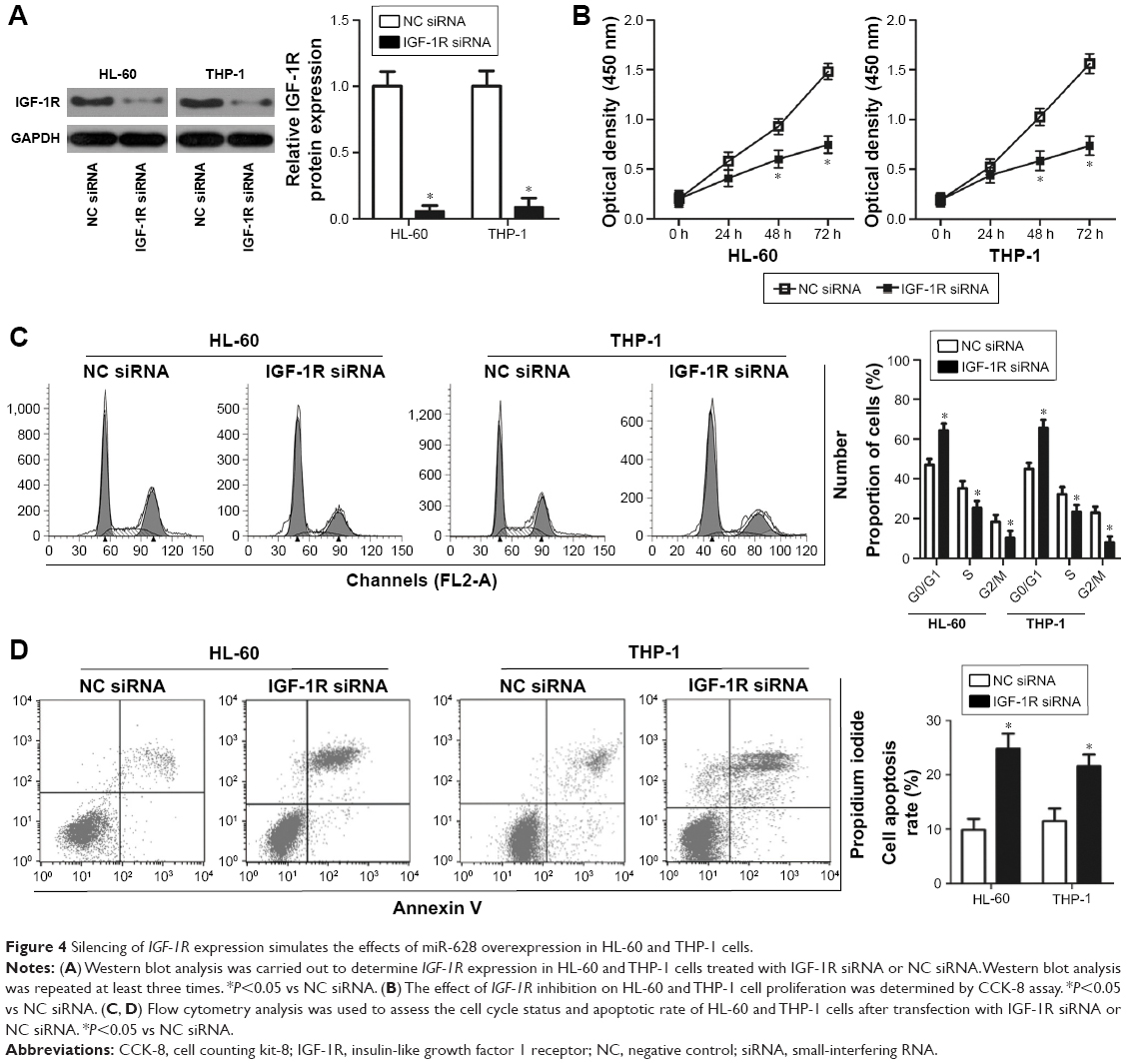 | Figure 4 Silencing of IGF-1R expression simulates the effects of miR-628 overexpression in HL-60 and THP-1 cells. |
Restoration of IGF-1R expression abrogates the effects of miR-628 in AML cells
Rescue experiments were performed to explore the role of IGF-1R in miR-628-regulated AML progression. IGF-1R expression was recovered in miR-628 mimics-transfected HL-60 and THP-1 cells after cotransfection with IGF-1R expression plasmid pcDNA3.1-IGF-1R (pc-IGF-1R), as confirmed by Western blot analysis (Figure 5A, P<0.05). Functional experiments showed that the effects of miR-628 upregulation on the proliferation (Figure 5B, P<0.05), cell cycle status (Figure 5C, P<0.05), and apoptosis (Figure 5D, P<0.05) of HL-60 and THP-1 cells were abrogated after the restoration of IGF-1R expression. These results suggest that miR-628 overexpression inhibits the growth of AML cells in vitro, at least in part, through the inhibition of IGF-1R expression.
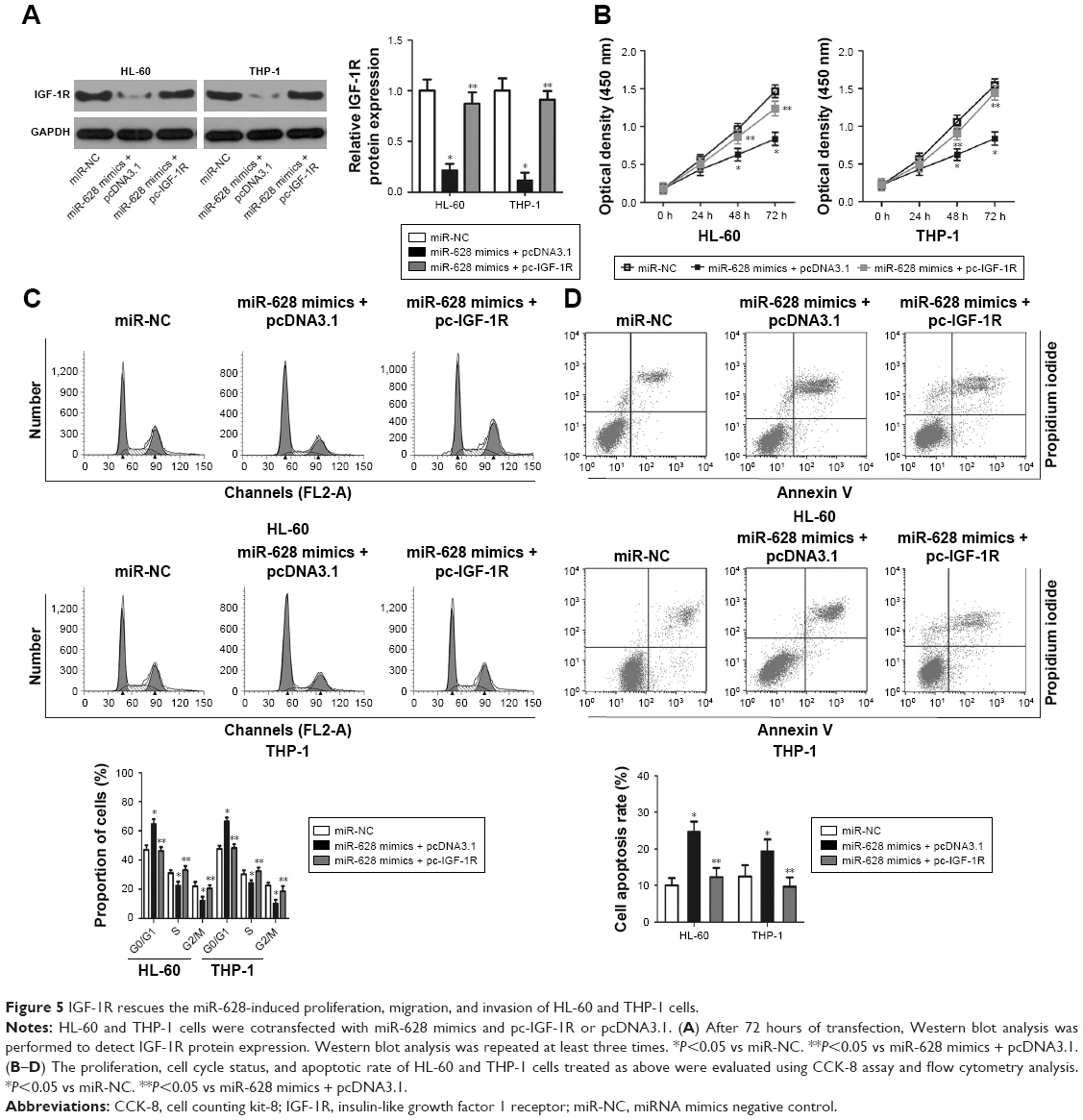 | Figure 5 IGF-1R rescues the miR-628-induced proliferation, migration, and invasion of HL-60 and THP-1 cells. |
miR-628 deactivates the PI3K/Akt pathway in AML cells by targeting IGF-1R
Multiple studies have reported the involvement of IGF-1R in the regulation of the PI3K/Akt signaling pathway.27–30 We investigated whether miR-628 targets IGF-1R to inhibit the activation of the PI3K/Akt pathway in AML cells. miR-628 mimics were cotransfected with pc-IGF-1R or pcDNA3.1 into HL-60 and THP-1 cells. Following transfection for 72 hours, Western blot analysis was performed, and the results revealed that miR-628 overexpression significantly decreased the protein levels of p-PI3K and p-Akt in HL-60 and THP-1 cells. However, the levels of total PI3K and Akt proteins were unaltered. Furthermore, IGF-1R restoration abolished the changes in p-PI3K and p-Akt protein expression levels induced in response to miR-628 overexpression (Figure 6, P<0.05). These results clearly indicate that miR-628 deactivates the PI3K/Akt signaling pathway in AML cells by targeting IGF-1R.
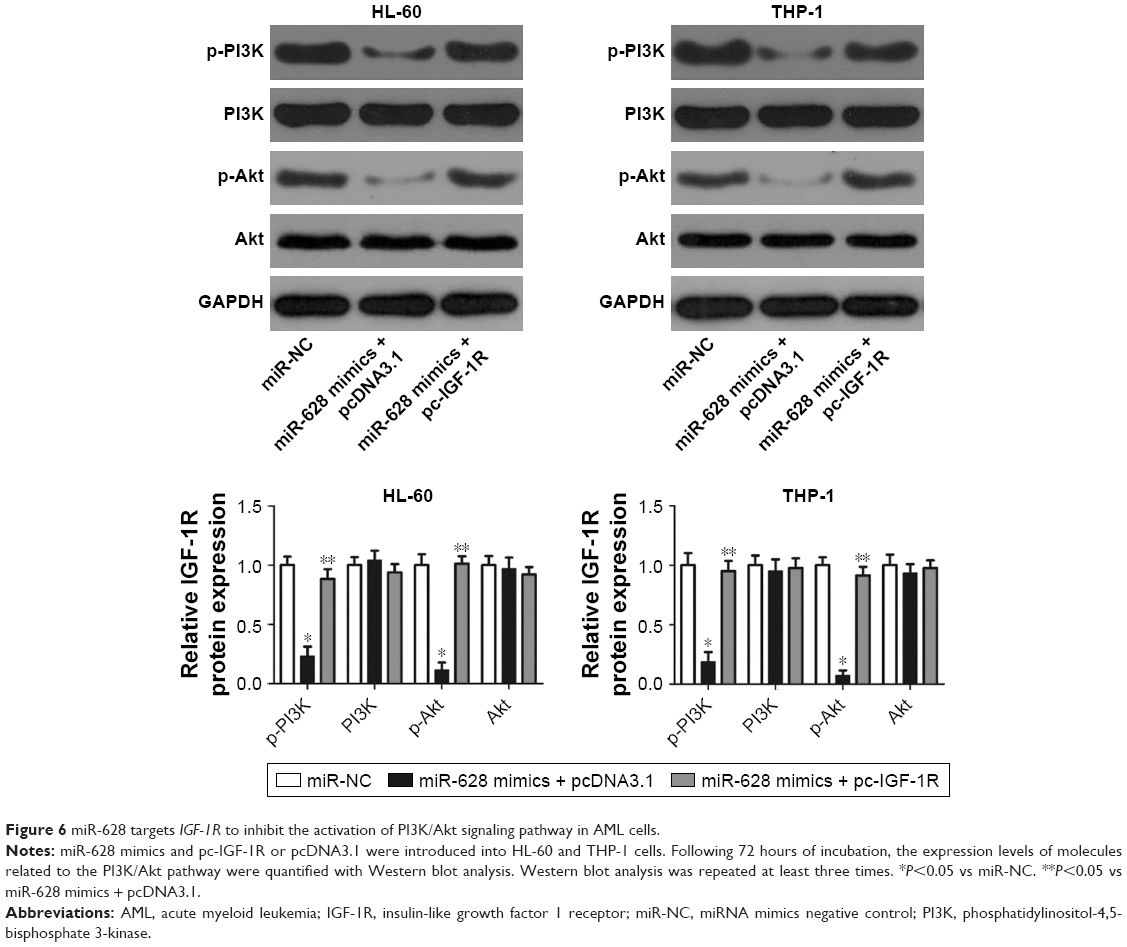 | Figure 6 miR-628 targets IGF-1R to inhibit the activation of PI3K/Akt signaling pathway in AML cells. |
miR-628 suppresses tumor growth in AML in vivo
Xenograft tumor assay was used to study the effect of miR-628 on the tumor growth of AML in vivo. HL-60 cells were transfected with miR-628 mimics or miR-NC and subcutaneously implanted into nude mice. The volume of xenografts formed was detected every 2 days. As a result, the tumor volume in miR-628 group was notably lower than that reported in miR-NC group (Figure 7A and B, P<0.05). In addition, the weight of tumor xenografts derived from miR-628 mimics-transfected HL-60 cells was significantly decreased (Figure 7C, P<0.05). RT-qPCR analysis was used to measure the expression level of miR-628 in tumor xenografts to confirm that miR-628 upregulation was responsible for the in vivo tumor growth suppression. The data showed that miR-628 was expressed at high levels in the tumor xenografts obtained from miR-628 group (Figure 7D, P<0.05). Furthermore, the protein levels of IGF-1R and molecules related to the PI3K/Akt pathway were quantified by Western blot analysis. An obvious downregulation in the protein levels of IGF-1R, p-PI3K, and p-Akt was observed in the mice treated with miR-628 mimics (Figure 7E, P<0.05). These observations suggest that miR-628 inhibits the growth of AML cells in vivo via direct targeting of IGF-1R and inhibition of the PI3K/Akt signaling pathway.
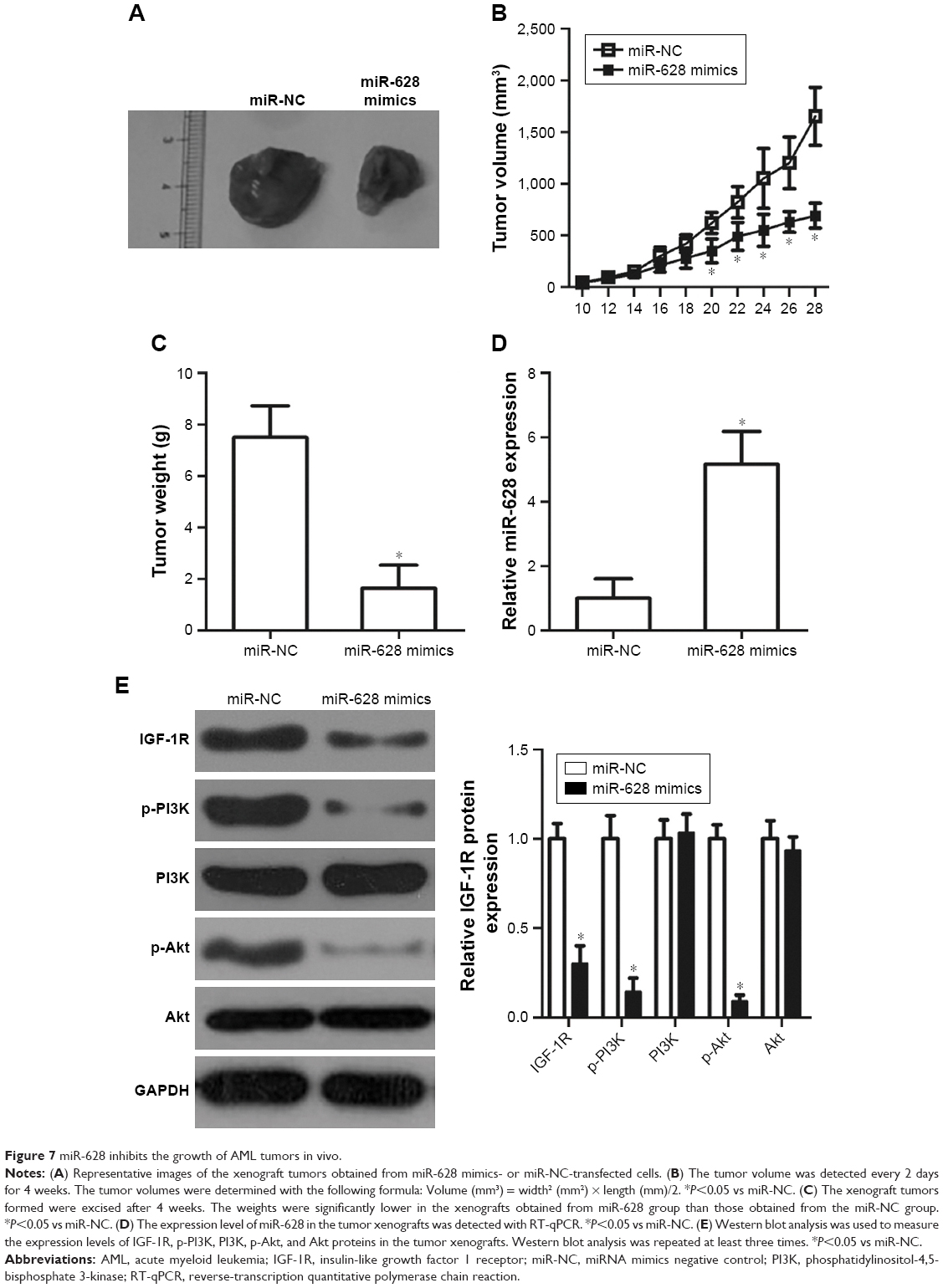 | Figure 7 miR-628 inhibits the growth of AML tumors in vivo. |
Discussion
A variety of miRNAs have been known to exhibit aberrant expression in AML.31–33 These dysregulated miRNAs perform crucial roles in tumorigenesis and tumor progression of AML and may regulate the major cancer-associated biological traits.34–36 Therefore, the comprehensive investigation of the regulatory mechanism underlying miRNA functions in AML occurrence and development is of significant importance for the development of therapeutic strategies to treat patients with AML. To the best of our knowledge, this is the first study to detect the expression level of miR-628 in AML. In addition, we explored the detailed roles and molecular mechanisms responsible for the action of miR-628 on AML progression.
In recent decades, the expression level of miR-628 was reported to be downregulated in colorectal cancer18 and pancreatic cancer.19 The expression of miR-628 was relatively low in bone metastatic breast cancer cells than in the primary breast cancer cells, suggestive of the close relationship between miR-628 expression and tumor metastasis.20 Functionally, miR-628 was identified as a tumor suppressor in breast cancer20 and lung cancer.21 In breast cancer, the restoration of miR-628 expression resulted in the inhibition of cell migration, invasion, and epithelial-to-mesenchymal transition in vitro.20 In lung cancer, resumption of miR-628 expression attenuated cell proliferation and promoted cell apoptosis in vitro.21 However, the expression pattern of miR-628 in AML remains unclear. Hence, RT-qPCR analysis was performed to determine the expression level of miR-628 in the bone marrow samples derived from the patients with AML as well as in a panel of AML cell lines. The data revealed that miR-628 expression was low in patients with AML and AML cell lines. These findings suggest that miR-628 may serve as an attractive biomarker for the diagnosis of patients with these human cancers.
Two genes, Son of Sevenless Homolog 120 and heat shock protein 90a,21 have been validated as direct targets of miR-628. Validation of the direct targets of miR-628 in AML is essential for the complete understanding of the molecular mechanisms underlying the functional roles of miR-628 in AML development and progression and may be useful for the identification of effective therapeutic target to treat patients with AML. We attempted to determine whether IGF-1R is a direct target of miR-628 in AML. Bioinformatic analysis predicted IGF-1R as a candidate target of miR-628. Furthermore, luciferase reporter assay revealed that miR-628 was able to recognize and directly target the 3′-UTR of IGF-1R. Moreover, IGF-1R expression was upregulated in patients with AML and inversely correlated with miR-628 expression. miR-628 upregulation decreased the expression of IGF-1R in AML cells at both mRNA and protein levels. Moreover, silencing of IGF-1R expression reproduced the miR-628-induced phenotype in AML cells. Besides, the restored IGF-1R expression abolished the tumor-suppressive roles of miR-628 in AML cells. Taken together, miR-628 may function as a tumor suppressor in AML by directly targeting IGF-1R.
IGF-1R, a transmembrane tyrosine kinase receptor of the insulin receptor family, has two extracellular α subunits and two transmembrane β subunits.37 IGF-1R was previously demonstrated to be upregulated in multiple human malignant tumors, such as colorectal cancer,38 gastric cancer,39 breast cancer,40 oral squamous cell carcinoma,41 and glioblastoma.42 IGF-1R activates multiple signaling pathways such as the PI3K/AKT pathway and is implicated in the carcinogenesis and progression of various human cancers.43–45 IGF-1R overexpression was reported in patients with AML as well as in AML cell lines.23 IGF-1R activation has been reported to promote the aggressive phenotypes of AML cells, including cell proliferation, survival, apoptosis, cycle, and chemotherapy resistance.23–26 Herein, we revealed that miR-628 targeted IGF-1R and restricted the malignant progression of AML cells in vitro and in vivo. These observations suggest that silencing of IGF-1R expression via restoration of miR-628 expression may be potentially effective for the treatment of patients with AML.
Conclusion
In conclusion, miR-628 expression was downregulated in patients with AML and in AML cell lines in this study. Ectopic miR-628 expression inhibited the growth of AML cells both in vitro and in vivo by directly targeting IGF-1R and deactivating PI3K/Akt pathway. The results of this study provide a theoretical basis for the role of miR-628/IGF-1R pathway in the management of patients with AML. However, this study included several limitations. First, we did not explore the association between miR-628 and prognosis of AML patients. We will collect the prognosis data and examine the association between miR-628 and prognosis of patients with AML. Second, the relationship between miR-628 expression and the subtype as well as mutations of AML patients was not examined. It is due to the small sample size, and we will collect more samples and resolve the limitation in the near future. Third, we did not perform loss-of-function assay to investigate the roles of miR-628 downregulation in AML progression. Fourth, the regulation effect of miR-628 in the PI3K/Akt signaling pathway in AML tissues was not clarified. The two limitations will also be resolved in our future investigations.
Disclosure
The authors report no conflicts of interest in this work.
References
Short NJ, Ravandi F. Acute myeloid leukemia: past, present, and prospects for the future. Clin Lymphoma Myeloma Leuk. 2016;16(Suppl):S25–S29. | ||
Medinger M, Lengerke C, Passweg J. Novel prognostic and therapeutic mutations in acute myeloid leukemia. Cancer Genomics Proteomics. 2016;13(5):317–329. | ||
Schlenk RF, Dohner K, Krauter J. German-Austrian acute myeloid Leukemia Study. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–1918. | ||
Woods BA, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Immunol Rev. 2015;263(1):22–35. | ||
Chiu CF, Weng JR, Jadhav A, Wu CY, Sargeant A, Bai LY. T315 decreases acute myeloid leukemia cell viability through a combination of apoptosis induction and autophagic cell death. Int J Mol Sci. 2016;17(8):1337. | ||
Hackl H, Astanina K, Wieser R. Molecular and genetic alterations associated with therapy resistance and relapse of acute myeloid leukemia. J Hematol Oncol. 2017;10(1):51. | ||
Yamauchi T. Treatment of acute myeloid leukemia under relapsed/refractory conditions or in older adults. Rinsho Ketsueki. 2016;57(10):1934–1943. | ||
Gabra MM, Salmena L. microRNAs and acute myeloid leukemia chemoresistance: a mechanistic overview. Front Oncol. 2017;7:255. | ||
Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15(8):509–524. | ||
Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16(7):421–433. | ||
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. | ||
Tutar L, Tutar E, Özgür A, Tutar Y. Therapeutic targeting of microRNAs in cancer: future perspectives. Drug Dev Res. 2015;76(7):382–388. | ||
di Leva G, Croce CM. miRNA profiling of cancer. Curr Opin Genet Dev. 2013;23(1):3–11. | ||
Torres S, Garcia-Palmero I, Bartolomé RA, et al. Combined miRNA profiling and proteomics demonstrates that different miRNAs target a common set of proteins to promote colorectal cancer metastasis. J Pathol. 2017;242(1):39–51. | ||
Huang Y, Zou Y, Lin L, Ma X, Chen H. Identification of serum miR-34a as a potential biomarker in acute myeloid leukemia. Cancer Biomark. 2018;22(4):799–805. | ||
Ding Q, Wang Q, Ren Y, Zhu HQ, Huang Z. MicroRNA-126 attenuates cell apoptosis by targeting TRAF7 in acute myeloid leukemia cells. Biochem Cell Biol. Epub 2018 Jan 25. | ||
Wang Z, Luo H, Fang Z, et al. MiR-204 acts as a potential therapeutic target in acute myeloid leukemia by increasing BIRC6-mediated apoptosis. BMB Rep. 2018;51(9):444–449. | ||
Hamfjord J, Stangeland AM, Hughes T, et al. Differential expression of miRNAs in colorectal cancer: comparison of paired tumor tissue and adjacent normal mucosa using high-throughput sequencing. PLoS One. 2012;7(4):e34150. | ||
Qadir MI, Faheem A. miRNA: a diagnostic and therapeutic tool for pancreatic cancer. Crit Rev Eukaryot Gene Expr. 2017;27(3):197–204. | ||
Lin C, Gao B, Yan X, et al. MicroRNA 628 suppresses migration and invasion of breast cancer stem cells through targeting Sos1. Onco Targets Ther. 2018;11:5419–5428. | ||
Pan J, Jiang F, Zhou J, Wu D, Sheng Z, Li M. Hsp90: a novel target gene of miRNA-628-3p in A549 cells. Biomed Res Int. 2018;2018:4149707. | ||
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. | ||
Zhang J, Huang FF, Wu DS, et al. Sumoylation of insulin-like growth factor 1 receptor, promotes proliferation in acute myeloid leukemia. Cancer Lett. 2015;357(1):297–306. | ||
He Y, Zhang J, Zheng J, et al. The insulin-like growth factor-1 receptor kinase inhibitor, NVP-ADW742, suppresses survival and resistance to chemotherapy in acute myeloid leukemia cells. Oncol Res. 2010;19(1):35–43. | ||
Chapuis N, Lacombe C, Tamburini J, Bouscary D, Mayeux P. Insulin receptor A and IGF-1R in AML – Letter. Cancer Res. 2010;70(17):7010. | ||
Wahner Hendrickson AE, Haluska P, Schneider PA, et al. Expression of insulin receptor isoform A and insulin-like growth factor-1 receptor in human acute myelogenous leukemia: effect of the dual-receptor inhibitor BMS-536924 in vitro. Cancer Res. 2009;69(19):7635–7643. | ||
Chapuis N, Tamburini J, Cornillet-Lefebvre P, et al. Autocrine IGF-1/IGF-1R signaling is responsible for constitutive PI3K/Akt activation in acute myeloid leukemia: therapeutic value of neutralizing anti-IGF-1R antibody. Haematologica. 2010;95(3):415–423. | ||
Xue M, Cao X, Zhong Y, et al. Insulin-like growth factor-1 receptor (IGF-1R) kinase inhibitors in cancer therapy: advances and perspectives. Curr Pharm Des. 2012;18(20):2901–2913. | ||
Wojtalla A, Salm F, Christiansen DG, et al. Novel agents targeting the IGF-1R/PI3K pathway impair cell proliferation and survival in subsets of medulloblastoma and neuroblastoma. PLoS One. 2012;7(10):e47109. | ||
Zhang Y, Moerkens M, Ramaiahgari S, et al. Elevated insulin-like growth factor 1 receptor signaling induces antiestrogen resistance through the MAPK/ERK and PI3K/Akt signaling routes. Breast Cancer Res. 2011;13(3):R52. | ||
Chang H, Gao Q, Ding W, Qing X. Identification of a robust subpathway-based signature for acute myeloid leukemia prognosis using an miRNA integrated strategy. PLoS One. 2018;13(3):e0194245. | ||
Ma QL, Wang JH, Yang M, Wang HP, Jin J. MiR-362-5p as a novel prognostic predictor of cytogenetically normal acute myeloid leukemia. J Transl Med. 2018;16(1):68. | ||
Krejcik Z, Belickova M, Hrustincova A, et al. MicroRNA profiles as predictive markers of response to azacitidine therapy in myelodysplastic syndromes and acute myeloid leukemia. Cancer Biomark. 2018;22(1):101–110. | ||
Bi L, Zhou B, Li H, et al. A novel miR-375-HOXB3-CDCA3/DNMT3B regulatory circuitry contributes to leukemogenesis in acute myeloid leukemia. BMC Cancer. 2018;18(1):182. | ||
Si X, Zhang X, Hao X, et al. Upregulation of miR-99a is associated with poor prognosis of acute myeloid leukemia and promotes myeloid leukemia cell expansion. Oncotarget. 2016;7(47):78095–78109. | ||
Wang X, Zuo D, Yuan Y, Yang X, Hong Z, Zhang R. MicroRNA-183 promotes cell proliferation via regulating programmed cell death 6 in pediatric acute myeloid leukemia. J Cancer Res Clin Oncol. 2017;143(1):169–180. | ||
Hu Q, Gong JP, Li J, et al. Down-regulation of miRNA-452 is associated with adriamycin-resistance in breast cancer cells. Asian Pac J Cancer Prev. 2014;15(13):5137–5142. | ||
Song Y, Zhao Y, Ding X, Wang X. microRNA-532 suppresses the PI3K/Akt signaling pathway to inhibit colorectal cancer progression by directly targeting IGF-1R. Am J Cancer Res. 2018;8(3):435–449. | ||
Ge J, Chen Z, Wu S, et al. Expression levels of insulin-like growth factor-1 and multidrug resistance-associated protein-1 indicate poor prognosis in patients with gastric cancer. Digestion. 2009;80(3):148–158. | ||
Ochnik AM, Baxter RC. Insulin-like growth factor receptor and sphingosine kinase are prognostic and therapeutic targets in breast cancer. BMC Cancer. 2017;17(1):820. | ||
Gao L, Wang X, Wang X, et al. IGF-1R, a target of let-7b, mediates crosstalk between IRS-2/Akt and MAPK pathways to promote proliferation of oral squamous cell carcinoma. Oncotarget. 2014;5(9):2562–2574. | ||
Maris C, D’Haene N, Trépant AL, et al. IGF-IR: a new prognostic biomarker for human glioblastoma. Br J Cancer. 2015;113(5):729–737. | ||
Singh P, Alex JM, Bast F. Insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-1R) signaling systems: novel treatment strategies for cancer. Med Oncol. 2014;31(1):805. | ||
Ma H, Zhang T, Shen H, Cao H, Du J. The adverse events profile of anti-IGF-1R monoclonal antibodies in cancer therapy. Br J Clin Pharmacol. 2014;77(6):917–928. | ||
Negi A, Ramarao P, Kumar R. Recent advancements in small molecule inhibitors of insulin-like growth factor-1 receptor (IGF-1R) tyrosine kinase as anticancer agents. Mini Rev Med Chem. 2013;13(5):653–681. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.