")
Back to Journals » Cancer Management and Research » Volume 11
microRNA-548b suppresses aggressive phenotypes of hepatocellular carcinoma by directly targeting high-mobility group box 1 mRNA
Authors Yun Z, Meng F, Jiang P, Yue M, Li S
Received 18 December 2018
Accepted for publication 7 April 2019
Published 25 June 2019 Volume 2019:11 Pages 5821—5834
DOI https://doi.org/10.2147/CMAR.S198615
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Harikrishna Nakshatri
This paper has been retracted.
Zhennan Yun, 1 Fanqi Meng, 1 Peiqiang Jiang, 2 Meng Yue, 1 Shiquan Li 1
1Department of Colorectal and Anal Surgery, The First Hospital of Jilin University, Changchun, Jilin 130021, People’s Republic of China; 2Department of Hepatobiliary and Pancreatic Surgery, The First Hospital of Jilin University, Changchun, Jilin 130021, People’s Republic of China
Background and purpose: An increasing number of studies have revealed that microRNAs (miRNAs) are the main drivers of hepatocarcinogenesis including progression to later stages of liver cancer. Recently, miR-548b was identified as a cancer-related miRNA in glioma and tongue squamous cell carcinoma. Nonetheless, the expression pattern and specific roles of miR-548b in hepatocellular carcinoma (HCC) have not yet been clarified.
Methods: Expression levels of miR-548b in HCC tissues and cell lines were measured by reverse-transcription quantitative PCR. In vitro and in vivo functional assays were performed to determine the effects of miR-548b on the malignant phenotypes of HCC cells. In addition, the molecular mechanisms by which miR-548b regulates the initiation and progression of HCC were investigated in detail.
Results: miR-548b expression was weak in HCC tissues and cell lines. The low miR-548b expression significantly correlated with tumor size, TNM stage, and venous infiltration of HCC. In addition, exogenous miR-548b expression suppressed HCC cell proliferation, colony formation, and metastasis and induced apoptosis in vitro. Silencing of miR-548b exerted an opposite effect on these characteristics of HCC cells. Furthermore, miR-548b overexpression hindered tumor growth in vivo. Mechanistic analysis identified high-mobility group box 1 (HMGB1) as a direct target gene of miR-548b in HCC cells. Moreover, an HMGB1 knockdown reproduced the effects of miR-548b upregulation on HCC cells. Recovered HMGB1 expression reversed the effects of miR-548b on HCC cells. Notably, miR-548b overexpression deactivated the PI3K–AKT pathway in HCC cells in vitro and in vivo.
Conclusion: Our findings provide the first evidence that miR-548b restrains HCC progression, at least partially, by downregulating HMGB1 and deactivating the PI3K–AKT pathway. Thus, miR-548b might be a novel target for the development of new therapies for HCC.
Keywords: microRNA-548b, hepatocellular carcinoma, PI3K–AKT pathway, high-mobility group box 1, HMGB1
Introduction
Hepatocellular carcinoma (HCC), the most prevalent primary liver cancer, is the fifth most common malignant tumor and the third main cause of cancer-related deaths worldwide.1 Currently, multiple therapeutic approaches, including surgical resection, percutaneous ablation, liver transplantation, radiotherapy, and chemotherapy, are available for the treatment of HCC.2 Owing to significant advances in the diagnosis and treatments, the clinical outcomes of patients with HCC have improved;3 unfortunately, HCC remains a deadly cancer, with a 5-year survival rate of less than 5%.4 Postoperative recurrence and high frequency of intrahepatic and extrahepatic metastases are the primary causes of the poor prognosis of patients with HCC.5 The pathogenesis of HCC is complicated, and various risk factors, such as infection with hepatitis virus B or C, exposure to aflatoxin B1, and genetic and epigenetic alterations, play crucial roles in HCC tumorigenesis;6,7 however, the detailed molecular mechanisms are still largely unclear; this situation is another important reason for the unsatisfactory therapeutic outcomes. Therefore, further exploration of the exact mechanisms is urgently necessary and will help to identify effective targets for antineoplastic therapy.
MicroRNAs (miRNAs) are a family of small endogenous noncoding RNA molecules of ~17–23 nucleotides.8 MiRNAs are widely expressed in animals, plants, and even viruses and perform crucial functions in almost all physiological and pathological processes.9 MiRNAs negatively regulate gene expression by binding to the 3′ untranslated region (UTR), thus triggering either messenger RNA (mRNA) degradation or translational suppression.10 Increasing numbers of reports have revealed that many miRNAs are aberrantly expressed in HCC and have indicated the involvement of miRNAs in hepatocarcinogenesis including progression to later stages of liver cancer.11–13 A growing body of experimental evidence suggests that miRNAs can exert oncogenic or tumor-suppressive actions in HCC and can regulate a wide range of biological phenomena, such as cell proliferation, apoptosis, cell cycle, metastasis, epithelial–mesenchymal transition, and chemoresistance.14–16 Thus, miRNAs may be promising targets for noninvasive treatment of HCC.
Recently, miR-548b was recognized as a cancer-related miRNA in glioma17 and tongue squamous cell carcinoma.18 Nevertheless, the expression pattern and specific involvement of miR-548b in HCC have not yet been clarified. In this study, we aimed to assess the miR-548b expression and to investigate its functions in HCC. In addition, the molecular mechanisms by which miR-548b regulate the progression of HCC were explored in detail.
Materials and methods
Clinical samples
HCC and normal paracarcinoma tissue samples were collected from 59 patients who were newly diagnosed as having HCC and were treated with surgical resection at The First Hospital of Jilin University between August 2015 and October 2017. None of these patients had received preoperative percutaneous ablation, radiotherapy, chemotherapy, or other anticancer therapy. Prior to experiments, all tissues were frozen in liquid nitrogen and stored at −80°C. The study protocol was approved by the Ethics Committee of The First Hospital of Jilin University and was in compliance with the guidelines of the Declaration of Helsinki. In addition, written informed consent was obtained from all the participants or their relatives.
Cell culture
Four HCC cell lines (Hep3B, Huh7, SK-HEP-1, and Bel-7402) and an immortalized normal human liver epithelial cell line (L02) were purchased from the Shanghai Cell Bank of the Chinese Academy of Sciences (Shanghai, China). All the cell lines were maintained at 37°C in a humidified atmosphere containing 5% of CO2 and were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% of the fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific, Inc.), 100 μg/mL streptomycin, and 100 U/mL penicillin (Sigma-Aldrich, Merck KGaA, Darmstadt, Germany).
A transfection assay
MiR-548b mimics, an miR-548b inhibitor, negative control mimics (NC mimics), and a negative control inhibitor (NC inhibitor) were purchased from Guangzhou RiboBio Co., Ltd. (Guangzhou, China), whereas synthetic small interfering RNA (siRNA) against HMGB1 expression (si-HMGB1) and negative control siRNA (si-NC) from Shanghai GenePharma Co., Ltd. (Shanghai, China). HMGB1 plasmid pCMV-HMGB1 and empty plasmid pCMV were synthesized by OriGene (Rockville, MD, USA). Cells were seeded in six-well plates at a density of 4 × 105 cells per well and grown to 70–80% confluence. A cell transfection assay was performed with Lipofectamine™ 2000 (Invitrogen, Thermo Fisher Scientific, Inc.). All the procedures of the transfection assay were based on the product specifications. After transfection for 6 hrs, the cells were washed with phosphate-buffered saline (PBS; Gibco, Thermo Fisher Scientific, Inc.) and cultured in fresh DMEM containing 10% of FBS.
RNA isolation and reverse-transcription quantitative PCR (RT-qPCR)
The miRcute Extraction and Separation of miRNAs Kit (DP501; Tiangen, Beijing, China) was used to extract miRNAs from cells and homogenized tissues. For evaluation of the miR-548b expression, cDNA synthesis was carried out with the miScript Reverse Transcription Kit (Qiagen GmbH, Hilden, Germany). After that, qPCR was performed on an Applied Biosystems 7500 Fast Real-Time PCR System (Thermo Fisher Scientific, Inc.) with the miScript SYBR Green PCR Kit (Qiagen GmbH). U6 small nuclear RNA served as an internal reference for the normalization of miR-548b expression.
To quantify HMGB1 mRNA expression, total RNA was isolated by means of the TRIzol reagent (Invitrogen, Thermo Fisher Scientific, Inc.). cDNA was synthesized from total RNA by reverse transcription using the PrimeScript™ RT reagent kit (TaKaRa, Dalian, China). Next, SYBR® Premix Ex Taq™ (TaKaRa) was employed to carry out qPCR. GAPDH mRNA served as the endogenous control for HMGB1 mRNA expression. Relative gene expression was calculated by the 2−∆∆Cq method.
A 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay
Transfected cells were seeded in 96-well plates at initial density 2× 103 cells/well and were cultured at 37°C and 5% CO2. After cultivation for 0, 1, 2, or 3 days, 20 µL of the MTT solution (5 mg/mL; Sigma-Aldrich, Merck KGaA) was added into each well and incubated with the cells for additional 4 h at 37°C and 5% CO2. Next, the culture medium was gently removed, and 200 µL of dimethyl sulfoxide (Beyotime Institute of Biotechnology, Inc., Shanghai, China) was added into each well to dissolve the formazan crystals. Finally, absorbance was read at 490 nm on a microplate reader (Bio-Rad, Hercules, CA, USA).
A clonogenic assay
This assay was conducted to determine the colony formation capacity of cells. Transfected cells were seeded in six-well plates at a density of 1,000 cells per well. The cells were allowed to grow at 37°C in a humidified atmosphere containing 5% of CO2 for 2 weeks. The culture medium was refreshed every 3 days. On day 15, the colonies were washed with PBS, fixed with 75% methanol, and stained with 0.1% crystal violet. The number of colonies formed was photographed under an inverted light microscope and then counted (Nikon, Tokyo).
Flow-cytometric analysis
The apoptosis rate was determined using an Annexin V-Fluorescein Isothiocyanate (FITC) Apoptosis Detection Kit (BioLegend, San Diego, CA, USA). Briefly, transfected cells were collected and washed with precooled PBS. The cells were then resuspended in 100 µL of 1× binding buffer, followed by staining with 5 µL of Annexin V-FITC and 5 µL of a propidium iodide solution. After 15 mins of incubation at room temperature in the dark, the percentage of apoptotic cells was determined on a flow cytometer (FACScan™, BD Biosciences, Franklin Lakes, NJ, USA).
Transwell migration and invasion assays
Transwell chambers (8 µm; Costar, Corning, NY, USA) were chosen to test the cell migration ability. A total of 200 µL of a cell suspension containing 5 × 104 cells was added into each upper chamber, while the lower chambers were filled with 500 µL of the culture medium containing 10% of FBS. After incubation for 24 hrs, nonmigratory cells were carefully scraped off, whereas the migratory cells were fixed with 75% methanol. Then, 0.05% crystal violet was used to stain the migratory cells. The experimental procedures of the invasion assay were similar to those of the migration assay, but the upper chambers were precoated with Matrigel (BD Biosciences). Images of the migratory and invading cells were captured by means of an inverted light microscope at ×200 magnification. Finally, the migratory and invasive abilities were assessed by counting the numbers of migratory and invading cells in five random visual fields per chamber.
A tumor xenograft model
Animal experimental protocols were approved by the Ethics Committee of The First Hospital of Jilin University and were performed in accordance with the Laboratory Animal Care and Use Guidelines of the National Institutes of Health. Briefly, Hep3B cells transfected with miR-548b mimics or NC mimics were inoculated into the flanks of nude mice (Shanghai Laboratory Animal Co. Ltd.; Shanghai, China) via subcutaneous injection. A total of eight nude mice were used and were subdivided into two groups. One group was injected with miR-548b mimic–transfected Hep3B cells, whereas the other group was treated with Hep3B cells transfected with NC mimics. Fifteen days after implantation, the tumor width and length were recorded every 3 days. Tumor volume was calculated via the following formula: Volume (mm3) =0.5× length (mm) × [width (mm)]2. All the mice were euthanized at the final time point. The xenograft was removed and weighed.
Hematoxylin-eosin (HE) staining
Tumor xenografts were fixed in 4% paraformaldehyde solution, dehydrated, and embedded in paraffin. Four-micrometer frozen sections were prepared and subjected to staining using hematoxylin and eosin kit (Beyotime Institute of Biotechnology, Inc.). The staining was analyzed and observed using a light microscope (magnification, × 400).
Bioinformatic analysis
Two miRNA target prediction software packages, namely, TargetScan (
A luciferase reporter assay
The fragments of the HMGB1 3′-UTR containing the wild-type or mutant miR-548b–binding site were amplified by Shanghai GenePharma Co., Ltd. The fragments were then cloned into the pmiR-RB-Report™ luciferase vector (Promega Corporation, Madison, WI, USA) to generate luciferase plasmids: pmiR-HMGB1-3ʹ-UTR wild-type and pmiR-HMGB1-3ʹ-UTR mut. Cells were seeded in 24-well plates and cotransfected with the wild-type or mutant luciferase plasmid and miR-548b mimics or the miR-548b inhibitor. The transfected cells were harvested 48 hrs after transfection, lysed, and assayed with a Dual-Luciferase reporter system (Promega Corporation, Madison, WI, USA). Renilla luciferase activity served for normalization.
Western blot analysis
Cells and tissues were lysed with radioimmunoprecipitation assay lysis buffer (Nanjing KeyGen Biotech Co., Ltd., Nanjing, China). The concentration of total protein was quantified using the Bradford protein assay kit (Bio-Rad). Equal amounts of protein were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis in a 10% gel and were transferred to polyvinylidene fluoride membranes (Beyotime Institute of Biotechnology, Inc.). Next, the membranes were blocked with 5% skimmed milk (dry basis) and incubated with primary antibodies (1:1,000 dilution) overnight at 4°C. The primary antibodies were as follows: a mouse monoclonal anti-human HMGB1 (cat. # ab77302; Abcam, Cambridge, UK), rabbit monoclonal anti-human phospho- (p-)PI3k (ab182651; Abcam), mouse monoclonal anti-human p85 PI3k (ab189403; phospho Y607; Abcam), mouse monoclonal anti-human p-Akt (sc-271964; Santa Cruz Biotechnology, Dallas, TX, USA), mouse monoclonal anti-human Akt (sc-56878; Santa Cruz Biotechnology), and mouse monoclonal anti-human GAPDH (sc-166574; Santa Cruz Biotechnology). After extensive washing with Tris-buffered saline containing 0.05% of Tween 20 (TBST), the membranes were incubated with a secondary antibody: a horseradish peroxidase-conjugated goat anti-mouse IgG antibody (ab97046) or a goat anti-rabbit IgG antibody (ab97051; Abcam) at a 1:5,000 dilution. Finally, the membranes were washed thrice with TBST, and the protein signals were detected with an enhanced chemiluminescence detection kit (Sigma-Aldrich, Merck KGaA).
Statistical analysis
All the results are expressed as the mean ± standard deviation and were analyzed in SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA). Differences between two groups were assessed by Student’s t test, whereas one-way analysis of variance (ANOVA) followed by the Student–Newman–Keuls post hoc test was conducted to evaluate differences among multiple groups. The correlation between the expression of miR-548b and the clinical features of HCC patients was studied by the χ2 test. The statistical significance of a positive correlation between miR-548b and HMGB1 mRNA was evaluated by Spearman’s correlation analysis. Data with P<0.05 were considered statistically significant.
Results
miR-548b expression is frequently low in HCC tissues and cell lines
The expression level of miR-548b was analyzed in 59 pairs of HCC and normal paracarcinoma tissue samples by RT-qPCR. Obviously, lower miR-548b expression in HCC tissue samples than in normal paracarcinoma tissues was observed (Figure 1A, P<0.05). In addition, we analyzed miR-548b expression in four HCC cell lines: Hep3B, Huh7, SK-HEP-1, and Bel-7402. An immortalized normal human liver epithelial cell line (L02) served as a control. The data from RT-qPCR showed that miR-548b expression was lower in HCC tissue samples than in L02 cells (Figure 1B, P<0.05). Thus, we concluded that miR-548b is frequently downregulated in HCC tumors and cell lines.
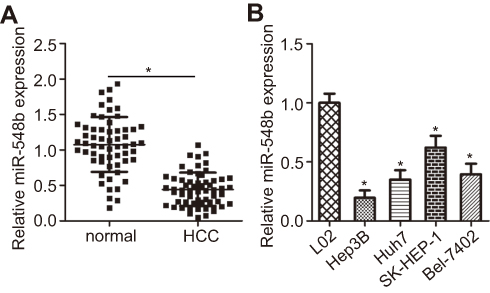 | Figure 1 miR-548b expression is low in hepatocellular carcinoma (HCC) tissue samples and cell lines. (A) Reverse-transcription-qPCR was carried out to measure miR-548b expression in 59 pairs of HCC and normal paracarcinoma tissue samples. *P<0.05 as compared with normal paracarcinoma tissues. (B) The expression levels of miR-548b in four HCC cell lines – Hep3B, Huh7, SK-HEP-1, and Bel-7402 – with an immortalized normal human liver epithelial cell line (L02) as a control. *P<0.05 as compared with L02 cells. |
Clinical significance of underexpressed miR-548b in patients with HCC
To clarify the clinical meaning of miR-548b underexpression in HCC, we distributed the 59 HCC patients into two subgroups – miR-548b low-expression group (<median, n=30) and miR-548b high-expression group (≥median, n=29) – based on the median value of the miR-548b expression in HCC tissue samples. The results of the χ2 test (Table 1) indicated that low miR-548b expression obviously correlated with tumor size (P=0.002), TNM stage (P=0.027), and venous infiltration of HCC (P=0.044). No significant association was observed between miR-548b expression and other clinical parameters, including patients’ age (P=0.5247), gender (P=0.514), α-fetoprotein (AFP) levels (P=0.520), or the tumor number (P=0.417). These data suggested that miR-548b underexpression may be related to HCC progression.
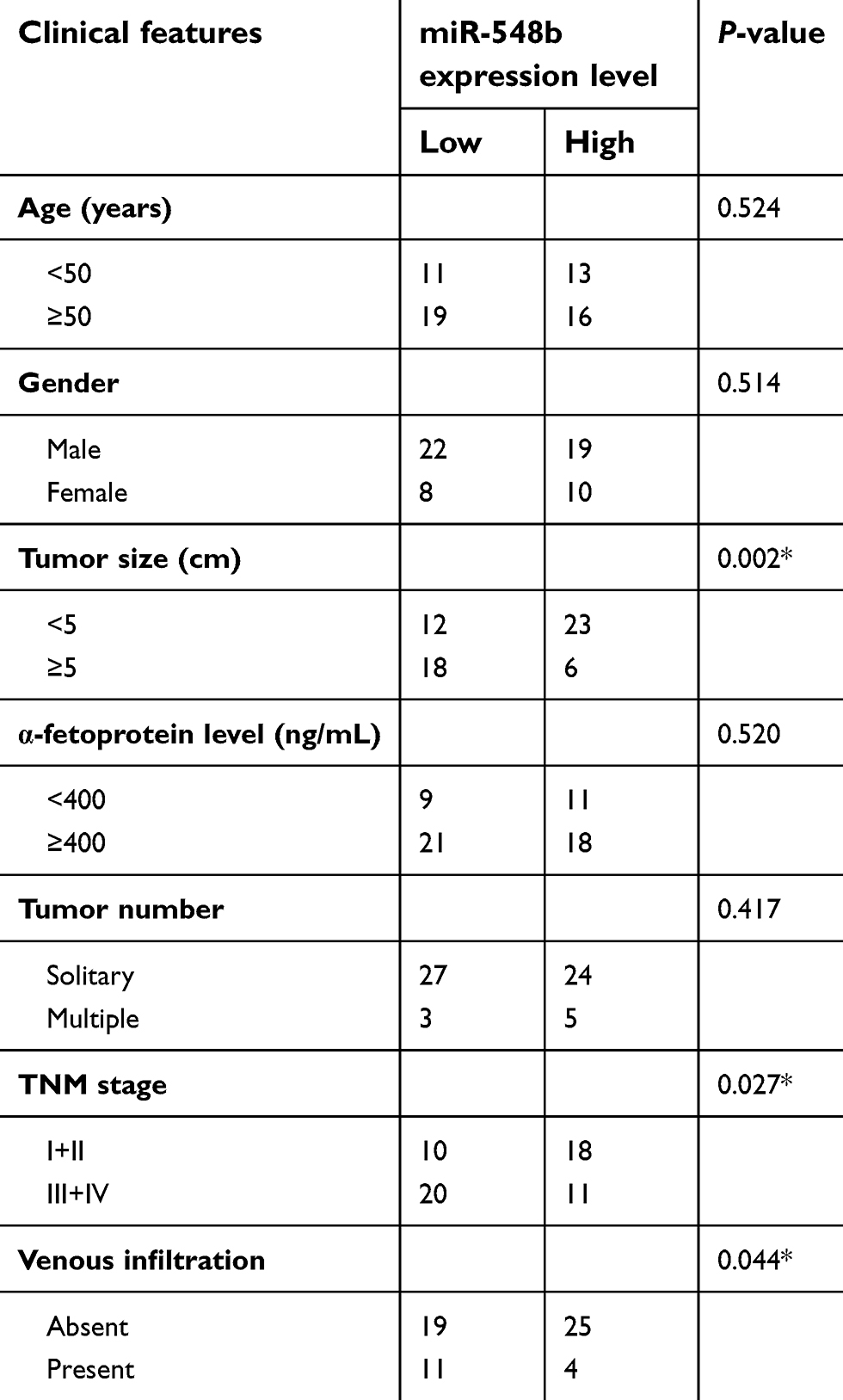 | Table 1 Clinical correlation of miR-548b expression in patients with hepatocellular carcinoma (HCC) |
miR-548b inhibits proliferation and colony formation and induces apoptosis of HCC cells in vitro
HCC cell line Hep3B, which had the lowest miR-548b expression, was transfected with miR-548b mimics, whereas the miR-548b inhibitor was transfected into SK-HEP-1 cells, which manifested the highest miR-548b expression among the four HCC cell lines. Transfection efficiency was confirmed by RT-qPCR (Figure 2A, P<0.05). MTT and clonogenic assays were performed to clearly determine the influence of miR-548b on HCC cell growth. The results revealed that artificial miR-548b upregulation decreased the proliferative and colony formation abilities of Hep3B cells; in contrast, the miR-548b knockdown enhanced the proliferative and colony formation abilities of SK-HEP-1 cells (Figure 2B and C, P<0.05). Given that miR-548b significantly affected HCC cell proliferation, we next examined the role of miR-548b in the regulation of apoptosis by flow-cytometric analysis. The data showed that ectopic miR-548b expression induced apoptosis of Hep3B cells, whereas the knockdown of miR-548b had the opposite effect on SK-HEP-1 cells (Figure 2D, P<0.05). These results indicate that miR-548b exerts a tumor-suppressive action in HCC cells in vitro.
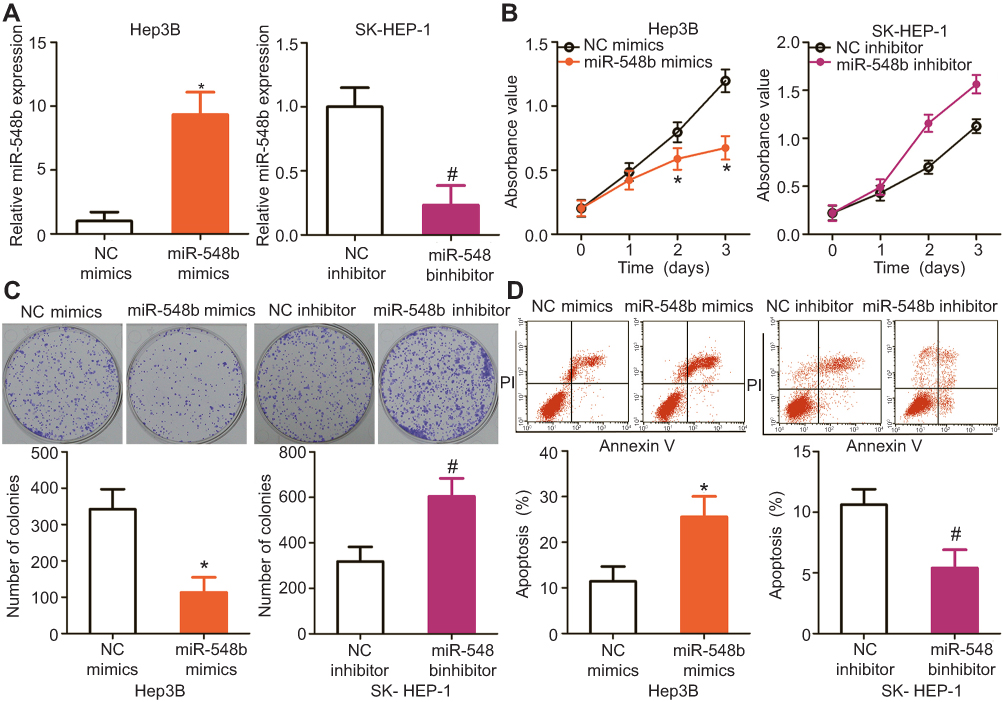 | Figure 2 miR-548b inhibits the proliferation, colony formation, and apoptosis of hepatocellular carcinoma (HCC) cells. (A) The expression levels of miR-548b in miR-548b mimic–transfected Hep3B cells and miR-548b inhibitor–transfected SK-HEP-1 cells were determined by reverse-transcription quantitative PCR. The negative control (NC) mimics and NC inhibitor served as controls for miR-548b mimics and the miR-548b inhibitor, respectively. *P<0.05 as compared with NC mimics. #P<0.05 as compared with the NC inhibitor. (B and C) MTT and clonogenic assays were performed to determine the effects of miR-548b on the proliferative and colony formation abilities of Hep3B and SK-HEP-1 cells. *P<0.05 as compared with NC mimics. #P<0.05 as compared with the NC inhibitor. (D) The apoptotic rates of Hep3B and SK-HEP-1 cells after transfection with miR-548b mimics or the miR-548b inhibitor were assessed by flow cytometry. *P<0.05 as compared with NC mimics. #P<0.05 as compared with the NC inhibitor. |
miR-548b plays an inhibitory part in the metastasis of HCC cells
Transwell migration and invasion assays were carried out to test whether miR-548b restrains or promotes migration and invasiveness of HCC cells. The results indicated that restoration of miR-548b expression in Hep3B cells remarkably reduced their migratory and invasive abilities, whereas artificial downregulation of miR-548b in SK-HEP-1 cells significantly increased their migration and invasion (Figure 3A and B, P<0.05). Thus, we preliminarily concluded that miR-548b might be a key suppressor of HCC metastasis.
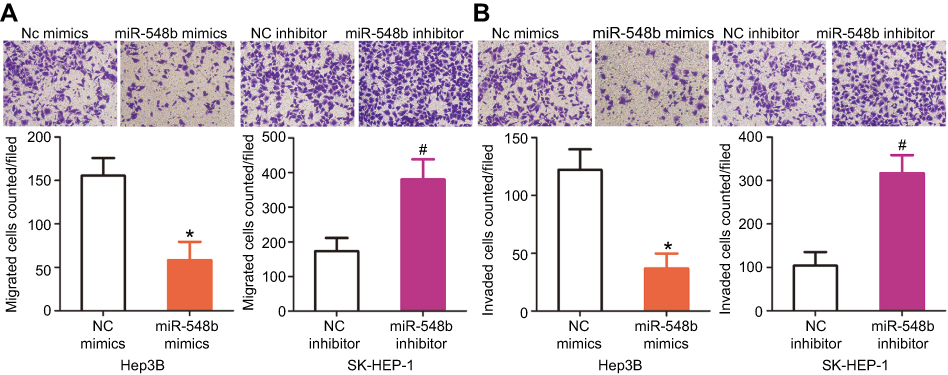 | Figure 3 MiR-548b inhibits the migration and invasiveness of hepatocellular carcinoma (HCC) cells. (A and B) Hep3B cells were transected with miR-548b mimics or negative control (NC) mimics, whereas the miR-548b inhibitor or NC inhibitor was introduced into SK-HEP-1 cells. After cultivation, Transwell migration and invasion assays were carried out to evaluate cellular migration and invasion. *P<0.05 as compared with NC mimics. #P<0.05 as compared with the NC inhibitor. |
HMGB1 mRNA is a target of miR-548b in HCC cells
To illustrate the potential molecular mechanisms by which miR-548b exerts its impact on the growth and metastasis of HCC cells, bioinformatic analysis was performed to predict a potential target of miR-548b. The 3′-UTR of HMGB1 mRNA contains a complementary site for miR-548b (Figure 4A), and this gene was chosen for validation because it has been implicated in hepatocarcinogenesis and cancer progression.19–31 To test whether miR-548b can directly target the 3′-UTR of HMGB1 mRNA, wild-type and mutant luciferase plasmids were chemically synthesized and cotransfected with miR-548b mimics or the miR-548b inhibitor into Hep3B and SK-HEP-1 cells. MiR-548b overexpression in Hep3B cells decreased, whereas the miR-548b knockdown in SK-HEP-1 cells increased the luciferase activity of the plasmid harboring a wild-type miR-548b–binding site (P<0.05). In contrast, alteration of miR-548b expression in Hep3B and SK-HEP-1 cells had no influence on the luciferase activity of the plasmid carrying the mutant binding site (Figure 4B).
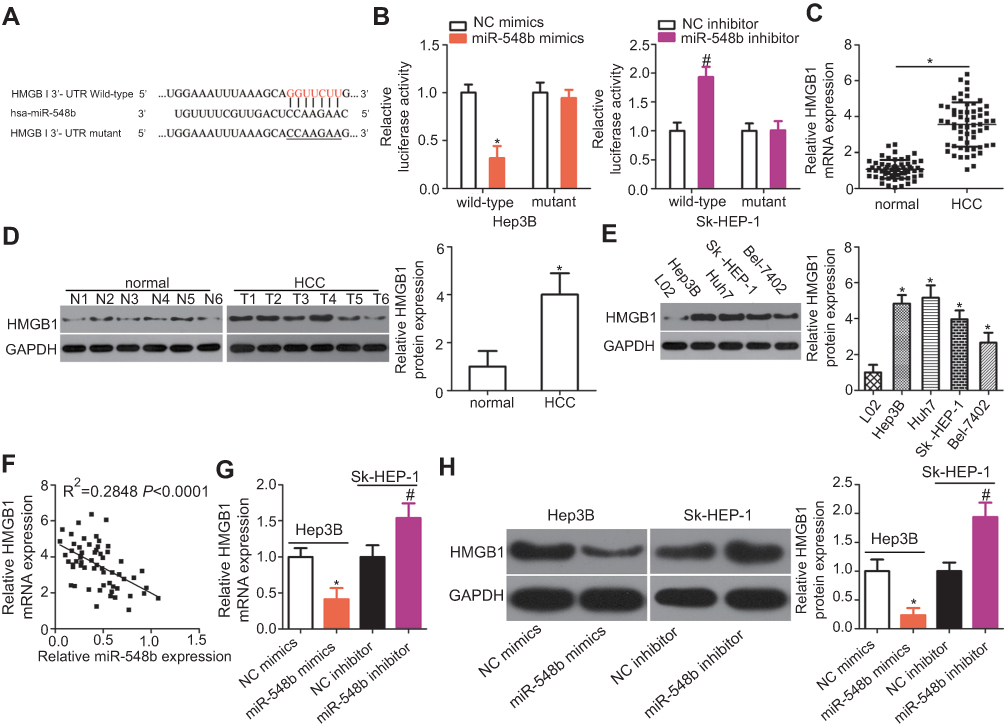 | Figure 4 HMGB1 mRNA is a direct target of miR-548b in hepatocellular carcinoma (HCC) cells. (A) The putative binding site of miR-548b in the 3′-UTR of HMGB1 mRNA. The mutant site is also presented. (B) Forced miR-548b expression reduced the luciferase activity yielded by the plasmid carrying the wild-type 3′-UTR in Hep3B cells. Downregulation of miR-548b in SK-HEP-1 cells increased the luciferase activity generated by the plasmid harboring the wild-type binding site. *P<0.05 as compared with NC mimics. #P<0.05 as compared with the negative control (NC) inhibitor. (C) The expression of HMGB1 mRNA was evaluated in 59 pairs of HCC and normal paracarcinoma tissue samples by reverse-transcription quantitative (RT-q) PCR. *P<0.05 as compared with normal paracarcinoma tissue samples. (D) Western blot analysis was conducted to measure HMGB1 protein expression in several pairs of HCC and normal paracarcinoma tissue samples. *P<0.05 as compared with normal paracarcinoma tissues. (E) Western blotting was applied to determine the HMGB1 protein expression in four HCC cell lines – Hep3B, Huh7, SK-HEP-1, and Bel-7402 – with an immortalized normal human liver epithelial cell line (L02) as a control. *P<0.05 as compared with L02 cells. (F) A negative correlation between HMGB1 mRNA and miR-548b levels among HCC tissue samples was confirmed by Spearman’s correlation analysis. R2=0.2848, P<0.001. (G) HMGB1 mRNA was quantified by RT-qPCR in Hep3B and SK-HEP-1 cells after transfection with miR-548b mimics or the miR-548b inhibitor, respectively. *P<0.05 as compared with NC mimics. #P<0.05 as compared with the NC inhibitor. (H) The protein expression of HMGB1 in Hep3B cells was obviously decreased by transfection with miR-548b mimics, whereas the miR-548b inhibitor significantly increased HMGB1 protein expression in SK-HEP-1 cells. *P<0.05 as compared with NC mimics. #P<0.05 as compared with the NC inhibitor. |
The expression levels of HMGB1 mRNA and protein in 59 pairs of HCC and normal paracarcinoma tissue samples were determined via RT-qPCR and Western blotting. The findings revealed that HMGB1 was highly expressed in HCC tissue samples at both mRNA (Figure 4C, P<0.05) and protein levels (Figure 4D, P<0.05) as compared with normal paracarcinoma tissue samples. In addition, the expression level of HMGB1 protein was increased in all the four tested HCC cell lines than that in L02 (Figure 4E, P<0.05). Furthermore, an inverse correlation between the expression levels of miR-548b and HMGB1 was identified among HCC tissue samples by Spearman’s correlation analysis (Figure 4F; R2=0.2848, P<0.0001). Moreover, HMGB1 mRNA and protein expression in Hep3B cells were decreased by transfection with miR-548b mimics, whereas transfection with the miR-548b inhibitor raised HMGB1 mRNA and protein levels in SK-HEP-1 cells (Figure 4G and H, P<0.05). Consequently, HMGB1 mRNA was demonstrated to be a target of miR-548b in HCC cells.
The HMGB1 knockdown and miR-548b upregulation exert similar effects in HCC cells
To elucidate the exact roles of HMGB1 in HCC tumorigenesis, an siRNA against HMGB1 (si-HMGB1) was utilized to knock down endogenous HMGB1 expression in Hep3B and SK-HEP-1 cells. Western blotting confirmed that HMGB1 expression was efficiently silenced in Hep3B and SK-HEP-1 cells that were transfected with si-HMGB1 (Figure 5A, P<0.05). MTT and clonogenic assays were conducted to assess cell growth after the HMGB1 knockdown, and the results indicated that Hep3B and SK-HEP-1 cell proliferation (Figure 5B, P<0.05) and colony formation (Figure 5C, P<0.05) were notably suppressed in the si-HMGB1 group. Additionally, the percentage of apoptotic cells was obviously higher among the si-HMGB1–transfected Hep3B and SK-HEP-1 cells than among cells transfected with si-NC (Figure 5D, P<0.05). Furthermore, the HMGB1 knockdown had an inhibitory influence on the metastasis (Figure 5E and F, P<0.05) of Hep3B and SK-HEP-1 cells. These findings implied that HMGB1 silencing can mimic the tumor-suppressive activity of miR-548b overexpression in HCC cells, further supporting the idea that HMGB1 mRNA is a direct target of miR-548b.
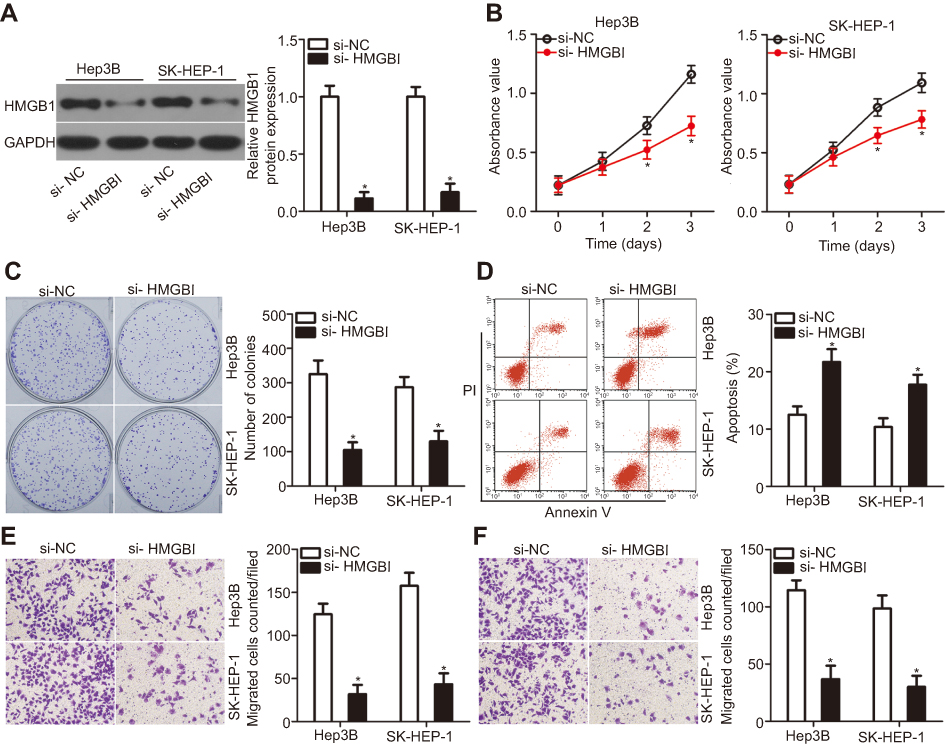 | Figure 5 The action of HMGB1 silencing is similar to that of miR-548b overexpression in hepatocellular carcinoma (HCC) cells. si-HMGB1 or si-negative control (NC) was transfected into Hep3B and SK-HEP-1 cells. The transfected cells were harvested after incubation and subjected to the following assays: (A) The protein amount of HMGB1 was quantified via Western blot analysis. *P<0.05 as compared with si-NC. (B and C) The proliferation and colony formation abilities were determined in MTT and clonogenic assays, respectively. *P<0.05 as compared with si-NC. (D) Flow-cytometric analysis of the percentage of apoptotic cells. *P<0.05 as compared with si-NC. (E and F) Transwell migration and invasion assays of HMGB1 knockdown Hep3B and SK-HEP-1 cells. *P<0.05 as compared with si-NC. |
Recovered HMGB1 expression reverses the effects of miR-548b in HCC cells
Rescue experiments were conducted to confirm that HMGB1 is involved in the anticancer activity of miR-548b in HCC cells. MiR-548b–overexpressing Hep3B cells were transfected with HMGB1 plasmid pCMV-HMGB1, whereas si-HMGB1 was transfected into SK-HEP-1 cells after the miR-548b knockdown. Seventy-two hours after transfection, miR-548b mimic–mediated HMGB1 downregulation in Hep3B cells was reversed after cotransfection with pCMV-HMGB1. Besides, miR-548b inhibitor-mediated upregulation of the HMGB1 protein in SK-HEP-1 cells was reversed by si-HMGB1 (Figure 6A, P<0.05). Functional assays revealed that HMGB1 neutralized the influence of miR-548b on the proliferation (Figure 6B, P<0.05), colony formation (Figure 6C, P<0.05), apoptosis (Figure 6D, P<0.05), migration (Figure 6E, P<0.05), and invasiveness (Figure 6F, P<0.05) of Hep3B and SK-HEP-1 cells. Taken together, these results suggested that the tumor-suppressive actions of miR-548b in HCC cells are partly mediated by HMGB1 downregulation.
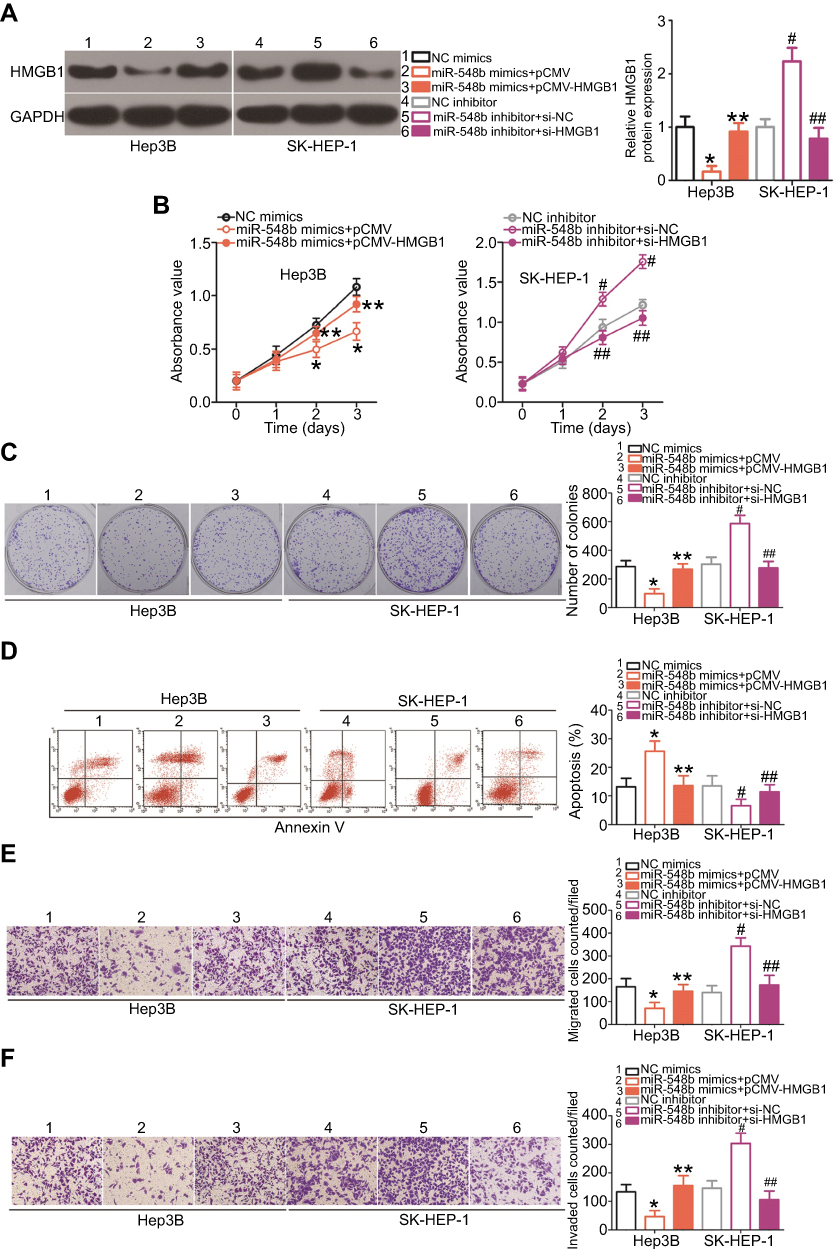 | Figure 6 A series of rescue experiments were conducted to confirm that HMGB1 downregulation mediates the activities of miR-548b in hepatocellular carcinoma (HCC) cells. Hep3B cells were cotransfected with miR-548b mimics and pCMV-HMGB1 or pCMV, whereas the miR-548b inhibitor was cotransfected with si-HMGB1 or si-negative control (NC) into SK-HEP-1 cells. (A) The protein amount of HMGB1 was determined by Western blotting with GAPDH as an internal reference. *P<0.05 as compared with NC mimics. **P<0.05 as compared with miR-548b mimics+pCMV. #P<0.05 relative to the NC inhibitor. ##P<0.05 as compared with miR-548b inhibitor+si-NC. (B–F) The proliferation, colony formation, apoptosis, migration, and invasion abilities of the aforementioned cells were assessed by the MTT assay, clonogenic assay, flow cytometry, and Transwell migration and invasion assays, respectively. *P<0.05 as compared with NC mimics. **P<0.05 as compared with miR-548b mimics+pCMV. #P<0.05 in comparison with the NC inhibitor. #P<0.05 as compared with miR-548b inhibitor+si-NC. |
miR-548b suppresses the PI3K–AKT pathway in HCC cells
HMGB1 has been reported to participate in the PI3K–AKT signaling pathway and thereby regulate carcinogenesis including cancer progression.32,33 Hence, we next determined whether miR-548b regulates the PI3K–AKT pathway in HCC cells through HMGB1. To test our hypothesis, we assayed the effects of miR-548b upregulation or knockdown on the cellular expression levels of PI3K–AKT pathway-related proteins. Firstly, we evaluated the transfection efficiency of pCMV-HMGB1 in Hep3B cells. As shown in Figure 7A, transfection of pCMV-HMGB1 notably increased the expression level of HMGB1 protein in Hep3B cells (P<0.05). The transfection efficiency of si-HMGB1 in SK-HEP-1 cells was already revealed in Figure 5A. Western blotting indicated that miR-548b overexpression decreased the protein levels of p-PI3K and p-AKT in Hep3B cells, whereas miR-548b downregulation increased the p-PI3K and p-AKT protein levels in SK-HEP-1 cells. Notably, normalization of HMGB1 expression reversed the changes in the p-PI3K and p-AKT amounts in Hep3B and SK-HEP-1 cells caused by the miR-548b upregulation or knockdown (Figure 7B). These data suggested that miR-548b inhibited the activation of the PI3K–AKT pathway in HCC cells.
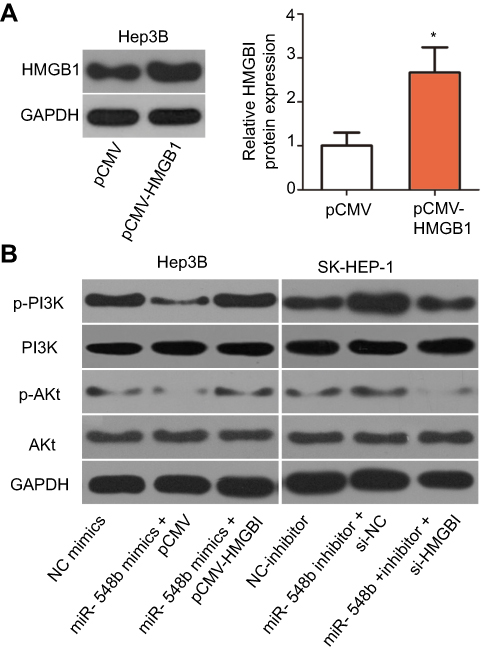 | Figure 7 miR-548b suppresses the PI3K–AKT signaling cascade in hepatocellular carcinoma (HCC) cells by downregulating HMGB1. Hep3B cells overexpressing miR-548b were next transfected with pCMV-HMGB1 or pCMV. MiR-548b inhibitor–transfected SK-HEP-1 cells were treated with si-HMGB1 or si-negative control (NC). (A) Hep3B cells were transfected with pCMV-HMGB1 or pCMV. Western blot analysis was used to determine the HMGB1 protein expression. *P<0.05 as compared with pCMV. (B) Seventy-two hours after transfection, Western blotting was carried out to measure p-PI3K, PI3K, p-AKT, and AKT protein levels. GAPDH served as a loading control. |
miR-548b inhibits tumor growth of HCC cells in vivo
Finally, a tumor xenograft model was set up to examine the impact of miR-548b on tumor growth in vivo. Hep3B cells transfected with miR-548b mimics or NC mimics were injected into nude mice. The tumor xenografts derived from miR-548b mimic transfectants obviously had smaller volume (Figure 8A and B, P<0.05) and lesser weight (Figure 8C, P<0.05) than did the tumor xenografts derived from the NC mimic transfectants. In addition, we measured miR-548b expression in the tumor xenografts by RT-qPCR. The tumor xenografts derived from miR-548b mimic–transfected Hep3B cells expressed more miR-548b relative to the cells transfected with the NC inhibitor (Figure 8D, P<0.05). After that, HE staining was performed to reveal the tumor xenografts were real tumor tissues (Figure 8E). Western blot analysis was used to evaluate the protein expression in tumor xenografts. The protein amounts of HMGB1, p-PI3K, and p-AKT turned out to be obviously lower in the miR-548b mimics group (Figure 8F). These results meant that miR-548b overexpression inhibits HCC tumor growth in vivo by repressing HMGB1 expression and PI3K–AKT signaling.
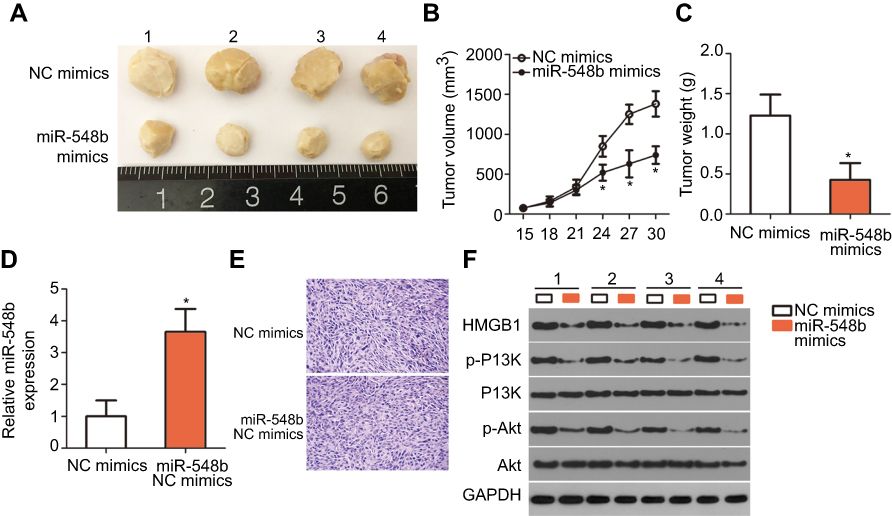 | Figure 8 miR-548b overexpression suppresses tumor growth of hepatocellular carcinoma (HCC) cells in vivo. (A) Photographs of tumor xenografts derived from Hep3B cells transfected with miR-548b mimics or negative control (NC) mimics. (B) The tumor volume in the miR-548b overexpression group was notably smaller than that in the NC mimics group. *P<0.05 as compared with NC mimics. (C) The weight of tumor xenografts in groups “miR-548b mimics” and “NC mimics” was quantitated. *P<0.05 in comparison with NC mimics. (D) Reverse-transcription quantitative PCR analysis was performed to determine miR-548b expression in the tumor xenografts derived from Hep3B cells transfected with miR-548b mimics or NC mimics. *P<0.05 relative to NC mimics. (E) H&E staining was utilized to determine whether the tumor xenografts were real tumor tissues. (F) Western blotting was carried out to measure the HMGB1, p-PI3K, PI3K, p-AKT, and AKT amounts in the tumors excised from the mice. |
Discussion
Dysregulation of miRNAs in HCC is reported frequently.34–36 An increasing number of studies have proven that miRNAs are the main drivers of HCC tumorigenesis because they function as oncogenes or tumor suppressor genes.37–39 Moreover, miRNA-based targeted therapies have been tested in several preclinical models, including HCC models.40 Therefore, identification of oncogenic or tumor-suppressive miRNAs and detailed elucidation of their roles in HCC pathogenesis may help identify effective biomarkers for HCC diagnosis and prognosis as well as therapeutic targets in this cancer. MiR-548b is underexpressed in glioma17 and highly expressed in tongue squamous cell carcinoma.18 Nonetheless, the expression status of miR-548b in HCC has been unclear. Hence, we measured the expression of miR-548b in HCC and found that miR-548b is downregulated in both HCC tissues and cell lines. Low miR-548b expression significantly correlated with tumor size, TNM stage, and venous infiltration of HCC. These findings suggest that miR-548b may serve as a biomarker for the diagnosis of HCC.
MiR-548b has been identified as a tumor suppressor in glioma. Ectopic miR-548b expression inhibits the proliferation and metastasis of glioma cells in vitro and decreases tumor growth in vivo.17 Mechanistic studies have revealed that metastasis tumor-associated protein 2 (MTA2) mRNA is a direct target of miR-548b in glioma cells, and MTA2 silencing is essential for the tumor-suppressive properties of miR-548b toward glioma.17 On the other hand, the participation of miR-548b in HCC has rarely been reported. Herein, miR-548b upregulation dramatically decreased cell proliferation, colony formation, migration, and invasiveness of HCC cells and induced their apoptosis, whereas an miR-548b knockdown exerted the opposite effects. Additionally, overexpression of miR-548b reduced HCC tumor growth in vivo. These observations suggest that miR-548b may be a therapeutic target in HCC.
In subsequent mechanistic experiments, bioinformatic analysis was first carried out to predict potential targets of miR-548b. The 3′-UTR of HMGB1 mRNA turned out to contain a highly conserved binding site for miR-548b. After that, a luciferase activity assay was performed to validate the targeting of miR-548b to the 3′-UTR of HMGB1 mRNA. Besides, HMGB1 expression was found to be high in HCC tissues and inversely correlated with miR-548b expression. By RT-qPCR and Western blotting, we detected a reduction in HMGB1 mRNA and protein levels in HCC cells after miR-548b overexpression, whereas the miR-548b knockdown had the opposite effect. Furthermore, silencing of HMGB1 reproduced the effects of miR-548b overexpression in HCC cells. Subsequent rescue experiments validated the notion that the expression changes of HMGB1 induced by miR-548b mediate the effects of miR-548b in HCC cells. Notably, miR-548b overexpression deactivated, whereas the miR-548b knockdown activated the PI3K–AKT pathway in HCC cells. Thus, to our knowledge, this study is the first to show that the miR-548b–HMGB1–PI3K–AKT pathway is implicated in the pathogenesis of HCC.
HMGB1 is a highly conserved DNA-binding protein that can relocate from the cytoplasm to nucleus and interact with transcription factors, nucleosomes, and histones.41 It is encoded in human chromosomal region 13q12 and is overexpressed in a variety of human cancers, including bladder cancer,42 breast cancer,43 gastric cancer,44 and esophageal squamous cell carcinoma.45 Furthermore, HMGB1 is upregulated in HCC, and its expression is associated with AFP expression, tumor size, vascular invasion, capsule invasion, and TNM stage.19–21 HCC patients with high HMGB1 levels in the tumor have shorter overall survival than do patients with low HMGB1 amounts.19 Multivariate analysis has identified high HMGB1 expression as an independent prognostic factor for the overall survival and disease-free survival of patients with HCC.19,20 Multiple pieces of evidence support the involvement of HMGB1 as an oncogene in HCC. HMGB1 performs crucial functions in the formation and progression of HCC by regulating a number of malignant properties, including rapid cell proliferation, immortality, suppressed apoptosis, an out-of-control cell cycle, angiogenesis, metastasis, epithelial–mesenchymal transition, and chemoresistance.22–31 In our current study, it was revealed that miR-548b directly targets HMGB1 mRNA and inhibits its protein expression, thereby inhibiting the aggressive characteristics of HCC cells. Our results, together with previous findings, suggest that silencing of HMGB1 via miR-548b restoration might be a promising therapeutic strategy against HCC.
Limitations
This study has several limitations. First, we did not examine the correlation between miR-548b and overall survival among patients with HCC. In addition, the effect of miR-548b inhibition on tumor growth in vivo and in vivo was not tested in rescue experiments. Furthermore, tumor growth (ie, Ki67 or PCNA staining) and cell death (ie, annexin V staining) were not investigated in the grafted tumor tissues in mice. We aim to fill these gaps in our further research.
Conclusions
We demonstrated that miR-548b acts as a tumor-suppressive miRNA in HCC by inhibiting cell proliferation, colony formation, migration, and invasion in vitro, by promoting apoptosis in vitro, and by slowing in vivo tumor growth. Mechanistic assays confirmed HMGB1 mRNA as a direct target of miR-548b in HCC cells. Of note, miR-548b deactivated the PI3K–AKT signaling cascade in HCC both in vitro and in vivo. Our identification of the miR-548b–HMGB1 axis can help researchers gain novel insights into the mechanisms underlying HCC initiation and progression and can facilitate the development of novel therapies for patients with this deadly disease.
Disclosure
The authors declare that they have no competing interests.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
2. Di Maio M, De Maio E, Perrone F, Pignata S, Daniele B. Hepatocellular carcinoma: systemic treatments. J Clin Gastroenterol. 2002;35(5 Suppl 2):S109–S114.
3. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–2576. doi:10.1053/j.gastro.2007.04.061
4. Hao K, Luk JM, Lee NP, et al. Predicting prognosis in hepatocellular carcinoma after curative surgery with common clinicopathologic parameters. BMC Cancer. 2009;9:389. doi:10.1186/1471-2407-9-389
5. Galun D, Basaric D, Zuvela M, et al. Hepatocellular carcinoma: from clinical practice to evidence-based treatment protocols. World J Hepatol. 2015;7(20):2274–2291. doi:10.4254/wjh.v7.i20.2274
6. Nishida N, Goel A. Genetic and epigenetic signatures in human hepatocellular carcinoma: a systematic review. Curr Genomics. 2011;12(2):130–137. doi:10.2174/138920211795564359
7. Zhang YJ. Interactions of chemical carcinogens and genetic variation in hepatocellular carcinoma. World J Hepatol. 2010;2(3):94–102. doi:10.4254/wjh.v2.i3.94
8. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297.
9. Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–355. doi:10.1038/nature02871
10. Engels BM, Hutvagner G. Principles and effects of microRNA-mediated post-transcriptional gene regulation. Oncogene. 2006;25(46):6163–6169. doi:10.1038/sj.onc.1209909
11. Vasuri F, Visani M, Acquaviva G, et al. Role of microRNAs in the main molecular pathways of hepatocellular carcinoma. World J Gastroenterol. 2018;24(25):2647–2660. doi:10.3748/wjg.v24.i25.2647
12. Xu J, Li J, Zheng TH, Bai L, Liu ZJ. MicroRNAs in the occurrence and development of primary hepatocellular carcinoma. Adv Clin Exp Med. 2016;25(5):971–975. doi:10.17219/acem/36460
13. Shen S, Lin Y, Yuan X, et al. Biomarker MicroRNAs for diagnosis, prognosis and treatment of hepatocellular carcinoma: a functional survey and comparison. Sci Rep. 2016;6:38311. doi:10.1038/srep38311
14. Xiao S, Yang M, Yang H, Chang R, Fang F, Yang L. miR-330-5p targets SPRY2 to promote hepatocellular carcinoma progression via MAPK/ERK signaling. Oncogenesis. 2018;7(11):90. doi:10.1038/s41389-018-0097-8
15. Li S, Li T, Li X, et al. MicroRNA-32 regulates development and progression of hepatocellular carcinoma by targeting ADAMTS9 and affects its prognosis. Med Sci Monit Basic Res. 2018;24:177–187. doi:10.12659/MSMBR.910522
16. Wang L, Lu J, Zhang H, Lyu X, Sun Z. MicroRNA8765p inhibits the progression of glioblastoma multiforme by directly targeting Forkhead box M1. Oncol Rep. 2019;41(1):702–710. doi:10.3892/or.2018.6804
17. Pan Y, Liang W, Zhao X, Liu L, Qing Y, Li Y. miR-548b inhibits the proliferation and invasion of malignant gliomas by targeting metastasis tumor-associated protein-2. Neuroreport. 2016;27(17):1266–1273. doi:10.1097/WNR.0000000000000690
18. Berania I, Cardin GB, Clement I, et al. Four PTEN-targeting co-expressed miRNAs and ACTN4- targeting miR-548b are independent prognostic biomarkers in human squamous cell carcinoma of the oral tongue. Int J Cancer. 2017;141(11):2318–2328. doi:10.1002/ijc.30915
19. Liu F, Zhang Y, Peng Z, Gao H, Xu L, Chen M. High expression of high mobility group box 1 (hmgb1) predicts poor prognosis for hepatocellular carcinoma after curative hepatectomy. J Transl Med. 2012;10:135. doi:10.1186/1479-5876-10-233
20. Xiao J, Ding Y, Huang J, et al. The association of HMGB1 gene with the prognosis of HCC. PLoS One. 2014;9(2):e89097. doi:10.1371/journal.pone.0089097
21. Jiang W, Wang Z, Li X, Fan X, Duan Y. High-mobility group box 1 is associated with clinicopathologic features in patients with hepatocellular carcinoma. Pathol Oncol Res. 2012;18(2):293–298. doi:10.1007/s12253-011-9442-3
22. Jiang W, Wang Z, Li X, et al. Reduced high-mobility group box 1 expression induced by RNA interference inhibits the bioactivity of hepatocellular carcinoma cell line HCCLM3. Dig Dis Sci. 2012;57(1):92–98. doi:10.1007/s10620-011-1944-z
23. Yan W, Chang Y, Liang X, et al. High-mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology. 2012;55(6):1863–1875. doi:10.1002/hep.25572
24. Chen RC, Yi PP, Zhou RR, et al. The role of HMGB1-RAGE axis in migration and invasion of hepatocellular carcinoma cell lines. Mol Cell Biochem. 2014;390(1–2):271–280. doi:10.1007/s11010-014-1978-6
25. Zhou RR, Kuang XY, Huang Y, et al. Potential role of High mobility group box 1 in hepatocellular carcinoma. Cell Adh Migr. 2014;8(5):493–498. doi:10.4161/19336918.2014.969139
26. Chen M, Liu Y, Varley P, et al. High-mobility group box 1 promotes hepatocellular carcinoma progression through miR-21-mediated matrix metalloproteinase activity. Cancer Res. 2015;75(8):1645–1656. doi:10.1158/0008-5472.CAN-14-2147
27. Liu Z, Dou C, Wang Y, et al. Highmobility group box 1 has a prognostic role and contributes to epithelial mesenchymal transition in human hepatocellular carcinoma. Mol Med Rep. 2015;12(4):5997–6004. doi:10.3892/mmr.2015.4182
28. Wang X, Xiang L, Li H, et al. The role of HMGB1 signaling pathway in the development and progression of hepatocellular carcinoma: a review. Int J Mol Sci. 2015;16(9):22527–22540. doi:10.3390/ijms160922527
29. Chen Y, Lin C, Liu Y, Jiang Y. HMGB1 promotes HCC progression partly by downregulating p21 via ERK/c-Myc pathway and upregulating MMP-2. Tumour Biol. 2016;37(4):4399–4408. doi:10.1007/s13277-015-4049-z
30. Xiao Y, Sun L, Fu Y, et al. High mobility group box 1 promotes sorafenib resistance in HepG2 cells and in vivo. BMC Cancer. 2017;17(1):857. doi:10.1186/s12885-017-3868-2
31. Kawahara N, Tanaka T, Yokomizo A, et al. Enhanced coexpression of thioredoxin and high mobility group protein 1 genes in human hepatocellular carcinoma and the possible association with decreased sensitivity to cisplatin. Cancer Res. 1996;56(23):5330–5333.
32. Ko YB, Kim BR, Nam SL, Yang JB, Park SY, Rho SB. High-mobility group box 1 (HMGB1) protein regulates tumor-associated cell migration through the interaction with BTB domain. Cell Signal. 2014;26(4):777–783. doi:10.1016/j.cellsig.2013.12.018
33. Sun Y, Tu Y, He LI, Ji C, Cheng BO. High mobility group box 1 regulates tumor metastasis in cutaneous squamous cell carcinoma via the PI3K/AKT and MAPK signaling pathways. Oncol Lett. 2016;11(1):59–62. doi:10.3892/ol.2015.3843
34. Hu Z, Wang P, Lin J, et al. MicroRNA-197 promotes metastasis of hepatocellular carcinoma by activating Wnt/beta-catenin signaling. Cell Physiol Biochem. 2018;51(1):470–486. doi:10.1159/000495242
35. Zhang Y, You W, Zhou H, et al. Downregulated miR-621 promotes cell proliferation via targeting CAPRIN1 in hepatocellular carcinoma. Am J Cancer Res. 2018;8(10):2116–2129.
36. Lou Z, Gong YQ, Zhou X, Hu GH. Low expression of miR-199 in hepatocellular carcinoma contributes to tumor cell hyper-proliferation by negatively suppressing XBP1. Oncol Lett. 2018;16(5):6531–6539. doi:10.3892/ol.2018.9476
37. Tricoli L, Niture S, Chimeh U, Kumar D. Role of microRNAs in the development of hepatocellular carcinoma and acquired drug resistance. Front Biosci. 2019;24:545–554. doi:10.2741/4734
38. Wong CM, Tsang FH, Ng IO. Non-coding RNAs in hepatocellular carcinoma: molecular functions and pathological implications. Nat Rev Gastroenterol Hepatol. 2018;15(3):137–151. doi:10.1038/nrgastro.2017.169
39. Wang Z, Wu Z, Huang P. The function of miRNAs in hepatocarcinogenesis induced by hepatitis B virus X protein (Review). Oncol Rep. 2017;38(2):652–664. doi:10.3892/or.2017.5716
40. Callegari E, Gramantieri L, Domenicali M, D‘Abundo L, Sabbioni S, Negrini M. MicroRNAs in liver cancer: a model for investigating pathogenesis and novel therapeutic approaches. Cell Death Differ. 2015;22(1):46–57. doi:10.1038/cdd.2014.136
41. Bianchi ME, Agresti A. HMG proteins: dynamic players in gene regulation and differentiation. Curr Opin Genet Dev. 2005;15(5):496–506. doi:10.1016/j.gde.2005.08.007
42. Wang W, Zhu H, Zhang H, Zhang L, Ding Q, Jiang H. Targeting HMGB1 inhibits bladder cancer cells bioactivity by lentivirus-mediated RNA interference. Neoplasma. 2014;61(6):638–646. doi:10.4149/neo_2014_079
43. Sun S, Zhang W, Cui Z, et al. High mobility group box-1 and its clinical value in breast cancer. Onco Targets Ther. 2015;8:413–419. doi:10.2147/OTT.S73366
44. Zhang J, Zhang R, Lu WW, et al. Clinical significance of hmgb1 expression in human gastric cancer. Int J Immunopathol Pharmacol. 2014;27(4):543–551. doi:10.1177/039463201402700410
45. Di X, He G, Chen H, et al. High-mobility group box 1 protein modulated proliferation and radioresistance in esophageal squamous cell carcinoma. J Gastroenterol Hepatol. 2019 Apr;34(4):728–735. doi:10.1111/jgh.14371. Epub 2018 Jul 24
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.