")
Back to Journals » OncoTargets and Therapy » Volume 10
MicroRNA 543 suppresses breast cancer cell proliferation, blocks cell cycle and induces cell apoptosis via direct targeting of ERK/MAPK
Authors Chen P, Xu W, Luo Y, Zhang Y, He Y, Yang S, Yuan Z
Received 28 July 2016
Accepted for publication 12 December 2016
Published 6 March 2017 Volume 2017:10 Pages 1423—1431
DOI https://doi.org/10.2147/OTT.S118366
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Jianmin Xu
Po Chen,1 Wentao Xu,2 Yi Luo,1 Yi Zhang,3 Yi He,1 Shuo Yang,1 Zhijun Yuan1
1Department of Medical Oncology, Hunan Cancer Hospital, Changsha, 2Clinical Medical College of An Hui Medical University, Hefei, 3Department of Breast Surgery, Hunan Cancer Hospital, Changsha, China
Background: Breast cancer affects millions of people with a high mortality rate throughout the world; microRNA 543 (miR-543) has been reported to suppress progression in some kinds of cancers, but has not been reported in breast cancer. Thus, the purpose of this study is to investigate the function of miR-543 in breast cancer cells.
Methods: Two cell lines, MCF-7 and MDA-MB-231, were selected to be the research objects; the miR-543 overexpression and knockdown models were established in the study by transforming miR-543 mimics and miR-543 inhibitor. Real-time polymerase chain reaction, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Western blot, clone formation and cell flow cytometer assay were used to test the miR-543’s function. Dual-luciferase assay was used for the detection of miR-543 and ERK2 targeting relationship.
Results: The results showed that the cell proliferation and cell cycle were inhibited, and the capability of cell apoptosis was upregulated when miR-543 was overexpressed; we found that there was a target relationship between ERK2 and miR-543. Furthermore, downstream factors of mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase-2 (ERK2) pathway, including RSK2 and MSK1, were decreased in miR-543 overexpression model.
Conclusion: This study provides series evidences to support that breast cancer progression was inhibited by miR-543 via direct targeting of ERK2 in MAPK/ERK signal pathway, which may provide a molecular basis for better treatment for patients.
Keywords: microRNA 543, breast cancer, ERK2, apoptosis, cell cycle, proliferation
Introduction
Breast cancer usually accompanies with a high mortality rate, which has been diagnosed in 200,000 females in the USA.1 The etiology of breast cancer is complicated and involves various factors.2 Although a large number of studies have reported that the metastasis and tolerance of tumor occurrence were closely related to intracellular signal transduction pathway (ISTP),2 the pathogenesis was not clear yet. In recent years, microRNA has attracted the attention of many researchers to explore the cancer therapy, and this noncoding RNA plays an essential role in regulating its target gene in ISTP.2 In this study, we aim to investigate how does this noncoding RNA affect breast cancer and achieve the goal of treatment.
MicroRNA is an endogenous RNA which is deemed to be a regulator that modulates the expression of targeting gene after mRNA transcription.3 Previous studies indicated that microRNA was closely associated with cell proliferation, apoptosis and differentiation,3,4 and it was reported that microRNAs were regarded as (a microRNA) was regarded as natural inhibitors of cancer-causing gene, which was able to inhibit the occurrence and metastasis of cancer.5 Thus, the abnormal expression of microRNA performed an important role in as an important element in diagnosis, therapy and prognosis stages of cancer.
The differential expression of microRNA was treated as a symbol, which is utilized to distinguish cancer occurrence.6,7 Moreover, the analysis of microRNA with its corresponding target gene in cancer site and normal tissues indicates that signal regulation in cells is influenced by the abnormally expressed microRNA and subsequently leads to the formation of breast cancer.8
miR-543 was characterized in a recent year as a newly found microRNA in gastric cancer cells9 and hepatoma cells.10 A research reported that there was a downregulation of miR-543 in breast cancer11 and endometrial cancer.12 Romero-Cordoba et al found that miR-543 was differentially expressed in breast cancer by using microarray analysis; the quantity of expressed miR-543 in breast cancer tissue was 2.87 times lower than that of the normal tissue. Furthermore, miR-543 was found related to MAPK signal pathway according to the function statistics.13 Mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) signal pathway was highly conserved in eukaryotes, and its abnormal expression was closely related to human diseases.14,15 Thus, we infer that miR-543 is a breast cancer suppressor that inhibits breast cancer cells, progression through MAPK/ERK signal pathway.
In this study, the influence of miR-543 in MAPK/ERK signal pathway was investigated. The cell proliferation and apoptosis were detected in miR-543 overexpression and knockdown models. Moreover, MAPK/ERK channel’s related factors and its downstream factors were tested.
Materials and methods
Reagents and cell culture
miR-543 mimics and miR-543 inhibitor were obtained from Gene Copopia (Rockville, MD, USA) to construct miR-543 overexpression and knockdown models. Breast cancer cell lines of MDA-MB-231 and MCF-7 were purchased from Shanghai Cell Bank of Chinese Academy of Science. The cells were cultured in RPMI 1640 medium (HyClone, Hudson, NH, USA) supplemented with 10% fetal bovine serum at 37°C and 5% CO2, and the cells were cultured for further experiments.
In situ hybridization assay (IsHyb)
The breast carcinoma and normal tissue chip were purchased from Auragene (Changsha, People’s Republic of China). The sequence of miR-543 probe was purchased from BGI Techcompany (Shenzhen, People’s Republic of China). Hybridization probe mixture (1:500) was added, and the operation followed the manufacturer’s instructions of IsHyb in situ Hybridization kit (Auragene). Finally, the stained chip was observed under optical microscope (Upototech, Changchun, People’s Republic of China).
Cell transfection
Cell transfection was performed as previously described.16 The cells were cultured in 6-well plates before being transfected with either miR-543 mimics or miR-543 inhibitor using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cells were collected for the following experiments after 48 h of transfecting, and a group of miR-543 mimics was added with 2.3 μM epidermal growth factor (EGF) and incubated at 37°C and 5% CO2 for another 24 h.
Real-time polymerase chain reaction (qPCR)
Total RNAs were isolated using TRIzol method. Revert Aid First Strand cDNA Synthesis Kit (Thermo Scientific Fermentas, K1622, Waltham, MA, USA) was utilized for reverse transcription. All reaction mixtures contain 10 μL Qpcr Master Mix (2×), 1 μL cDNA (5× first-strand buffer, dNTP, RNasin, M-MLV), and 9 μL ddH2O. RT-PCR reactions were performed in a qPCR quantification system (ABI 7300, Applied Biosystems, Foster City, CA, USA), which is set to the following conditions: initial denaturation at 95°C for 3 min, 39 cycles of denaturation at 95°C for 10 s and annealing at 60°C for 15 s. miR-543 and U6 primer were obtained from Gene Copopia, the other primers were as follows: ERK2: sense, TAC GAC CCG AGT GAC GAG; antisense, TTC CTT AGG CAA GTC ATC CA and β-actin: sense, AGG GGC CGG ACT CGT CAT ACT, antisense, GGC GGC ACC ACC ATG TAC CCT. All the qPCR data were processed using 2−ΔΔCT method.
Western blot analysis
The extracted protein from cells was separated on 10% sodium dodecyl sulfate polyacrylamide gels and transferred to nitrocellulose membranes at 100 mA for 2 h. The membranes were blocked in skimmed milk for 1 h at room temperature and overnight at 4°C in anti-CyclinD1, anti-Bcl-2, anti-Bax (1:1,000) and anti-ERK2, anti-p-ERK1/2, anti-RSK2, anti-p-RSK2, anti-MSK1 and anti-p-MSK1 (1:800; Cell Signaling Technology, Danvers, MA, USA), respectively, before they were conjugated with the secondary antibody. Signals were observed using Cano Scan (LiDE110, Tokyo, Japan).
MTT assay
The cells were collected, and the concentration was adjusted to 5×103 cell/well before they were cultured at 5% CO2, 37°C for 24, 48 and 72 h. After that, 20 μL of MTT was added to the cells (5 mg/mL) and maintained at 5% CO2, 37°C for another 4 h before MTT was removed, while optical density value was measured at 570 nm when cells were suspended in 150 μL of dimethyl sulfoxide.
Colony formation assay
The clonogenicity of single cell was detected by colony assay. Cells were collected by 0.25% trypsin and adjusted the concentration to 300 cell/petri dish, which was loaded with 2 mL of preheated culture media before they were cultured at 5% CO2, 37°C for 2–3 weeks. Colony formation was terminated until the colony was visible to naked eye. After that, cells were washed twice with phosphate-buffered saline (PBS), then 4% paraformaldehyde was added for 30 min to fix the cell before they were stained with Gimsa for 10 min. After that, the number of cloning was counted, and colony formation rate was calculated as follows: Colony Formation Rate = (Number of Colony/Inoculation Cell Number) ×100%.
Cell apoptosis and cell cycle
Cell apoptosis and cell cycle were measured by flow cytometer. Cells were collected and washed twice with PBS before suspended in 500 μL of binding buffer. Five microliters of Annexin V-fluorescein isothiocyanate and 5 μL of propidium iodide (PI) were added for cell apoptosis assay, whereas 500 μL of PBS containing 50 μg/mL PI was added for cell cycle assay before they were incubated in dark at room temperature for 15 min. The expression of the fluorescent was detected by flow cytometer.
Luciferase reporter assay
The possibility of ERK2 and miR-543 target binding was predicted using online software microrna.org (http://www.microrna.org/microrna/home.do). Wild-type or mutant type of human ERK2 mRNA 3′-untranslated region (UTR) regions was cloned into psi-CHECK2 vector. MCF-7 and MDA-MB-231 cells were cotransfected with 3′-UTR vectors and miR-543 mimics or scramble control. Forty-eight hours later the supernatants were collected, then the luciferase activities were measured using a dual-luciferase reporter assay system (Promega, Fitchburg, WI, USA).
Statistical analyses
Statistical analyses were performed using GraphPad Prism 6 software (GraphPad Software, Inc., La Jolla, CA, USA). The data are shown as the mean ± standard deviation. An unpaired two-tailed Student’s t-test or analysis of variance with Bonferroni t post-test was used to analyze the data depending on conditions. P-value of <0.05 was considered to indicate a statistically significant difference.
Results
The expression of miR-543 was lower in breast cancer
To explore the potential role of miR-543 in breast cancer, 20 pairs of cancer site tissues with their adjacent tissues were collected from clinical operations. The expression of miR-543 was measured by qPCR and IsHyb (Figure 1). qPCR demonstrates that the gene level of miR-543 decreases in cancer site when compared with adjacent site (Figure 1A), the same as the result of IsHyb (Figure 1B and C).
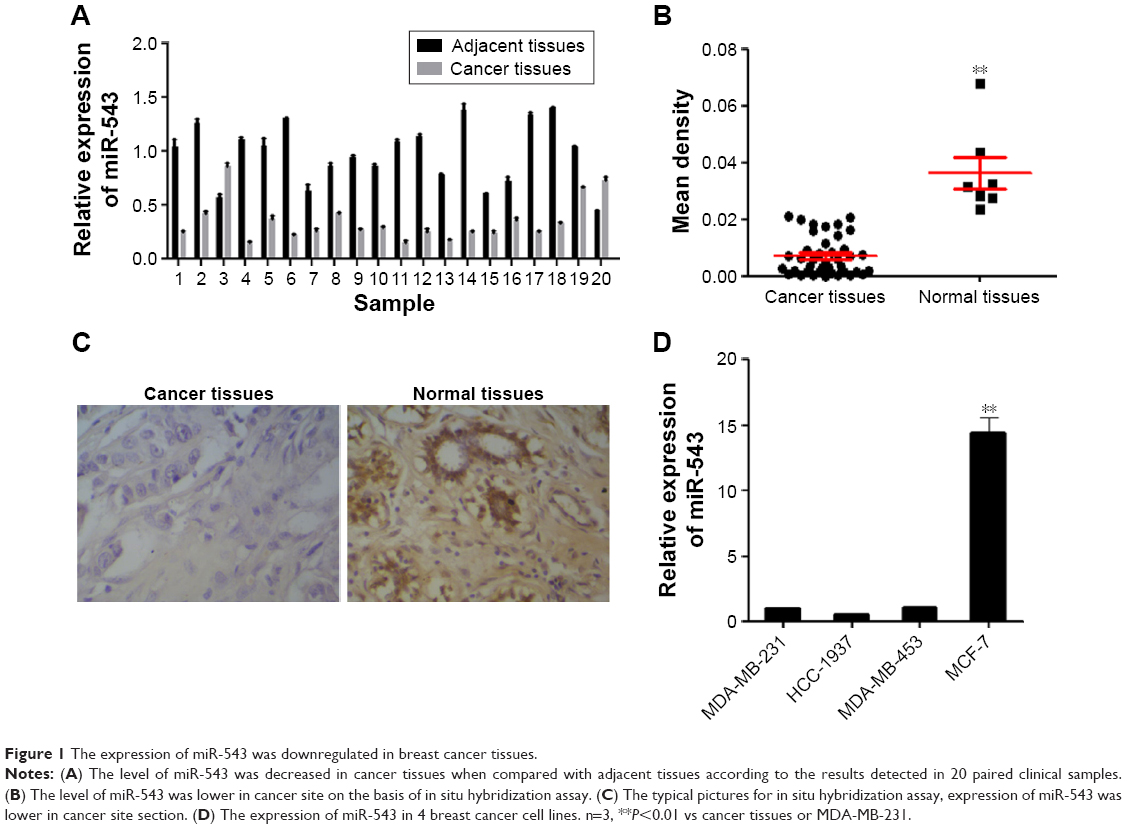 | Figure 1 The expression of miR-543 was downregulated in breast cancer tissues. |
In an attempt to further investigate the role of miR-543 in breast cancer. The expression level in the cell lines of MDA-MB-231, HCC-1937, MDA-MB-453 and MCF-7 was detected. As shown in Figure 1D, the expression of miR-543 in MCF-7 was the highest among others. According to this result, MCF-7 was chosen for further study, as well as MDA-MB-231.
miR-543 decreases cell proliferation, induces cell apoptosis and inhibits cell cycle of breast cancer cells
Cell models of miR-543 overexpression and knockdown were established by transfecting miR-543 mimics and miR-543 inhibitor. The results between the cell lines of MDA-MB-231 and MCF-7 shared the same tendency in the following tests. To begin with, the gene expression of miR-543 was detected by qPCR. As shown in Figure 2A, the expression of miR-543 in miR-543 mimics group witnessed a dramatic increase when contrasted to miR-543 inhibitor. This indicates that the miR-543 overexpression and knockdown models were constructed successfully. To further evaluate the role of miR-543 in breast cancer cells, the capability of proliferation was tested by MTT (Figure 2B) and colony formation (Figure 2C). The result of MTT indicates that the proliferation capacity was suppressed after the cell lines were transfected with miR-543 mimics, while it was increased in miR-543 inhibitor. Moreover, the clone formation rate was increased in miR-543 inhibitor when compared with control group, while it was almost doubled when contrasted to miR-543 mimics in both cell lines. Furthermore, cell apoptosis and cell cycle were measured by flow cytometer assay in this study. Figure 2D illustrates that apoptosis rate was upregulated when cell lines were transfected with miR-543 mimics, while it was declined in miR-543 inhibitor group. With regard to cell cycle (Figure 2E), it demonstrates that the cell proliferation was slowed down in miR-543 mimics group, however, IT’s faster in miR-543 inhibitor group. For instance, the cell proportion at G2+S stage in miR-543 mimics group was decreased to ~30% in MDA-MB-231, while it was increased to ~60% in miR-543 inhibitor group.
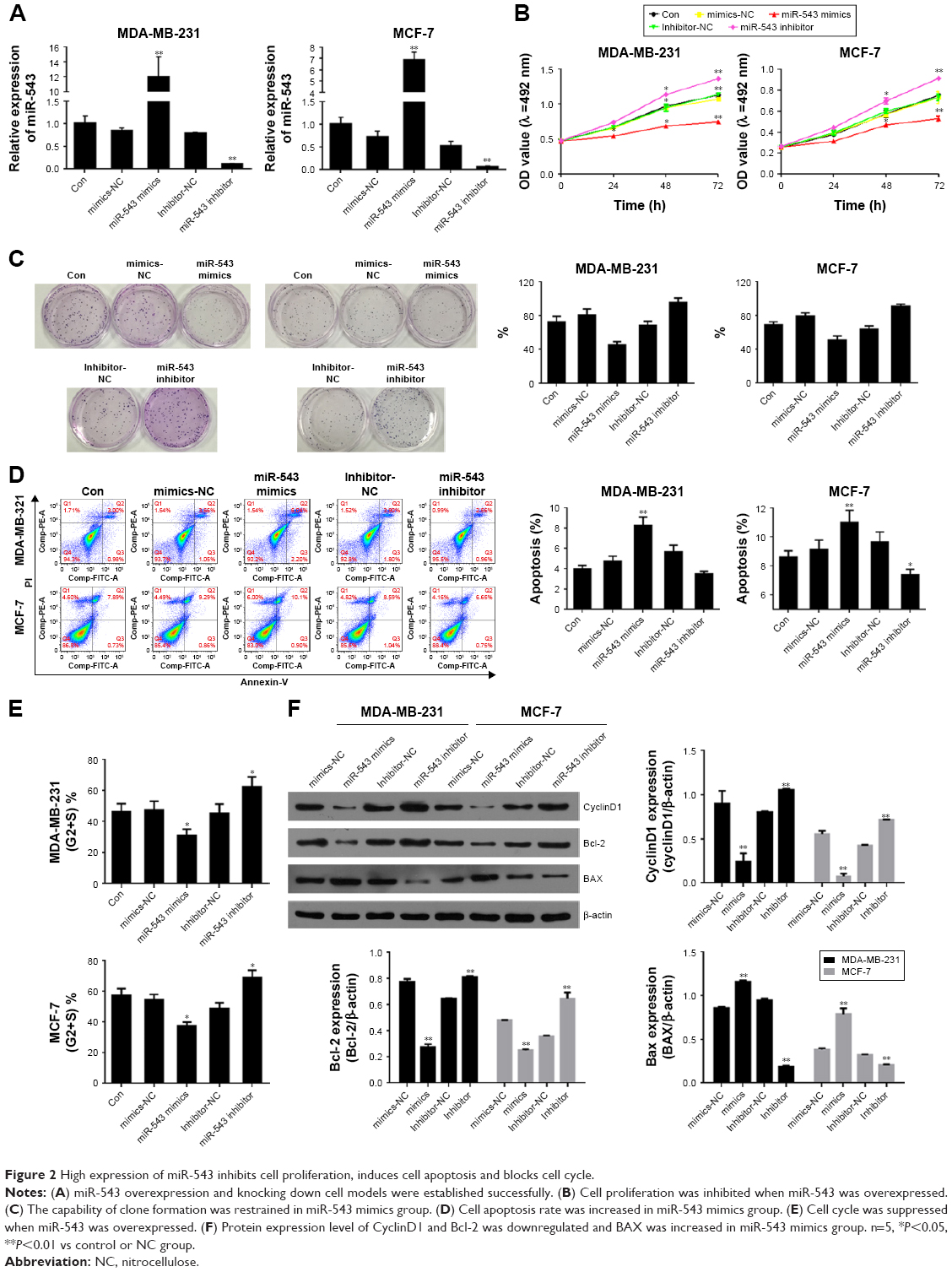 | Figure 2 High expression of miR-543 inhibits cell proliferation, induces cell apoptosis and blocks cell cycle. |
The factors related to cell cycle, such as cyclinD1, and the factors related to cell proliferation, including Bcl-2 and BAX, were measured by Western blot assay. As shown in Figure 2F, the levels of Bcl-2 and cyclinD1 were downregulated in miR-543 mimics group, which shared the opposite tendency with miR-543 inhibitor group. However, the expression of BAX was increased in miR-543 mimics group, while decreased in the inhibitor model. To conclude, miR-543 is an important element which can help inhibiting breast cancer development.
ERK2 is the target gene of miR-543
The protein level of ERK2 was measured by Western blot analyses among the cell lines of MDA-MB-231, HCC-1937, MDA-MB-453 and MCF-7 (Figure 3A). The level of ERK2 was highly expressed in MDA-MB-453, whereas the lowest level was witnessed in MCF-7, which almost shares the opposite tendency with the expression level of miR-543 in those cell lines (Figure 1D). To explore whether there is a target binding relationship between miR-543 and ERK2, we first used online software microRNA.org to predict the possibility of ERK2 and miR-543 target binding. The score was very high, and these two sites were highly conserved in several species. As shown in Figure 3B, we located potential binding sites for miR-543 at the 3′-UTR of ERK2 mRNAs. This suggested that there may exist a target relationship between miR-543 and ERK2. MiR-543 function interaction with the ERK2 may be performed via binding the predicted site positions. Then, we used dual-luciferase experiment to verify the targeting effect relationship. Cotransfected ERK2-3′-UTR-psi-CHECK2 plasmid and miR-543 mimics in MDA-MB-231 and MCF-7 cell samples, and the luciferase activity significantly decreased compared to the other groups, indicating that miR-543 and ERK2 in MDA-MB-231 and MCF-7 cells had targeting effect relationship (Figure 3C).
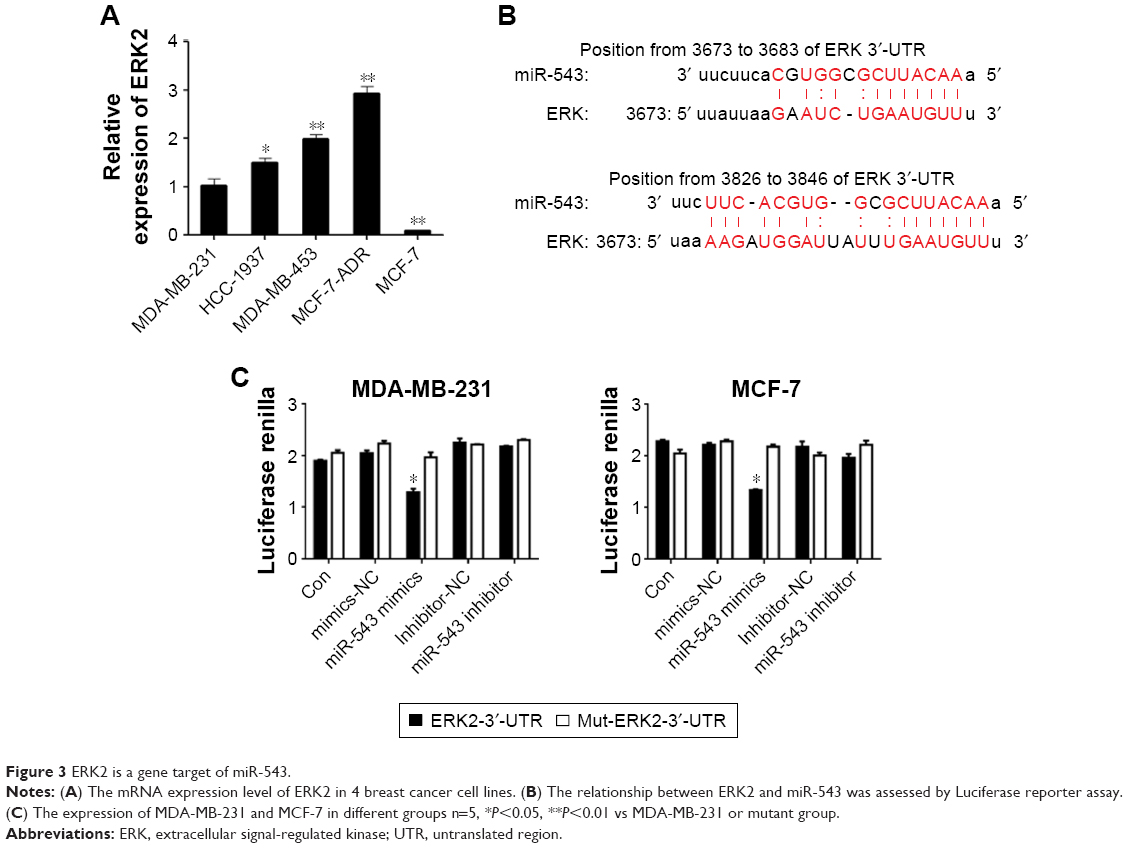 | Figure 3 ERK2 is a gene target of miR-543. |
MiR-543 regulates breast cancer via MAPK/ERK signal pathway
To verify the hypothesis that miR-543 regulate the progression of breast cancer via MAPK signal pathway, the gene expression and protein level of related factors including ERK2 and its phosphorylation site p-ERK1/2 were detected, as well as the pathway downstream factors such as RSK2, MSK1, p-RSK2 and p-MSK1 in the group of miR-543 mimics and MAPK/ERK activated group (miR-543 mimics treated with EGF). Figure 4A indicates that the level of ERK2 and its phosphorylation sites were decreased, while it was upregulated when miR-543 mimics were treated with EGF, a MAPK signaling agonist. Moreover, downstream factors were deregulated in miR-543 mimics group as well, while reversed by EGF.
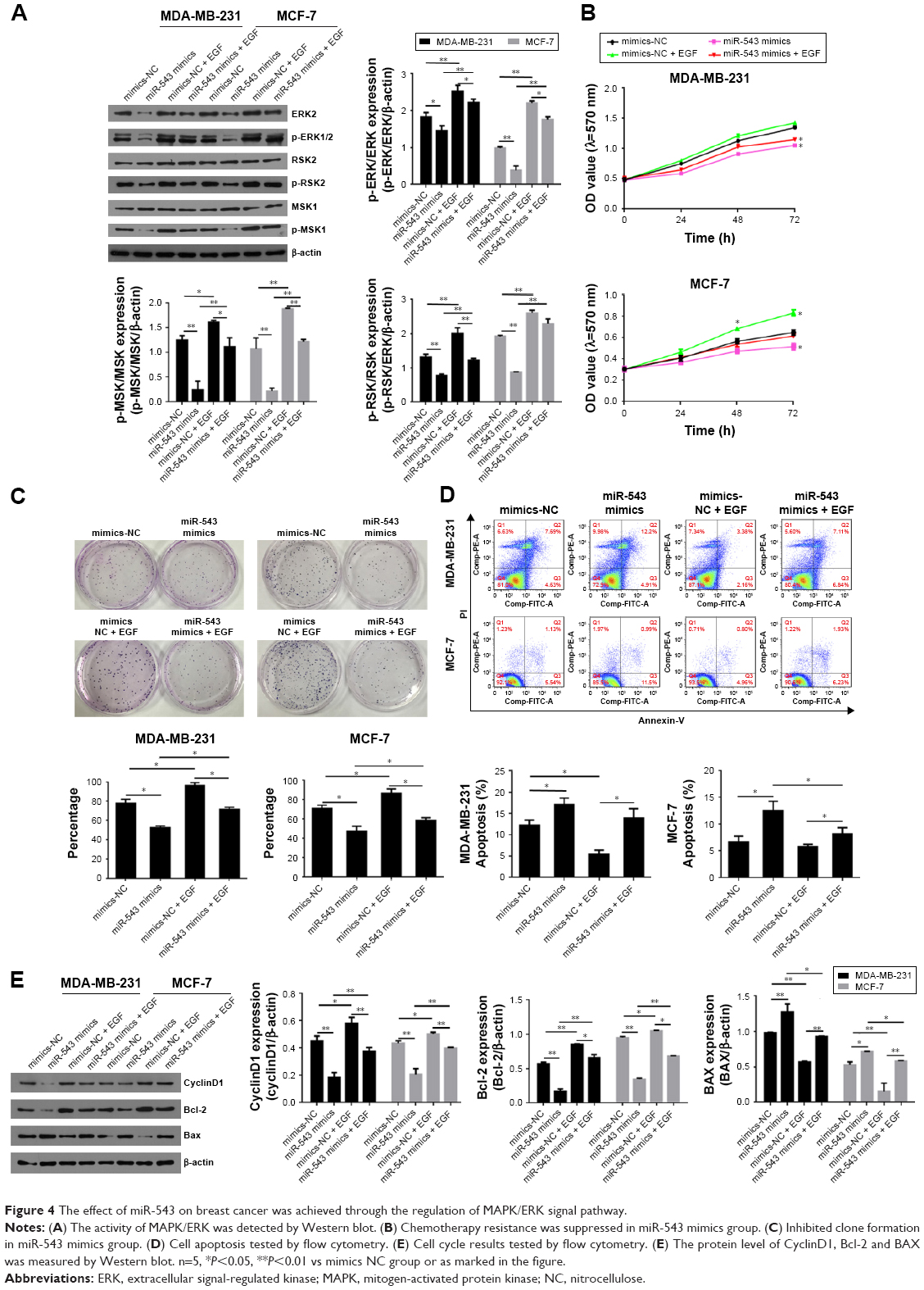 | Figure 4 The effect of miR-543 on breast cancer was achieved through the regulation of MAPK/ERK signal pathway. |
To confirm the potential role of miR-543 in breast cancer through MAPK/ERK pathway, cell proliferation, cell cycle and its apoptosis were tested. The results of MTT (Figure 4B) and clone formation (Figure 4C) revealed that cell proliferation capability was lower in miR-543 mimics groups when compared with miR-543 mimics + EGF. Figure 4D indicates that the apoptosis cell rate, including early apoptosis stage and later apoptosis stage, was higher in miR-543 overexpressed cell model when compared with MAPK/ERK activated group. When it comes to cell cycle (Figure 4E), it showed that cell proliferation ability in miR-543 mimics group was lower than that of miR-543 mimics + EGF group. For example, the G2+S stage in miR-543 mimics was ~30% in the cell line of MB-MDA-231, while it was upregulated to ~40% in miR-543 mimics + EGF group. Furthermore, the expression of apoptosis marker genes consisting of Bcl-2, BAX and the cell cycle marker gene CyclinD1 were measured using Western blot. As shown in Figure 4F, the level of BAX was increased in miR-543 mimics group, whereas Bcl-2 and cyclinD1 declined; they presented the opposite trend in miR-543 mimics + EGF group in both the cell lines of MB-MDA-231 and MCF-7. Consequently, the above data demonstrated that the inhibition function of miR-543 was performed through the inhibition of MAPK/ERK signal pathway in breast cancer.
Discussion
The effects of miR-543 in breast cancer were investigated in this study. Low expression of miR-543 in breast cancer tissues was detected by qPCR, which was corresponding to the result of microarray IsHyb. Thus, we assumed that the expression of miR-543 performed an important role in the development of breast cancer, which may help to inhibit tumor deterioration with its high expression. To confirm this presumption, miR-543 overexpression and knockdown models were established in MDA-MB-231 and MCF-7 cell lines. After modeling, cell proliferation was restrained in the group of miR-543 mimics according to the results of MTT and clone formation, whereas cell apoptosis was elevated and cell cycle was suppressed. Moreover, we found that there is a target relationship between miR-543 and ERK2, and the factors related to MAPK/ERK pathway were influenced by the expression of miR-543. Thus, we can conclude that miR-543 inhibits the development of breast cancer by targeting ERK2 via MAPK/ERK signal pathway.
miR-543 was regarded as an inhibitor in many cancer developments according to previous research. Scientists found that TWIST1 signal pathway was regulated by miR-543, which interacted with its target gene, resulting in restrained Emergency Medical Technician activity in colon cancer.11 Moreover, it was proven that the expression of miR-543 in endometrial cancer cell was downregulated, whereas the development of endometrial cancer cell lines of RL95-2 and AN3 was suppressed when this microRNA was overexpressed.12 In addition, a low level of miR-543 was found in osteosarcoma tissues as well.17 In this study, the results demonstrated that the proliferation and cell cycle of MDA-MB-231 and MCF-7 were inhibited under the condition of overexpressed miR-543 with an upregulated cell apoptosis. Whereas, the results presented the opposite state when miR-543 expression was inhibited. Consequently, we inferred that miR-543 can inhibit the progression of breast cancer.
MAPK/ERK signal pathway interacts with cellular signals and is strongly linked to cell growth, proliferation, survival and differentiation.18 As is known that EGF is a simulation inducing cell growth and proliferation,19 according tho the results, the cell proliferation and cell cycle were increased accompanying with decreased cell apoptosis in EGF-treated miR-543 mimics group (Figure 4). Thus, we conclude that the inhibition influence of miR-543 in breast cancer was achieved through the suppression of MAPK/ERK signal pathway. RSK2, a highly conserved sequence placed on the downstream of ERK pathway in S6 ribosome kinase, was activated and phosphorylated via ERK1/2, responding to growth factors and chemokines that were implicated with essential cell processes consisting of cell proliferation, survival and cell movement-related cytoplasm.20 With regard to MSK1, a nuclear histone modification enzyme placed on the downstream of ERK pathway was triggered through MAPK/ERK signal channel and closely associated with underlying genetic changes in chromatin structure.21 Thus, downstream factors, such as RSK2 and MSK1, were measured to further understand the potential role of miR-543 inhibition in breast cancer deterioration and the relationship with MAPK/ERK pathway. In addition, the protein levels, as well as the phosphorylation proteins of ERK2, RSK2 and MSK1, were detected by Western blot, indicating that the phosphorylation of ERK2 was restrained by miR-543, whereas MAPK/ERK was inhibited. Moreover, the phosphorylation of downstream factors including RSK2 and MSK1 was impeded. Consequently, miR-543 was regarded as an upstream regulatory factor in MAPK/ERK pathway and initiated changes in downstream factors, while influencing the cell functions.
In summary, this study indicates the potential role of miR-543 in breast cancer. The inhibited MAPK/ERK signal pathway was found in miR-543 overexpressed cell model, which comprehensively explains the mechanism of miR-543 regulated pathway that suppresses the development of breast cancer deterioration. Furthermore, it was verified that MAPK/ERK channel was inhibited via miR-543 targeting ERK2, which eventually inhibited tumor development. This study provides a novel targeted therapy for The Breast Cancer Research Foundation.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62(4):220–241. | ||
Nicoloso MS, Spizzo R, Shimizu M, Rossi S, Calin GA. MicroRNAs – the micro steering wheel of tumour metastases. Nat Rev Cancer. 2009;9(4):293–302. | ||
Maqbool RM, Ul Hussain M. MicroRNAs and human diseases: diagnostic and therapeutic potential. Cell Tissue Res. 2014;358(1):1–15. | ||
Sandhu S, Garzon R. Potential applications of microRNAs in cancer diagnosis, prognosis, and treatment. Semin Oncol. 2011;38(6):781–787. | ||
Le XF, Merchant O, Bast RC, Calin GA. The roles of microRNAs in the cancer invasion-metastasis cascade. Cancer Microenviron. 2010;3(1):137–147. | ||
Iorio MV, Ferracin M, Liu CG, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65(16):7065–7070. | ||
Blenkiron C, Goldstein LD, Thorne NP, et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol. 2007;8(10):R214. | ||
O’Day E, Lal A. MicroRNAs and their target gene networks in breast cancer. Breast Cancer Res. 2010;12(2):201. | ||
Li J, Dong G, Wang B, Gao W, Yang Q. miR-543 promotes gastric cancer cell proliferation by targeting SIRT1. Biochem Biophys Res Commun. 2016;469(1):15–21. | ||
Yu L, Zhou L, Cheng Y, et al. MicroRNA-543 acts as an oncogene by targeting PAQR3 in hepatocellular carcinoma. Am J Cancer Res. 2014;4(6):897–906. | ||
Haga CL, Phinney DG. MicroRNAs in the imprinted DLK1-DIO3 region repress the epithelial-to-mesenchymal transition by targeting the TWIST1 protein signaling network. J Biol Chem. 2012;287(51):42695–42707. | ||
Bing L, Hong C, Li-Xin S, Wei G. MicroRNA-543 suppresses endometrial cancer oncogenicity via targeting FAK and TWIST1 expression. Arch Gynecol Obstet. 2014;290(3):533–541. | ||
Romero-Cordoba S, Rodriguez-Cuevas S, Rebollar-Vega R, et al. Identification and pathway analysis of microRNAs with no previous involvement in breast cancer. PLoS One. 2012;7(3):e31904. | ||
Whelan JT, Hollis SE, Cha DS, Asch AS, Lee MH. Post-transcriptional regulation of the Ras-ERK/MAPK signaling pathway. J Cell Physiol. 2012;227(3):1235–1241. | ||
Thum T, Gross C, Fiedler J. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456(7224):980–984. | ||
Chen QY, Zheng Y, Jiao DM. Curcumin inhibits lung cancer cell migration and invasion through Rac1-dependent signaling pathway. J Nutr Biochem. 2014;25(2):177–185. | ||
Song N, Liu H, Ma X, Zhang S. Placental growth factor promotes metastases of ovarian cancer through MiR-543-regulated MMP7. Cell Physiol Biochem. 2015;37(3):1104–1112. | ||
Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68(2):320–344. | ||
Groenen LC, Nice EC, Burgess AW. Structure-function relationships for the EGF/TGF-alpha family of mitogens. Growth Factors. 1994;11(4):235–257. | ||
Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol. 2008;9(10):747–758. | ||
Hauge C, Frodin M. RSK and MSK in MAP kinase signalling. J Cell Sci. 2006;119(Pt 15):3021–3023. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.