")
Back to Journals » OncoTargets and Therapy » Volume 13
MicroRNA-375 Targets ATG14 to Inhibit Autophagy and Sensitize Hepatocellular Carcinoma Cells to Sorafenib
Authors Yang S, Wang M, Yang L, Li Y, Ma Y, Peng X, Li X, Li B, Jin H, Li H
Received 29 January 2020
Accepted for publication 30 March 2020
Published 28 April 2020 Volume 2020:13 Pages 3557—3570
DOI https://doi.org/10.2147/OTT.S247655
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
Shuo Yang,1,* Minggang Wang,2,* Liang Yang,1 Yan Li,1 Yingbo Ma,1 Xueqiang Peng,1 Xinyu Li,1 Bowen Li,1 Hongyuan Jin,1 Hangyu Li1
1Department of General Surgery, The Fourth Affiliated Hospital, China Medical University, Shenyang 110000, People’s Republic of China; 2Department of General Surgery, Shandong Provincial Qianfoshan Hospital, Shandong University, Jinan, Shandong Province 250014, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hangyu Li
Department of General Surgery, The Fourth Affiliated Hospital, China Medical University, Shenyang 110000, People’s Republic of China
Tel +86 24 6204 2117
Fax +86 24 6257 1119
Email [email protected]
Purpose: Sorafenib has revolutionized treatment of hepatocellular carcinoma (HCC), but its efficacy is limited by drug resistance. Autophagy is the process by which cellular components are transported to lysosomes for degradation, which promotes energy production and production of macromolecular precursors. Studies have suggested that the cytoprotective function of autophagy may contribute to chemoresistance or targeted drug resistance in cancer cells. We investigated the effects of miR-375 and autophagy-related protein 14, and their interrelationships, on sorafenib efficacy.
Methods: Cell viability was measured using the MTT assay, and apoptosis was evaluated using flow cytometry. Colony formation assay was performed to determine changes in cell number. Real-time PCR and Western blotting were performed to quantify the expression of key genes and proteins. Immunofluorescence and transmission electron microscopy were used to detect autophagy. Dual-luciferase reporter assays were used to verify a direct target.
Results: We characterized the relationship between sorafenib and autophagy. We showed that inhibition of autophagy enhanced sensitivity of HCC to sorafenib and showed that miR-375 was important in this process. Finally, we showed that miR-375 affected sensitivity of HCC cells to sorafenib through regulation of ATG14.
Conclusion: We showed that miR-375 sensitized HCC cells to sorafenib by blocking sorafenib-induced autophagy. We also showed that ATG14 was a direct autophagy-related target of miR-375. These findings indicated that miR-375-ATG14 was important in the development of sorafenib resistance in HCC.
Keywords: autophagy, hepatocellular carcinoma, sorafenib, miR-375, therapy, drug resistance
Introduction
Hepatocellular carcinoma (HCC) is the fourth leading cause of cancer death and is the fourth most common malignant tumor in China.1 Current treatments are limited and do not improve survival rates.2 Despite recent breakthroughs in treatment and surgical removal, the 5-year survival rate remains poor.3 In addition, use of anticancer drugs to treat HCC is limited by primary and acquired drug resistance.4,5 Therefore, elucidation of the molecular mechanisms of hepatocellular carcinoma and identification of prognostic indicators are critical to the development of effective treatments for hepatocellular carcinoma.
Autophagy is a catabolic pathway characterized by degradation of cellular components. Autophagy removes misfolded proteins, damaged organelles, and lipid droplets, plays a crucial role in energy balance and cytoplasmic quality control, and promotes liver homeostasis.6,7 Increasing numbers of studies have shown that autophagy plays an important role in HCC. Autophagy is associated with risk factors for HCC such as oxidative stress, chronic inflammation, viral infection, metabolic dysfunction, liver alcohol disorders, and fatty liver disease.8–10 Therefore, a comprehensive understanding of the role of autophagy in HCC may result in the development of new diagnostic and therapeutic techniques. Furthermore, many recent studies have identified genes that promote drug resistance through the regulation of autophagy.
Sorafenib is a multi-kinase inhibitor that affects cell surface tyrosine kinase receptors and intracellular serine/threonine kinases.11 Representative Phase III trials have shown that sorafenib significantly improved overall survival in patients with advanced HCC.12 Furthermore, sorafenib has been shown to activate autophagy and apoptosis.13,14 Interactions between non-coding RNA and autophagy have received increased attention with regard to hepatocellular carcinoma. MicroRNAs are a class of endogenous, short non-coding RNAs that post-transcriptionally regulate gene expression.15 MicroRNAs can affect many biological processes, such as cell development, infection, immunity, and carcinogenesis.16 MicroRNAs are involved in various stages of autophagy, including phagophore induction, nucleation, expansion, and maturation of autolysosomes and autophagosomes.17 In a previous study, we performed bioinformatics analysis using RT-PCR to evaluate the effects of sorafenib. MicroRNA 375 was identified for further study. The role of miR-375 in regulation of sorafenib resistance in HCC cells and the underlying mechanisms of this resistance have not been characterized. In this study, we showed that miR-375 sensitized HCC cells to sorafenib by blocking sorafenib-induced autophagy. We also showed that a key autophagic protein, autophagy-related protein 14 (ATG14), was a direct autophagy-related target of miR-375. These findings indicated that the miR-375-ATG14 axis was heavily involved in the development of sorafenib resistance in HCC.
Materials and Methods
Cell Culture and Reagents
Hepatocellular cell lines (Huh7 and HepG2) were purchased from Shanghai Institute of Cell Bank (Shanghai, China) and grown in Dulbecco’s modified Eagle’s medium (BioWhittaker, Walkersville, MD, USA) supplemented with 10% fetal bovine serum (HyClone, Logan, UT, USA), streptomycin (100 μg/mL), and penicillin (100 U/mL) at 37°C in 5% CO2.
Cell Transfection
The expression plasmids containing ATG14 cDNA, pcDNA-3.1, miR-375 mimics and miR-NC were purchased form Genechem (Shanghai, China). The siRNA and the negative control (NC) oligonucleotides were purchased from Sigma (Shanghai, China). The plasmids and siRNA were transfected into cells using Lipofectamine 3000 according to the manufacturer’s protocol. Diluted appropriate amounts of Lipo3000 and miR-375 mimics or inhibitor with opti-MEM in proportion. Then, drip the mix evenly into the medium and shake it slowly. Put it in CO2 incubator, and change the DMEM after about 8 h. After 48 h, cells were harvested for further assays.
Total RNA Isolation and qRT-PCR
We used Trizol reagent (Invitrogen, Thermo Fisher Scientific, Inc.) to isolate total RNA from HCC tissues and cells. We used a DNA synthesis kit (Takara, Dalian, China) to synthesize DNA according to the manufacturer’s instructions. The expression of RNA was detected by qRT-PCR using SYBR Premix Ex Taq II kit (Takara, Dalian, China). The expression of miR-375 was determined using the Taqman miRNA kit (Applied Biosystems, Foster City, CA). The levels of RNU6B mRNA and GAPDH mRNA were used for normalization. Data were analyzed using CT values, then converted to fold-changes. The primer sequences used in this study are summarized in Supplementary Table 1.
Western Blot
Total protein from cultured Huh7 cells was extracted on ice using RIPA Lysis Buffer (Beyotime) supplemented with a protease and phosphatase inhibitor cocktail (Roche, Basel, Switzerland). Protein concentration was determined using a BCA Protein Assay kit (Generay, Shanghai, China), and proteins were separated using sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE), then transferred onto PVDF membranes (Millipore). The membranes were incubated with primary antibodies against LC3, P62 (1:1000, Cell Signaling Technology, MA, USA), ATG14, Caspase-3, PARP, C-PARP, or β-actin (1:1000, Proteintech Group, Chicago, IL, USA), then blocked with 5% fat-free milk powder in TBST buffer at 4°C overnight. The membranes were incubated with secondary antibody (CWBIO; 1:5000 dilution) and bands were detected by enhanced chemiluminescence (Thermo Fisher Scientific) using an Automatic Chemiluminescence Imaging Analysis System (Tanon-4200; Tanon Science & Technology, Shanghai, China).
Colony Formation
Cells were inoculated in 6-well plates at a density of 1.5 –2 x 103 cells/well. The cells were then cultured for two weeks. Then, the cells were fixed with 4% paraformaldehyde (Beyotime Biotechnology, Beijing, China) for 15–20 min, stained with 0.5% crystal violet (Beyotime Biotechnology, Beijing, China) for 20 min, washed with PBS three times (5 min each), and cell colonies were counted. The experiment was repeated three times.
Analysis of Cell Viability
The inhibitory effects of sorafenib on the viability of the HCC cells were measured by an MTT assay. Cells were seeded in 96-well dishes at 1 x104 cells per well and incubated overnight. They were then treated with the indicated dose of sorafenib (2, 4, 6, 8,10μmol/mL) for 24 h. Subsequently, MTT reagent (Sigma-Aldrich) was added to each well at a final concentration of 0.5 mg/mL, and the cells were incubated for a further 2 h under the same conditions. The culture plate was centrifuged for 5 min at 25°C and the supernatant was removed. Dimethyl sulfoxide was added to dissolve the formazan crystals, and optical density was determined at 490 nm using a microplate reader (Tecan Biotechnology, Shanghai, China). The experiments were conducted three times independently, in triplicate each time, and the average values of the three independent experiments were calculated.
Analysis of Apoptosis Using Flow Cytometry
Cell apoptosis was determined using a FITC AnnexinV/Dead Cell Apoptosis Kit (Life Technologies Corporation 29851 Willow Creek Road Eugene, Oregon 97402) that quantitatively measures the percentage of apoptotic cells using flow cytometry.
Immunofluorescence
Cells were seeded and cultured in 6-well plates, then washed with PBS and fixed in 4% paraformaldehyde for 15 min. The cells were then permeabilized using 0.5% TritonX-100 for 30 min at 37°C and blocked with 1% BSA (bovine serum albumin) for 1 h. The cells were incubated with rabbit polyclonal LC3 antibody (1:100; MBL, Beijing, China) at 4°C overnight. The cells were washed, then incubated with fluorescent secondary antibody (1:100, Abbkine, Wuhan, China) for 1 h at 37°C in the dark. After washing three times, the cells were stained with DAPI (Beyotime, Beijing, China) for 5 min at 37°C and visualized using a fluorescence microscope.
Luciferase Reporter Assay
The luciferase activity assay was performed as previously described.18 Wild-type ATG14 (ATG14-wt) and a mutant devoid of the miR-375 binding site (ATG14-mut) were cloned downstream of the luciferase gene coding sequence. Cells were co-transfected with 100 ng of ATG14-miR-375-UTR-WT or ATG14-miR-375-UTR-Mut in the presence of 50 nM Lipofectamine2000. After 48 h, cells were assayed using a Dual Luciferase Reporter Assay Kit (Promega) according to the manufacturer’s instructions. Firefly luciferase activity was normalized to Renilla luciferase activity.
Transmission Electron Microscopy
Cells were fixed in 0.2% glutaraldehyde for 2 h at 37°C, then post-fixed in 0.1 M sodium cacodylate buffer. The cells were dehydrated using an ethanol gradient, then embedded in Durcupan ACM. Then, the cells were sliced into 80-nm sections and stained with uranyl acetate and lead citrate. The cells were visualized using a transmission electron microscope.
Statistical Analysis
All analyses were performed using SPSS 17.0 (SPSS, Chicago, IL, USA) and GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA). Statistical analysis was performed using Student’s t-test or one-way ANOVA. Spearman correlation was used to evaluate expression levels. Data are presented as means ± SD, and p values <0.05 were considered statistically significant.
Results
Sorafenib Promoted Autophagy in HCC
Autophagy has been shown to be associated with cancer progression,19 and increasing evidence has shown that autophagy can suppress or promote the growth of cancer cells.20,21 Stress-induced autophagy can promote cell survival, but excessive autophagy can result in apoptosis.22 Many factors contribute to drug resistance in HCC, including autophagy. We used a geodatabase (GSE10921123) and gene-set enrichment analysis to verify the relationship between sorafenib and autophagy. Using R software, we concluded that autophagy and sorafenib use were highly correlated in patients with HCC (Figure 1A). Studies have shown that microtubule-associated protein 1 light chain 3 (MAP1LC3/LC3) is a specific marker of autophagy initiation. In addition, p62 (a bridge between LC3B and ubiquitinated substrates to be degraded) also serves as a marker of autophagy induction. Autophagy is often detected using MAP1LC3/LC3 and P62 as markers. We determined that Huh7 cells were the most sensitive HCC cells to sorafenib (Figure S1A). HepG2 cells were used to verify conclusions made using Huh7 cells. Levels of LC3-II were significantly higher after 24 h of treatment with different concentrations of sorafenib than those in control cells, and p62 levels were lower in cells treated with sorafenib than those in control cells (Figure 1B). The expression of LC3-II was further increased and the expression of P62 was further decreased in cells treated with bafilomycin A1, which suggested that autophagic flux was increased in Huh7 cells treated with sorafenib (Figure 1C). Three principal methods are typically used to monitor the number of autophagosomes, including electron microscopy, light microscopic determination of subcellular localization of LC3, and biochemical detection of the membrane-associated form of LC3. The proportion of LC3 puncta in cells treated with sorafenib was significantly higher than that in the control group (Figure 1D). In addition, we observed characteristic autophagosomes in Huh7 cells treated with sorafenib using transmission electron microscopy (Figure 1E). These results indicated that sorafenib treatment promoted autophagy, and these results were verified in HepG2 cells (Figure S1B and C).
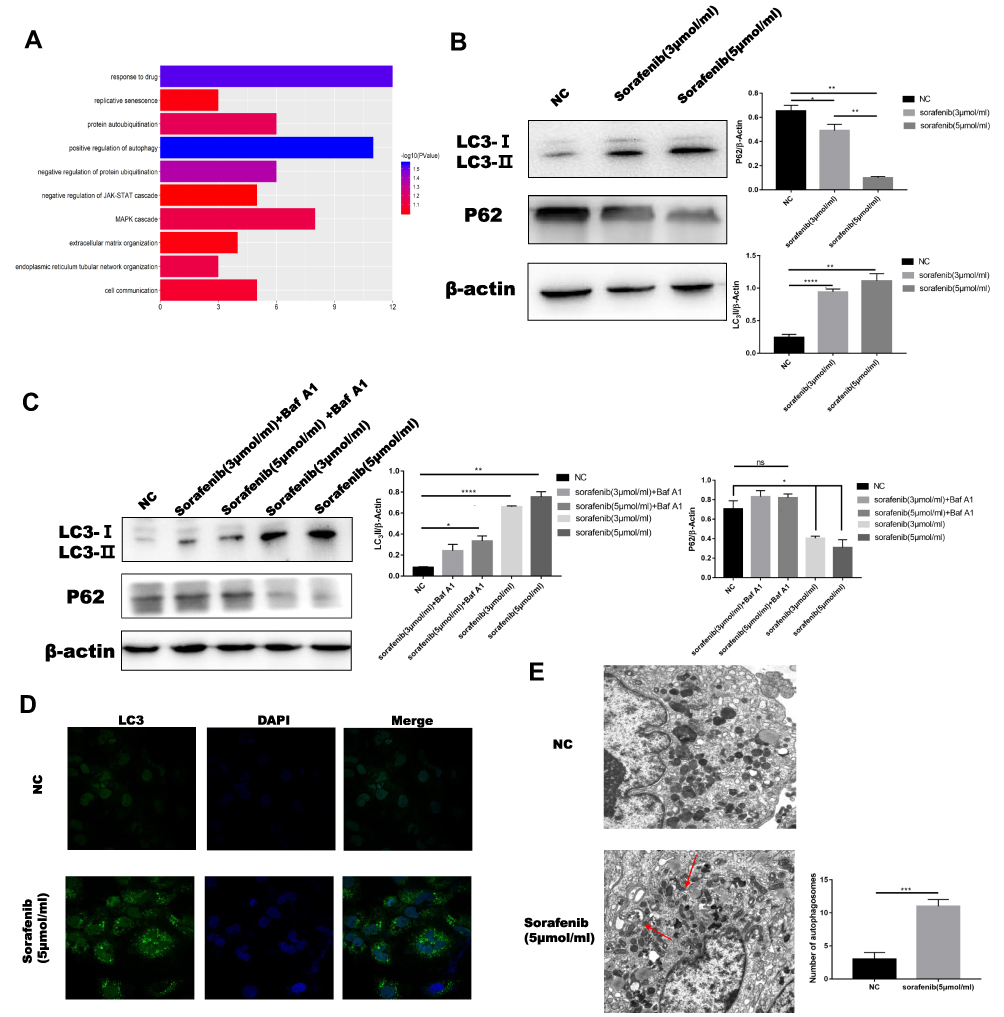 |
Figure 1 Autophagy in HCC was increased by treatment with sorafenib. (A) The positive correlation between autophagy in patients with HCC and the use of sorafenib was determined using R software. (B) Western blotting was performed to determine levels of autophagy-related proteins in HCC cells treated with increasing concentrations of sorafenib. (C) Western blotting was performed to measure the levels of autophagy-related proteins in HCC cells treated with different concentrations of sorafenib, with or without Baf-A1. (D) The effect of sorafenib on relative green fluorescence of LC3 puncta in Huh7 cells was evaluated using immunofluorescence. (E) The effect of sorafenib on a relative number of autophagosomes in Huh7 cells was evaluated using electron microscopy. Red arrows indicate autophagic double membrane structure. Data are expressed as mean ± SD of three independent experiments. The p-values represent comparisons between groups (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns means no Statistical significance). |
Inhibition of Autophagy Enhanced the Sensitivity of HCC Cells to Sorafenib
To determine whether autophagy modulated the effects of sorafenib on HCC cells, we used an autophagy inhibitor (Baf-A1) and small interfering RNA (siRNA). Unk-51-like kinase (ULK) is a key gene in autophagy. Studies have shown that si-ULK inhibited autophagy.24 Cell viability was significantly decreased in a sorafenib dose-dependent manner in cells treated with Baf-A1 and si-ULK (Figure 2A and B). Inhibition of autophagy in HepG2 cells resulted in increased cell death (Figure S2A). In addition, treatment with Baf-A1 and si-ULK, with or without sorafenib, resulted in a decreased capacity for colony formation, and increased apoptosis (Figure 2C and D). These results indicated that inhibition of autophagy increased the effects of sorafenib on HCC.
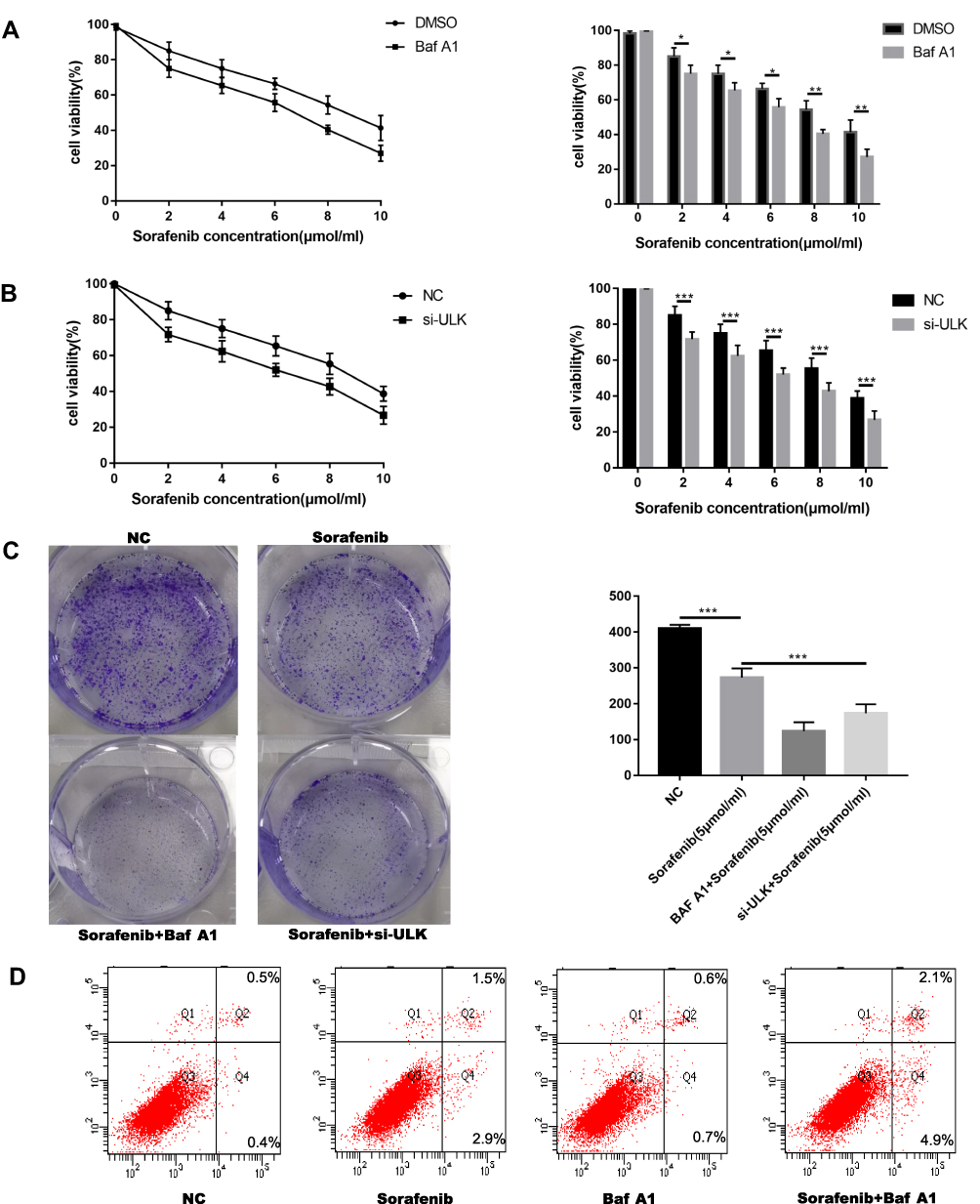 |
Figure 2 Inhibition of autophagy enhanced the sensitivity of HCC cells to sorafenib. (A, B) The MTT assay was used to measure the viability of Huh7 cells treated with sorafenib following treatment with Baf-A1 and si-ULK1. (C) Colony formation assay was performed to analyze cell proliferation following treatment with Baf-A1 and si-ULK1 (sorafenib: 5 μM, 12 h). (D) Flow cytometry was performed to measure apoptosis (sorafenib: 5 μM, 12 h). Data are expressed as mean ± SD of three independent experiments. The p-values represent comparisons between groups (*P < 0.05, **P < 0.01, ***P < 0.001). |
MicroRNA 375 Inhibited Sorafenib-Induced Autophagy
Our previous studies evaluated the functions and effects of non-coding RNA.25,26 MicroRNAs have the potential to revolutionize diagnosis, treatment, and monitoring of diseases. Previous studies have shown that miRNAs are involved in various aspects of autophagy.27–30 Furthermore, studies have shown that miRNAs contribute to tumorigenesis through regulation of cell proliferation, differentiation, and invasion.31,32 We used the GEO database, GSE20077, to identify aberrantly expressed miRNAs involved in HCC using comparative miRNA expression profiling of cancerous hepatocytes and normal primary human hepatocytes. This analysis resulted in identification of 37 dysregulated miRNAs in HCC.33 The GSE109211 dataset is comprised of a total of 67 samples from individuals treated with sorafenib (Sor) and 73 from patients who received placebo (Plac). We used R software to show the expression of several miRNAs by selecting the miRNA data and selecting the intersection between them (Figure 3A). Analysis of these two sets of data resulted in the selection of five microRNAs (hsa-miR-375, hsa-miR-146b, hsa-miR-338, hsa-miR-221, and hsa-miR-99b), with the most obvious differentiation and statistically significant to perform RT-PCR experiments under sorafenib treatment. We selected miR-375 for subsequent studies because it was most associated with sorafenib-treated HCC cells (Figure 3B). Analysis of TCGA using GSEA resulted in confirmation of a correlation between miR-375 and autophagy (Figure 3C). To verify the role of miR-375 in sorafenib-induced autophagy, we used Huh7 cells transiently transfected with miR-375 or miR-NC mimics. The results showed that cells transfected with miR-375 mimic and treated with sorafenib exhibited reduced LC3II expression and increased P62 expression (Figure 3D). In addition, we also found that miR-375 mimics inhibited sorafenib-induced autophagy, as determined using immunofluorescence and electron microscopy. The numbers of sorafenib-induced LC3 puncta or autophagic vacuoles, as determined using immunofluorescence or electron microscopy, were decreased following treatment with miR-375 mimics (Figure 3E and F). These results suggested that miR-375 negatively regulated sorafenib-induced autophagy in HCC. These results were verified in HepG2 cells (Figure S2C).
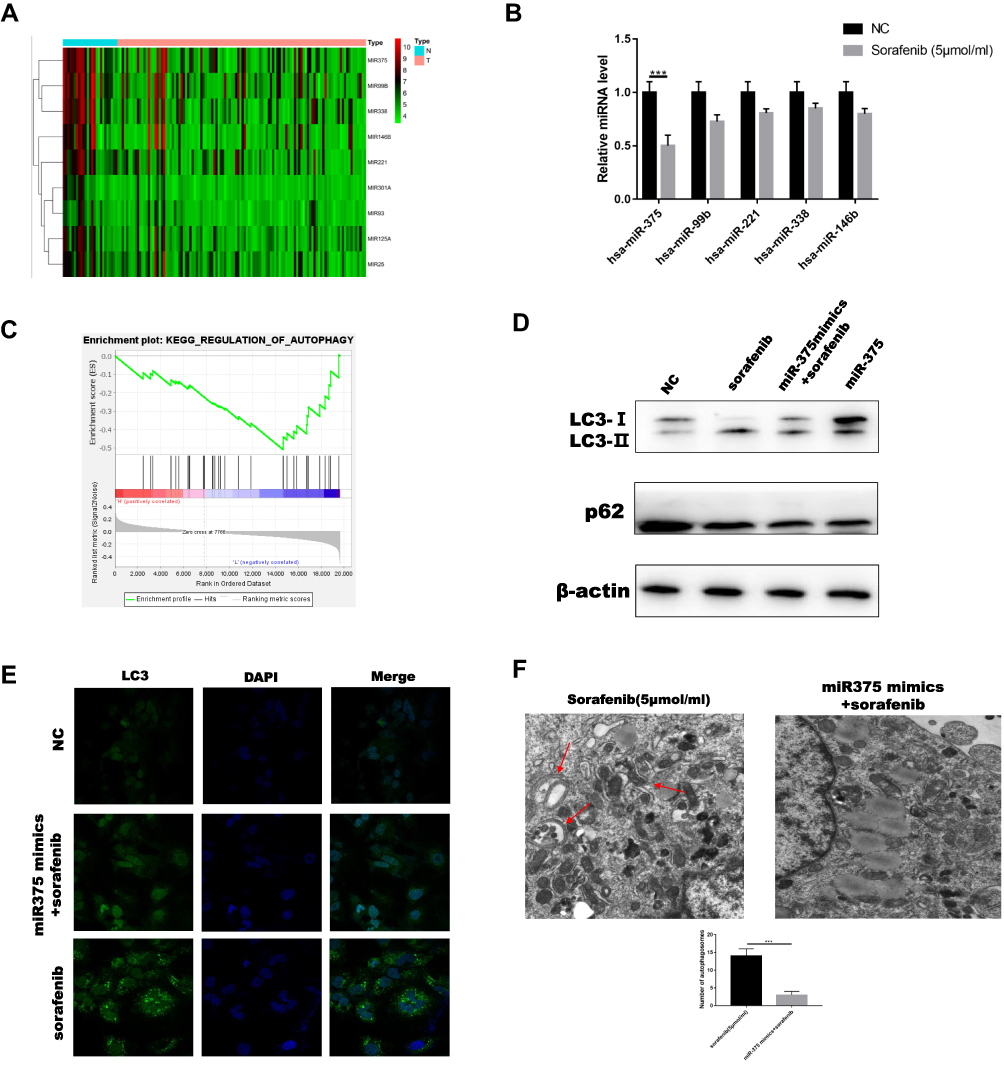 |
Figure 3 MicroRNA 375 inhibited sorafenib-induced autophagy. (A) R software was used to generate a heat map of miRNA expression following screening. (B) Real-time PCR was used to quantitate miRNA levels in Huh7 cells treated with sorafenib. (C) Results from GSEA showed that miR-375 was expressed at low levels in HCC, and was negatively correlated with autophagy. (D) LC3 and p62 levels were measured in cells treated with miR-375 mimics following sorafenib treatment (sorafenib: 5 μM, 12 h). (E) The levels of sorafenib-induced LC3 puncta were evaluated using immunofluorescence following treatment with miR-375 mimics. (F) The number of sorafenib-induced autophagic vacuoles was determined using electron microscopy following treatment with miR-375 mimics. Red arrows indicate autophagic double membrane structure. Data are expressed as mean ± SD of three independent experiments. The p-values represent comparisons between groups (***P < 0.001). |
Increased Expression of miR-375 Re-Sensitized HCC Cells to Sorafenib
Autophagy is associated with sensitivity to many drugs.34,35 Therefore, we evaluated the mechanisms by which miR-375 modulated the effects of sorafenib on HCC. The effects of miR-375 mimics on the relative viability of Huh7 cells in response to treatment with different concentrations of sorafenib were evaluated using the MTT assay, and transfection with miR-375 mimics and inhibitors had contrasting effects (Figure 4A and B). In addition, colony growth was significantly restricted in the miR-375 mimic group treated with sorafenib (Figure 4C). Flow cytometry analysis showed that miR-375 re-expression resulted in increased apoptosis in a sorafenib dose-dependent manner in Huh7 cells (Figure 4D). In addition, we showed that miR-375 re-expression increased the protein expression levels of caspase-3 in sorafenib-treated Huh7 cells compared with those in control cells. In addition, transfection of miR-375 mimic significantly decreased the expression of VEGF in sorafenib-treated Huh7 cells compared with control cells (Figure 4E). This can explain the effect of miR-375 may play an important role in sorafenib of HCC treatment. These results indicated that miR-375 enhanced the sensitivity of HCC to sorafenib.
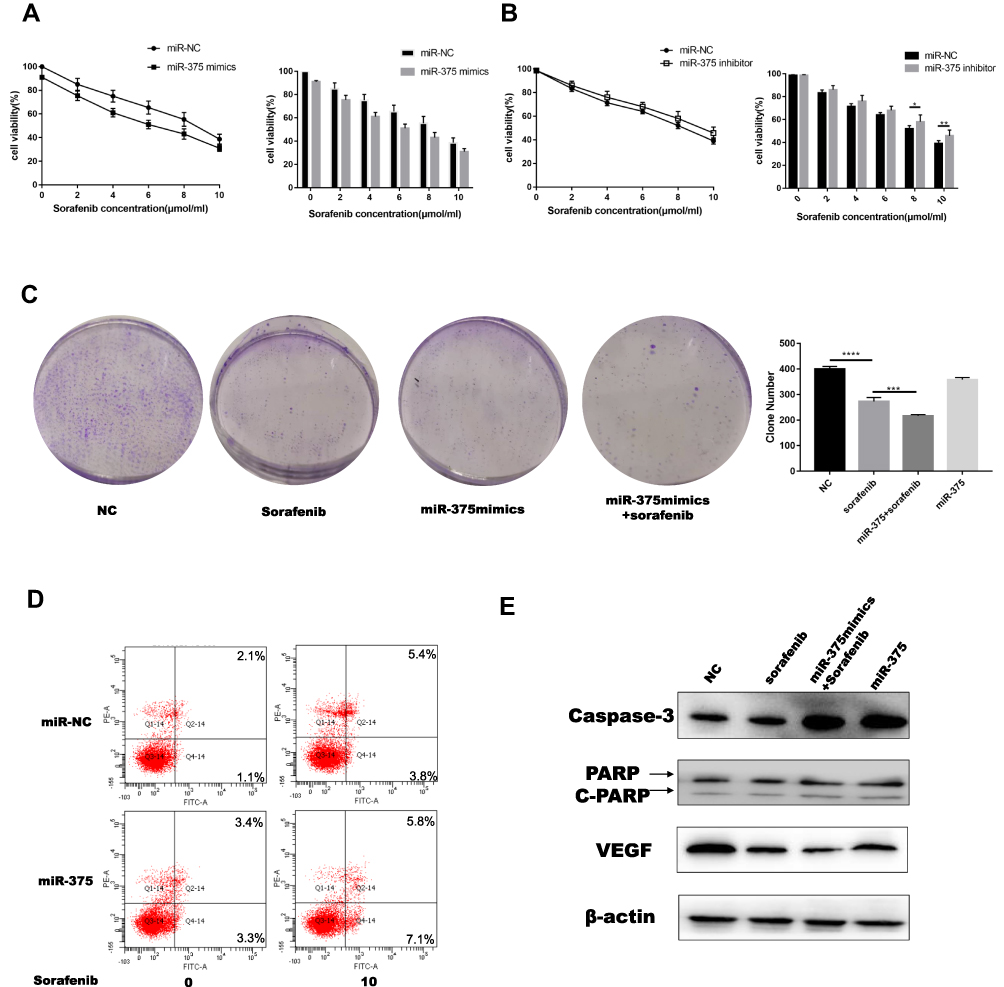 |
Figure 4 Increased expression of miR-375 re-sensitized HCC cells to sorafenib. (A, B) The influence of miR-375 mimics or inhibitors on the relative viability of Huh7 cells in response to different concentrations of sorafenib was evaluated using the MTT assay. (C) Colony formation assay was performed to analyze the effects of miR-375 mimics on cell proliferation following sorafenib treatment (sorafenib: 5 μM, 12 h). (D) Flow cytometry was performed to measure apoptosis (sorafenib: 10 μM, 12 h). (E) Western blotting was used to measure apoptosis-related protein levels. Data are expressed as mean ± SD of three independent experiments. The p-values represent comparisons between groups (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns means no Statistical significance). |
ATG14 Was a Direct Target of miR-375
To determine the mechanism by which miR-375 inhibited autophagosome formation, we used two online bioinformatics websites (Starbase and TargetScan) to identify binding sites between miR-375 and the 3ʹ-UTR regions of ATG2B, ATG14, ATG7, and ATG10. The results showed that the expression of ATG14 was highest in sorafenib-treated HCC cells, as determined using RT-PCR (Figure 5A). We then constructed GFP-RFP-LC3 lentivirus that produced red light in response to sorafenib treatment (Figure 5B). Green light was quenched by acid in lysosomes, which indicated that autophagy occurs in the fusion phase with lysosome. One study showed that ATG14 promoted membrane tethering and fusion of autophagosomes to endolysosomes.36 This finding agreed with our finding that ATG14 was involved in sorafenib-induced autophagy. The predicted interactions between miR-375 and the 3′-UTRs of ATG14 are shown in Figure 5C. We constructed 3′-UTR luciferase reporters of ATG14 that contained putative miR-375 binding sites or mutant binding sites downstream of the luciferase reporters to characterize ATG14 as a target gene of miR-375. We then co-transfected Huh7 cells with the reporter constructs and a miR-375-mimic or miR-NC-mimic. We performed luciferase activity assay 48 h after transfection. Luciferase reporter assay showed a binding affinity between miR-375 and the 3′-UTR of ATG14 mRNA (Figure 5D). Transfection of a miR-375 mimic decreased the expression of ATG14 mRNA (Figure 5E) and protein (Figure 5F) in Huh7 cells treated with or without sorafenib. These results showed that miR-375 downregulated ATG14 mRNA by directly interacting with 3′-UTRs.
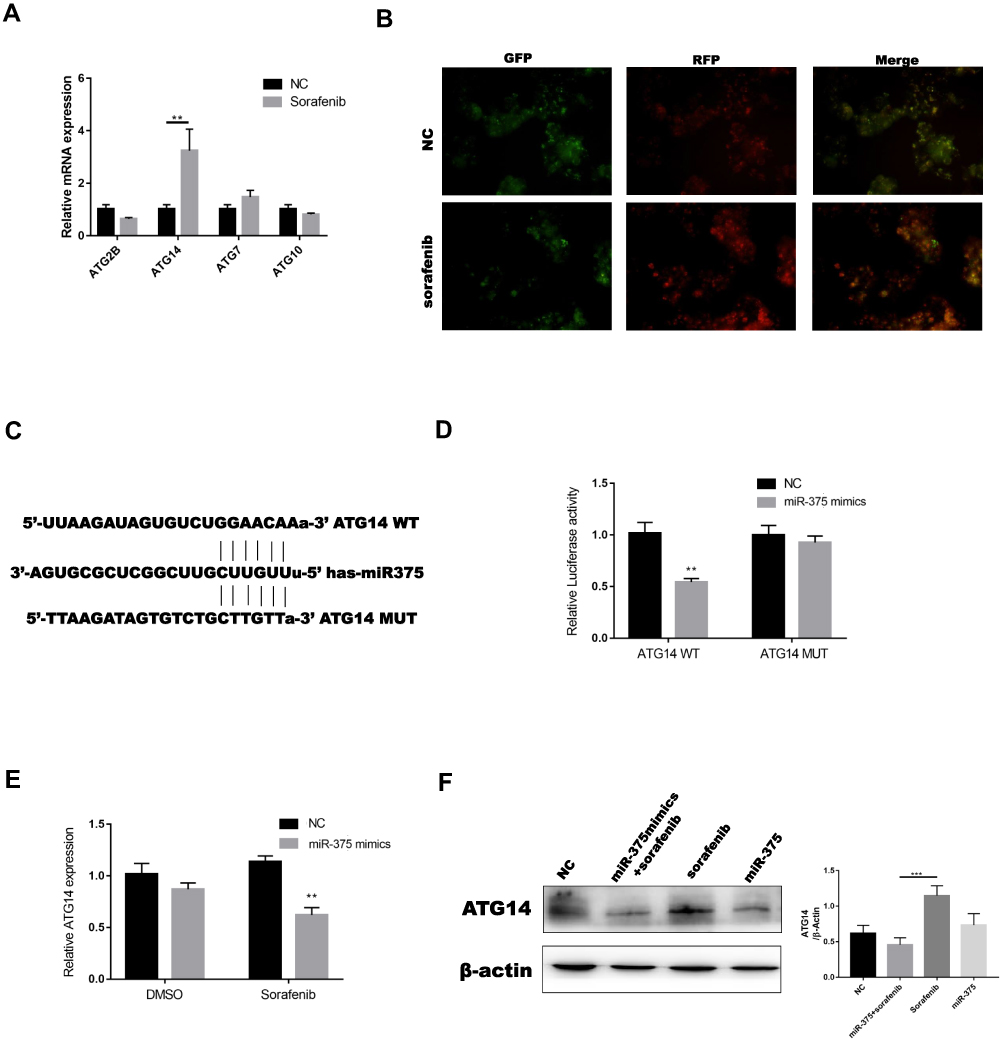 |
Figure 5 Autophagy-related protein 14 was a direct target of miR-375. (A) Real-time PCR was used to evaluate ATG14 levels in Huh7 cells treated with sorafenib. (B) We constructed a GFP-RFP-LC3 lentivirus that emitted green and red light in response to sorafenib treatment (sorafenib: 5 μM, 12 h). (C, D) The binding sites for miR-375 in the 3ʹ-UTRs of ATG14 were determined, and luciferase reporter assays were performed to confirm the relationships between miR-375 and ATG14. (E) Real-time PCR was used to measure ATG14 mRNA levels in Huh7 cells transfected with miR-375 mimics and treated with or without sorafenib (sorafenib: 5 μM, 12 h). (F) Western blotting was used to measure ATG14 protein levels in Huh7 cells transfected with miR-375 mimics and treated with or without sorafenib (sorafenib: 5 μM, 12 h). Data are expressed as mean ± SD of three independent experiments. The p-values represent comparisons between groups (**P < 0.01, ***P < 0.001). |
The Effect of miR-375 on Sensitivity of HCC Cells to Sorafenib Was Dependent on ATG14 Regulation
To confirm that miR-375-mediated downregulation of ATG14 resulted in sensitivity of HCC cells to sorafenib, we co-cultured miR-375 mimic with an ATG14 over-expressor plasmid. Reintroduction of ATG14 in the presence of miR-375 reversed the inhibition of Huh7 cell growth induced by the combination of miR-375 and sorafenib (Figure 6A). In addition, colony formation assay also showed that ATG14 reduced miR-375-induced apoptosis (Figure 6B). Furthermore, flow cytometry showed that increased miR-375-mediated apoptosis was partially abolished by ATG14 in HCC cells treated with sorafenib (Figure 6C). Western blot results showed that increased expression of apoptosis-related proteins (c-caspase-3 and c-PARP) induced by miR-375 was partially abrogated by increased expression of ATG14. Simultaneously, compared with group miR-NC and group Sorafenib in Figure 4D, the expression of autophagy was partially reduced when ATG14 was added (Figure 6D). In addition, the expression of VEGF was partially increased when ATG14 was added compared with control cells. This may indicate a link between ATG14, miR-375 and sorafenib. These results indicated that miR-375-induced sensitivity of HCC cells to sorafenib was dependent on ATG14 regulation. This result was verified in HepG2 cells (Figures S2B and S3).
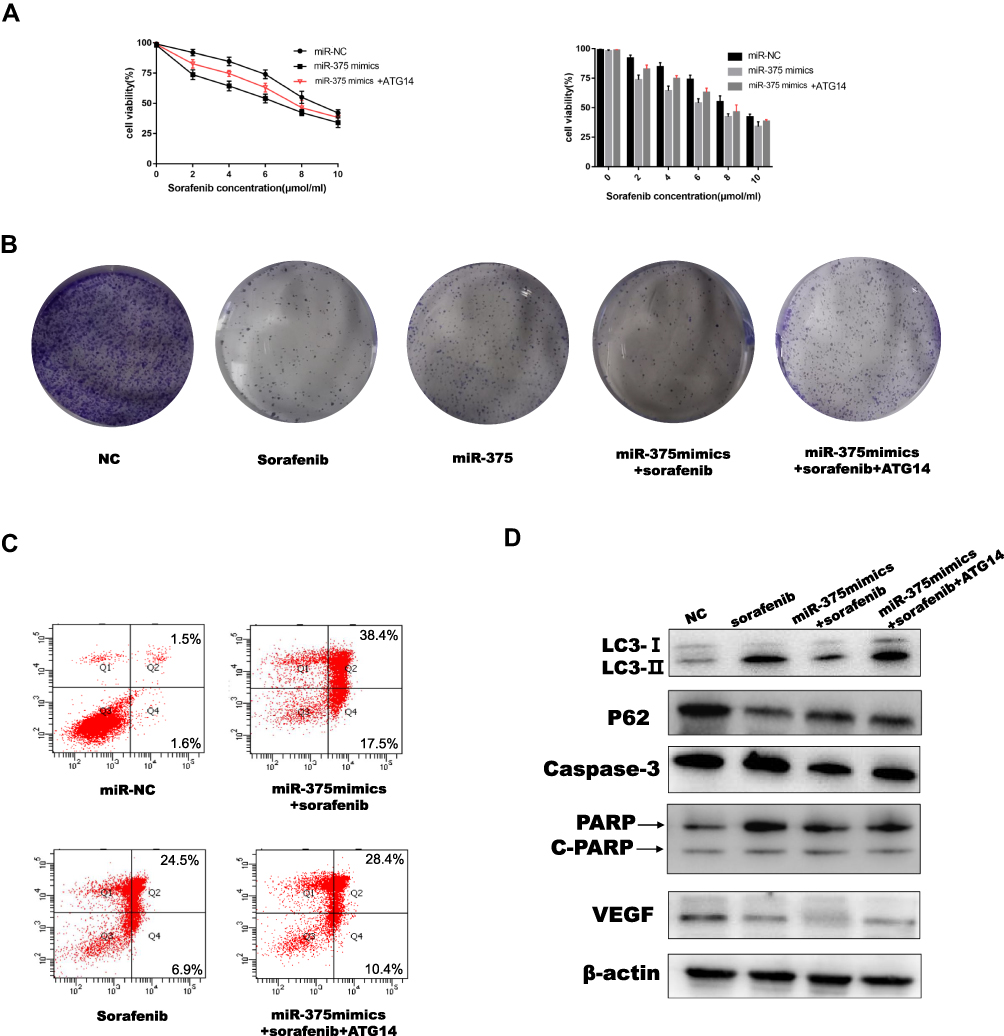 |
Figure 6 MicroRNA 375 affected sensitivity of HCC cells to sorafenib through ATG14. (A) The sensitivity of Huh7 cells co-transfected with miR-375 mimics and ATG14 to sorafenib was determined using the MTT assay. (B) Colony formation assay was performed to determine proliferation ability of Huh7 cells co-transfected with miR-375 mimics and ATG14, then treated with sorafenib. (C) Flow cytometry was used to measure sorafenib-induced apoptosis in Huh7 cells co-transfected with miR-375 mimics and ATG14 (sorafenib: 10 μM, 12 h). (D) Western blotting was used to determine the levels of the apoptosis-related proteins and autophagy-related proteins in Huh7 cells co-transfected with miR-375 mimics and ATG14 and treated with sorafenib (sorafenib: 5 μM, 12 h). |
The Expression of miR-375 Was Negatively Correlated with ATG14 Expression
Studies have shown that the expression of miR-375 was significantly reduced in liver cancer tissues.28 The GSE20077 dataset was analyzed to identify aberrantly expressed miRNAs involved in HCC through comparison of miRNA expression profiles in cancerous hepatocytes to those in normal primary human hepatocytes. This analysis resulted in the identification of 37 dysregulated miRNAs in HCC. The expression profile of miR-375 in the GEO database GSE20077 is shown in Figure 7A. The expression level of miR-375 in HCC tissues was significantly lower than that in normal tissues. The expression levels of ATG14 in normal or HCC tissues and matched TCGA normal and GTEx data are shown in Figure 7B. Data sources are referenced in GEPIA (http://gepia.cancer-pku.cn/index.html). The expression level of ATG14 in HCC tissues was higher than that in normal tissues. Correlation analysis showed that miR-375 and ATG14 expression was negatively correlated, as determined using Starbase (Figure 7C). We also investigated the relationship between miR-375 and ATG14 expression, and prognosis in patients with HCC. Kaplan-Meier survival analysis showed that the overall survival time of patients with high ATG14 expression was significantly shorter than that of patients with low ATG14 expression (Figure 7D). Data sources are referenced in OncoLnc (http://www.oncolnc.org/). Both of them are clinically significant and worth discussing. Collectively, this section is intended to illustrate the correlation between miR-375 and ATG14. Figure 7E shows the miR-375/ATG14 schematic overview of regulatory signal.
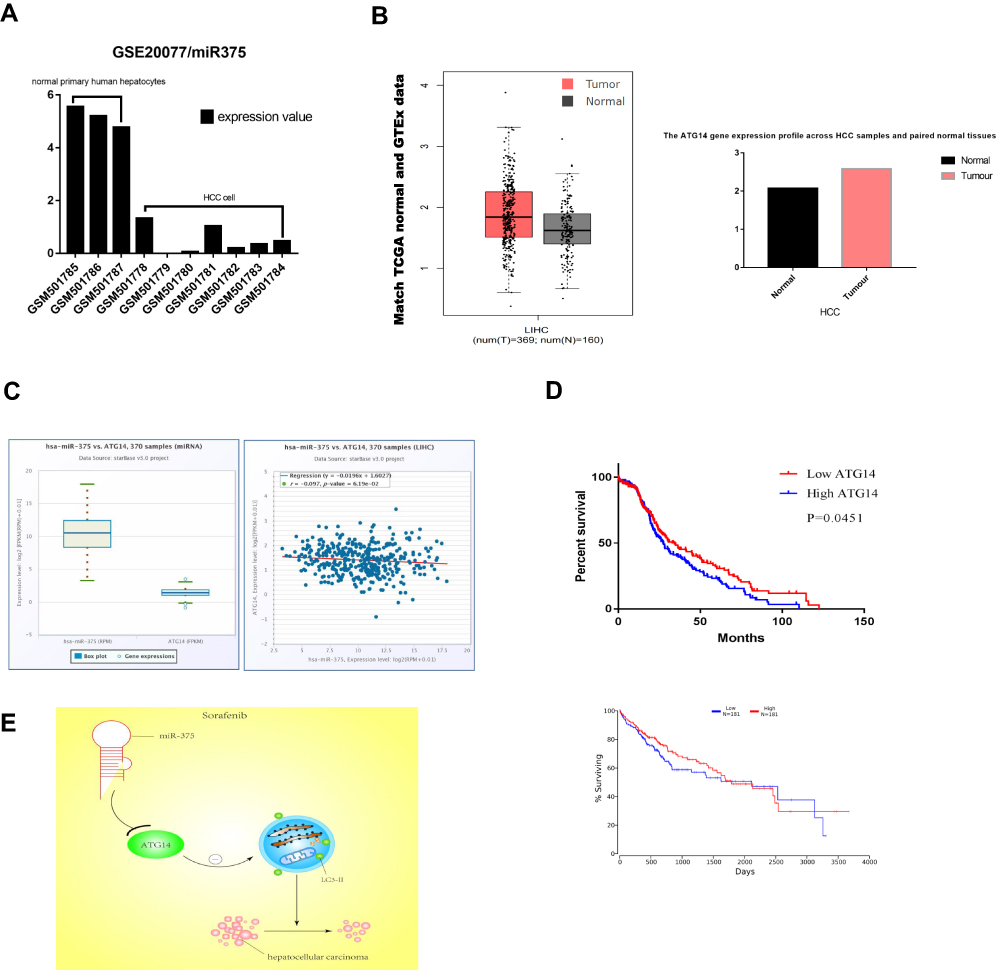 |
Figure 7 The expression of miR-375 in HCC tissues was negatively correlated with ATG14 expression. (A) The expression profile of miR-375 in the GEO database, GSE20077. (B) The expression of ATG14 in normal and HCC tissues and matched TCGA normal and GTEx data. Data sources are referenced in GEPIA (http://gepia.cancer-pku.cn/index.html). (C) The correlation between miR-375 and ATG14 in HCC obtained from Starbase. (D) Analysis of the correction among ATG14, miR-375, and survival probability in HCC. (E) Schematic overview of miR-375/ATG14 regulatory signalling. |
Discussion
The most effective current treatments for HCC are surgical resection, interventional radiotherapy, or liver transplantation.37 Sorafenib was the first systemic therapy approved for patients with advanced-stage HCC, following a landmark study that showed an improvement in median overall survival time from 8 to 11 months.5 However, the long-term value of sorafenib is limited due to primary and acquired resistance, which are related to activation of autophagy.14,38 Many studies have described abnormalities in autophagy in many human tumors. Autophagy plays a critical role in all stages of tumor development. Furthermore, autophagy plays dual roles in HCC, in which it protects cells from carcinogenesis during the early stages, and promotes tumor progression at advanced stages.39–41 These dual roles illustrate the complexity of targeting autophagy to treat HCC. Autophagy-related genes, non-coding RNA, and related signaling pathways, are involved in autophagy and the regulation of onset and progression of HCC. Yang et al showed that HOTAIR increased autophagy by increasing the expression levels of ATG3 and ATG7.42 MiR-375 is downregulated in HCC. It can decrease HCC cell growth, invasion, and apoptosis. What is more, there is a study showed that miR-375 inhibits tumor growth of hepatoma xenografts in nude mice.33 On this basis, we focus on its ability to improve the efficacy of sorafenib, and try to contribute and explore in terms of drug resistance. Autophagy and microRNAs are important regulators of cancer cell tumorigenesis, and microRNAs have potential as targets for the treatment of cancer.43,44
In this study, we evaluated the role of miRNA in autophagy-mediated regulation of sorafenib resistance in HCC cells. First, we showed that sorafenib promoted autophagy. In addition, we used RT-PCR to verify that the expression of multiple miRNAs differed in response to treatment with sorafenib, and identified miR-375, which was abnormally expressed in numerous types of cancers, as the focus of this study.45–47 The results showed that miR-375 attenuated sorafenib-induced autophagy and increased sensitivity of HCC to sorafenib. We demonstrated that miR-375 acted on target sequences located in the 3ʹ-UTR regions of ATG14 mRNA. Finally, we showed that miR-375 regulated sorafenib-induced autophagy through ATG14. Previous studies have shown that ATG14 participated in autophagosome nucleation and promoted fusion of autophagosomes and lysosomes.36,48,49 Furthermore, ATG14 may play a fundamental role in vesicle nucleation of the phosphatidylinositol (PtdIns) 3-kinase complex I.50
In conclusion, miR-375 inhibited sorafenib-induced autophagy in HCC. Furthermore, miR-375 enhanced the sensitivity of HCC to sorafenib. These results provided new insights into the mechanism of survival of HCC cells following sorafenib treatment. Further studies are needed to explore additional mechanisms of action of miR-375 and to elucidate the relationship between autophagy and other drug mechanisms.
Abbreviations
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PVDF, Poly(vinylidene fluoride); LC3, Microtubule associated protein light chain 3; P62, SQSTM1 (Sequestosome 1); PARP, poly ADP-ribose polymerase; C-PARP, c-poly ADP-ribose polymerase; TBST, Tris-Buffered Saline Tween; PBS, Phosphate buffered saline; DMSO, dimethyl sulfoxide; DAPI, 4ʹ,6-diamidino-2-phenylindole.
Funding
This study was supported by grants from the National Natural Science Foundation of China (No.81472302/No.81572425/No.81871983).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
2. Bertuccio P, Turati F, Carioli G, et al. Global trends and predictions in hepatocellular carcinoma mortality. J Hepatol. 2017;67(2):302–309. doi:10.1016/j.jhep.2017.03.011
3. Portolani N, Coniglio A, Ghidoni S, et al. Early and late recurrence after liver resection for hepatocellular carcinoma: prognostic and therapeutic implications. Ann Surg. 2006;243(2):229–235. doi:10.1097/01.sla.0000197706.21803.a1
4. Asghar U, Meyer T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? J Hepatol. 2012;56(3):686–695. doi:10.1016/j.jhep.2011.07.031
5. Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol. 2018;15(10):599–616. doi:10.1038/s41571-018-0073-4
6. Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9(10):1102–1109. doi:10.1038/ncb1007-1102
7. Allaire M, Rautou PE, Codogno P, Lotersztajn S. Autophagy in liver diseases: time for translation? J Hepatol. 2019;70(5):985–998. doi:10.1016/j.jhep.2019.01.026
8. White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12(6):401–410. doi:10.1038/nrc3262
9. Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19(8):983–997. doi:10.1038/nm.3232
10. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(19):1845–1846. doi:10.1056/NEJMra1205406
11. Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099–7109. doi:10.1158/0008-5472.CAN-04-1443
12. Wang H, Wang H, Yu Z, Liu H. Alternative treatment strategies to sorafenib in patients with advanced hepatocellular carcinoma: a meta-analysis of randomized Phase III trials. Onco Targets Ther. 2018;11:5195–5201. doi:10.2147/OTT.S171918
13. Fischer TD, Wang JH, Vlada A, Kim JS, Behrns KE. Role of autophagy in differential sensitivity of hepatocarcinoma cells to sorafenib. World J Hepatol. 2014;6(10):752–758. doi:10.4254/wjh.v6.i10.752
14. Chen KF, Chen HL, Tai WT, et al. Activation of phosphatidylinositol 3-kinase/Akt signaling pathway mediates acquired resistance to sorafenib in hepatocellular carcinoma cells. J Pharmacol Exp Ther. 2011;337(1):155–161. doi:10.1124/jpet.110.175786
15. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi:10.1016/j.cell.2009.01.002
16. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi:10.1016/S0092-8674(04)00045-5
17. Zhang J, Wang P, Wan L, Xu S, Pang D. The emergence of noncoding RNAs as Heracles in autophagy. Autophagy. 2017;13(6):1004–1024. doi:10.1080/15548627.2017.1312041
18. Li H, Li Y, Liu D, Sun H, Liu J. miR-224 is critical for celastrol-induced inhibition of migration and invasion of hepatocellular carcinoma cells. Cell Physiol Biochem. 2013;32(2):448–458. doi:10.1159/000354450
19. Lee YJ, Jang BK. The role of autophagy in hepatocellular carcinoma. Int J Mol Sci. 2015;16(11):26629–26643. doi:10.3390/ijms161125984
20. He C, Bassik MC, Moresi V, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481(7382):511–515. doi:10.1038/nature10758
21. Katheder NS, Khezri R, O’Farrell F, et al. Microenvironmental autophagy promotes tumour growth. Nature. 2017;541(7637):417–420. doi:10.1038/nature20815
22. Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–293. doi:10.1016/j.molcel.2010.09.023
23. Pinyol R, Montal R, Bassaganyas L, et al. Molecular predictors of prevention of recurrence in HCC with sorafenib as adjuvant treatment and prognostic factors in the Phase 3 STORM trial. Gut. 2019;68(6):1065–1075. doi:10.1136/gutjnl-2018-316408
24. Nazio F, Cecconi F. Autophagy up and down by outsmarting the incredible ULK. Autophagy. 2017;13(5):967–968. doi:10.1080/15548627.2017.1285473
25. Li X, Zhou Y, Yang L, et al. LncRNA NEAT1 promotes autophagy via regulating miR-204/ATG3 and enhanced cell resistance to sorafenib in hepatocellular carcinoma. J Cell Physiol. 2019.
26. Yang L, Peng X, Li Y, et al. Long non-coding RNA HOTAIR promotes exosome secretion by regulating RAB35 and SNAP23 in hepatocellular carcinoma. Mol Cancer. 2019;18(1):78. doi:10.1186/s12943-019-0990-6
27. Hall DP, Cost NG, Hegde S, et al. TRPM3 and miR-204 establish a regulatory circuit that controls oncogenic autophagy in clear cell renal cell carcinoma. Cancer Cell. 2014;26(5):738–753. doi:10.1016/j.ccell.2014.09.015
28. Chang Y, Yan W, He X, et al. miR-375 inhibits autophagy and reduces viability of hepatocellular carcinoma cells under hypoxic conditions. Gastroenterology. 2012;143(1):177–187 e178. doi:10.103/j.gastro.2012.04.009
29. Zhang H, Zhang Y, Zhu X, et al. DEAD box protein 5 inhibits liver tumorigenesis by stimulating autophagy via interaction with p62/SQSTM1. Hepatology. 2019;69(3):1046–1063. doi:10.1002/hep.30300
30. Yang S, Yang L, Li X, et al. New insights into autophagy in hepatocellular carcinoma: mechanisms and therapeutic strategies. Am J Cancer Res. 2019;9(7):1329–1353.
31. Ladeiro Y, Couchy G, Balabaud C, et al. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology. 2008;47(6):1955–1963. doi:10.1002/hep.22256
32. Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10(10):704–714. doi:10.1038/nrg2634
33. He XX, Chang Y, Meng FY, et al. MicroRNA-375 targets AEG-1 in hepatocellular carcinoma and suppresses liver cancer cell growth in vitro and in vivo. Oncogene. 2012;31(28):3357–3369. doi:10.1038/onc.2011.500
34. Li YJ, Lei YH, Yao N, et al. Autophagy and multidrug resistance in cancer. Chin J Cancer. 2017;36(1):52. doi:10.1186/s40880-017-0219-2
35. An Y, Zhang Z, Shang Y, et al. miR-23b-3p regulates the chemoresistance of gastric cancer cells by targeting ATG12 and HMGB2. Cell Death Dis. 2015;6:e1766. doi:10.1038/cddis.2015.123
36. Diao J, Liu R, Rong Y, et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature. 2015;520(7548):563–566. doi:10.1038/nature14147
37. Villanueva A, Hernandez-Gea V, Llovet JM. Medical therapies for hepatocellular carcinoma: a critical view of the evidence. NAT REV GASTRO HEPAT. 2013;10(1):34–42. doi:10.1038/nrgastro.2012.199
38. Amaravadi RK, Lippincott-Schwartz J, Yin XM, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clinical Cancer Res. 2011;17(4):654–666. doi:10.1158/1078-0432.CCR-10-2634
39. Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169(3):425–434. doi:10.1083/jcb.200412022
40. Ni HM, Woolbright BL, Williams J, et al. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J Hepatol. 2014;61(3):617–625. doi:10.1016/j.jhep.2014.04.043
41. Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. doi:10.1016/j.ccr.2006.06.001
42. Yang L, Zhang X, Li H, Liu J. The long noncoding RNA HOTAIR activates autophagy by upregulating ATG3 and ATG7 in hepatocellular carcinoma. Mol Biosyst. 2016;12(8):2605–2612. doi:10.1039/C6MB00114A
43. Ren Y, Chen Y, Liang X, Lu Y, Pan W, Yang M. MiRNA-638 promotes autophagy and malignant phenotypes of cancer cells via directly suppressing DACT3. Cancer Lett. 2017;390:126–136. doi:10.1016/j.canlet.2017.01.009
44. Kong P, Zhu X, Geng Q, et al. The microRNA-423-3p-bim axis promotes cancer progression and activates oncogenic autophagy in gastric cancer. Mol Ther. 2017;25(4):1027–1037. doi:10.1016/j.ymthe.2017.01.013
45. Tsukamoto Y, Nakada C, Noguchi T, et al. MicroRNA-375 is downregulated in gastric carcinomas and regulates cell survival by targeting PDK1 and 14-3-3zeta. Cancer Res. 2010;70(6):2339–2349. doi:10.1158/0008-5472.CAN-09-2777
46. Shen Y, Wang P, Li Y, et al. miR-375 is upregulated in acquired paclitaxel resistance in cervical cancer. Br J Cancer. 2013;109(1):92–99. doi:10.1038/bjc.2013.308
47. Yan JW, Lin JS, He XX. The emerging role of miR-375 in cancer. Int j Cancer. 2014;135(5):1011–1018. doi:10.1002/ijc.28563
48. He C, Wei Y, Sun K, et al. Beclin 2 functions in autophagy, degradation of G protein-coupled receptors, and metabolism. Cell. 2013;154(5):1085–1099. doi:10.1016/j.cell.2013.07.035
49. Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6(4):463–477. doi:10.1016/S1534-5807(04)00099-1
50. Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152(3):519–530. doi:10.1083/jcb.152.3.519
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.