")
Back to Journals » OncoTargets and Therapy » Volume 10
MicroRNA-320 inhibits invasion and induces apoptosis by targeting CRKL and inhibiting ERK and AKT signaling in gastric cancer cells
Authors Zhao Y, Dong Q, Wang E
Received 27 September 2016
Accepted for publication 17 December 2016
Published 20 February 2017 Volume 2017:10 Pages 1049—1058
DOI https://doi.org/10.2147/OTT.S123324
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ingrid Espinoza
Yue Zhao, Qianze Dong, Enhua Wang
Department of Pathology, The First Affiliated Hospital and College of Basic Medical Sciences, China Medical University, Shenyang, People’s Republic of China
Abstract: MicroRNA-320 (miR-320) downregulation has been reported in several human cancers. Until now, its expression pattern and biological roles in human cancer remain unknown. This study aims to clarify its clinical expression pattern and biological function in gastric cancers. We found miR-320 level was downregulated in gastric cancer tissues. miR-320 mimic was transfected in SGC-7901 cells with low endogenous expression. miR-320 inhibitor was used in BGC-823 cells with high endogenous expression. We found that miR-320 inhibited SGC-7901 proliferation and invasion, with decreased expression of cyclin D1 and MMP9 at both mRNA and protein levels. We also found that miR-320 mimic downregulated chemoresistance and cell survival of gastric cancer cells when treated with 5-fluorouracil. miR-320 inhibitor displayed the opposite effects in BGC-823 cell line. In addition, we discovered that miR-320 mimic could inhibit AKT and ERK activity. By using luciferase reporter assay, we found that CRKL serves as the target of miR-320. miR-320 mimic downregulated CRKL expression, whereas miR-320 inhibitor upregulated CRKL expression. miR-320 suppressed CRKL-3'-untranslated region reporter intensity in SGC-7901 cells. Furthermore, CRKL depletion abrogated the effects of miR-320. In gastric cancer tissues, we observed a negative correlation between CRKL and miR-320. In conclusion, our study demonstrated that downregulation of miR-320 was closely related with malignant progression of gastric cancer. miR-320 inhibits proliferation, invasion, and chemoresistance through ERK and AKT signaling by targeting CRKL.
Keywords: gastric cancer, miR-320, CRKL, proliferation, invasion
Introduction
Gastric cancer is one of the most frequent cancers, and in many countries, its incidence is rising in recent years.1 Tumor size, lymph node metastasis, and distant metastasis influence the prognosis of gastric cancer patients.2,3 To develop better treatment strategy, it is important to identify effective molecular targets which can influence tumor invasion and survival.
MicroRNA (miRNA) exerts its biological function by post-transcriptional downregulation of its genes.4,5 Many studies showed that miRNA dysregulation contributed to gastric cancer pathogenesis and progression.2,3 miR-320 is a member of the miR-200 family, which plays a very important role in tumor progression. Emerging evidence showed that miR-320 dysregulation level promotes tumor progression via modifying cell proliferation, invasion, and stem cell-like characteristics in colon cancer, prostate cancer, and glioma.6–8 However, expression pattern of miR-320 in human gastric cancer and its biological mechanism remain obscure.
We examined miR-320 expression in 25 cases with paired human gastric cancer/normal tissues using real-time RT-PCR. We also explored the role of miR-320 on biological behaviors of tumor cells and its molecular mechanisms. Our results demonstrated that miR-320 acted as a tumor suppressor in gastric cancer.
Materials and methods
Tissue samples
This study was conducted with the approval of the Ethics Committee at Institutional Review Board of China Medical University. Fresh gastric cancer samples and normal adjacent gastric tissue were obtained from patients in the First Affiliated Hospital of China Medical University from 2013 to 2016 with informed consent. Written informed consent was obtained from all patients.
Cell culture and transfection
AGS, SGC-7901, and BGC-823 cell lines were obtained from American Type Culture Collection (Manassas, VA, USA). GES-1 cell line was obtained from Shanghai Cell Bank (Shanghai, People’s Republic of China). Cells were cultured in Dulbecco’s Modified Eagle’s Medium (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA). Cells were cultured on sterilized dishes.
miR-320 mimic and miR-320 inhibitor were purchased from RiboBio (Guangzhou, People’s Republic of China) with corresponding controls. CRKL siRNA and non-targeting siRNA were purchased from Dharmacon (GE Healthcare, Rockville, MD, USA). Cells were transfected with miR-320 mimic, miR-320 inhibitor, mimic/inhibitor control, non-targeting siRNA, and CRKL siRNA with DharmaFECT transfection reagent (GE Healthcare). pCMV6 and pCMV-CRKL plasmids were from Origene (Lafayette, CO, USA). Plasmid transfection was carried out using Attractene (Qiagen, Hilden, Germany).
Quantitative PCR of miR-320
Total RNA was extracted from samples with RNAiso reagent (Takara, Dalian, People’s Republic of China). Real-time PCR was performed using SYBRGreen Master mix from ABI (Applied Biosystems, Foster City, CA, USA). miR-320 primers and U6 primers were purchased from RiboBio. PCR analysis was performed on an ABI 7900 system (Applied Biosystems). The relative levels of gene expression were represented as ΔCt=Ct gene–Ct reference, and the fold change of gene expression was calculated by the 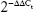 method.
method.
Quantitative PCR of target genes
Quantitative real-time PCR was carried out with SYBRGreen PCR Mastermix (Applied Biosystems) in a total volume of 20 μL using ABI 7500 Real-Time PCR (Applied Biosystems) as follows: 95°C for 30 s, 40 cycles of 95°C for 5 s, and 60°C for 30 s. β-Actin was used as the reference gene. A dissociation step was performed to generate a melting curve to confirm the specificity of the amplification. The relative levels of gene expression were represented as ΔCt=Ct gene–Ct reference, and the fold change of gene expression was calculated by the method. Experiments were repeated in triplicate. The primer sequences are listed as follows: cyclin D1 forward, 5′-GCTGGAGGTCTGCGAGGA-3′, cyclin D1 reverse, 5′-ACAGGAAGCGGTCCAGGTAGT-3; MMP9 forward, 5′-CCTCTGGAGGTTCGACGTGA-3′, MMP9 reverse, 5′-TAGGCTTTCTCTCGGTACTGGAA-3′; Bcl-2 forward, 5′-ACGGTGGTGGAGGAGCTCTT-3′, Bcl-2 reverse, 5′-CGGTTGACGCTCTCCACAC-3′; CRKL forward, 5′-CCTTTGCCATCCACACAGAAT-3′, CRKL reverse, 5′-TTTCACGATGTCACCAACCTCTA-3′; β-actin forward, 5′-ATAGCACAGCCTGGATAGCAACGTAC-3′; and β-actin reverse, 5′-CACCTTCTACAATGAGCTGCGTGTG-3′.
Western blot analysis
Protein was extracted using cell lysis buffer (Thermo, Rockford, IL, USA). Protein quantification was performed using Bradford method. A quantity of 50 μg protein was transferred to polyvinylidene difluoride membranes after separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Then, membranes were incubated at 4°C overnight with primary antibodies against cyclin D1, MMP9, CRKL, p-ERK, ERK, and GAPDH (1:1,000; Cell Signaling Technology, Boston, MA, USA). After incubation with HRP-coupled antimouse/rabbit IgG (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 37°C for 2 h, bound proteins were visualized using electrochemiluminescence (Pierce, Rockford, IL, USA) and detected using a DNR BioImaging System (DNR, Jerusalem, Israel).
Colony formation assay
Cells were planted on 6-cm culture dishes. Then, the plates were cultured for ~3 weeks. Plates were washed with phosphate-buffered saline and stained with Giemsa. The number of colonies with >50 cells was counted manually.
CCK8 assay
CCK8 assay was performed using Cell Counting Kit-8 solution (Dojindo, Gaithersburg, MD, USA) according to the manufacturer’s protocol. Cells were incubated with 10 μL/well of Cell Counting Kit-8 solution during the last 4 h of the culture. The cell culture plate was measured at 490 nm spectrophotometrically.
Matrigel invasion assay
Cell invasion assay was performed using a 24-well Transwell chamber with a pore size of 8 μm, and the inserts were coated with 20 μL Matrigel (1:3 dilution; BD Biosciences, San Jose, CA, USA). Forty-eight hours after transfection, cells were trypsinized, transferred to the upper Matrigel chamber in 100 μL serum-free medium, and incubated for 18 h. Medium supplemented with 10% FBS was added to the lower chamber. Then, the non-invaded cells on the upper membrane surface were moved out. Cells that invaded through the filter were fixed in 4% paraformaldehyde and stained with hematoxylin.
Luciferase reporter assay
Reporter gene transfection and luciferase activity assay were performed as follows: cells in confluent growth on a 24-well plate were co-transfected with the firefly luciferase reporter (0.2 μg) along with the Renilla luciferase reporter (0.02 μg), which was used for normalization, using an Attractene reagent (Qiagen) according to the protocols provided by manufacturers. The luciferase activity was measured in cellular extracts using a dual luciferase reported gene assay kit (Promega, San Luis Obispo, CA, USA). The relative activity of the reporter gene was calculated by dividing the signals from firefly luciferase reporter by the signals obtained from Renilla luciferase reporter.
Target gene validation
Luciferase reporter vector was used for validation of the interaction between miR-320 and CRKL. TargetScan 7.0 database was used to predict the binding site. We found that wild-type miR-320 target site at CRKL 3′-untranslated region (UTR) was CCAGCUU (position 108–114 of CRKL 3′-UTR). The mutant miR-320 target site was CCAAAUU. miR-320 mimic and reporter plasmid were transfected into cells. Attractene reagent (Qiagen) was used for plasmid transfection.
Statistical analysis
SPSS version 16 for Windows was used for all statistical analyses. Student’s t-test was used to compare densitometry data on focus numbers and invading cell numbers between control and treated cells. P<0.05 was considered to be statistically significant.
Results
miR-320 downregulation in gastric cancer tissues
We evaluated miR-320 levels in 33 cases of paired gastric cancers with adjacent normal tissues. Expression of miR-320 levels in cancer tissues was significantly lower than that in normal tissues. Downregulation of miR-320 was defined when its levels in tumor tissue was less than 50% of that in its corresponding non-tumor tissue. We found that miR-320 expression was significantly downregulated in 17 out of 33 gastric cancer tissues (Figure 1A). We compared the mean value of miR-320 expression between cancer and normal tissues and found that the mean value of miR-320 in normal tissue (2.591±0.615) was higher than that in cancer tissue (1.353±0.761) (mean ± SD, P<0.05). As seen in Figure 1B, Box plot showed that median/highest/lowest miR-320 levels of gastric cancer tissues were lower than those in corresponding normal tissues. In addition, we analyzed the relationship between miR-320 downregulation with clinical factors. As shown in Table 1, miR-320 downregulation significantly correlated with deeper local invasion (higher T stage, P=0.0488) and advanced TNM stage (P=0.038).
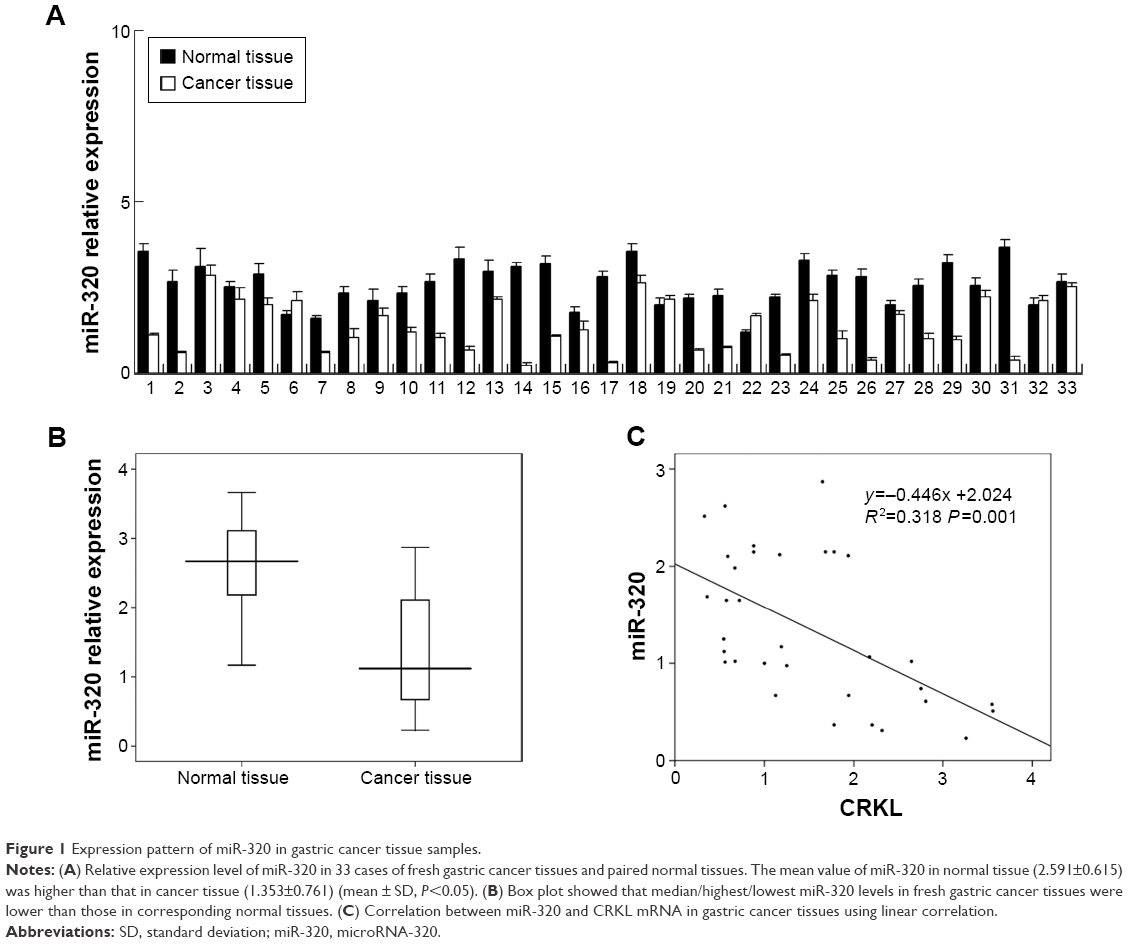 | Figure 1 Expression pattern of miR-320 in gastric cancer tissue samples. |
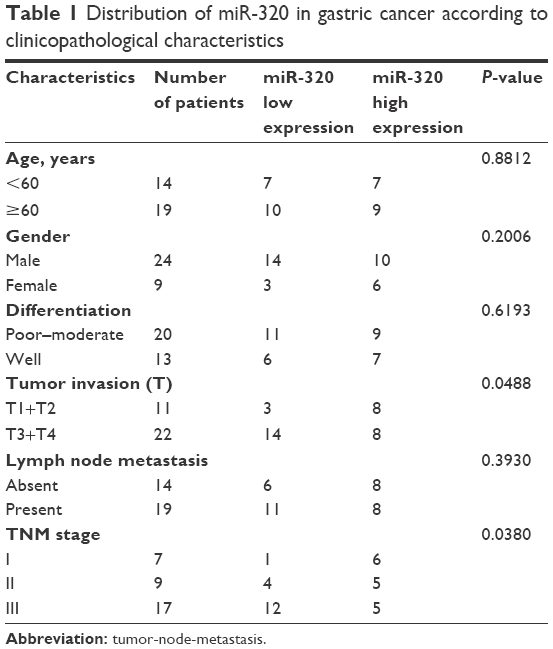 | Table 1 Distribution of miR-320 in gastric cancer according to clinicopathological characteristics |
miR-320 inhibits cell proliferation and invasion in gastric cancer
We applied real-time RT-PCR to check the expression level of miR-320 in normal cell line GES-1 and three gastric cancer cell lines including AGS, SGC-7901, and BGC-823. We observed that miR-320 expression in cancer cell lines was lower than that in GES-1 cell line. SGC-7901 has the lowest miR-320 level, and BGC-823 has the relatively high miR-320 expression. Thus, SGC-7901 was selected for miR-320 mimic transfection and BGC-823 was chosen for miR-320 inhibitor transfection. Efficiency was confirmed in both cell lines (Figure 2A). Using CCK8 assay, we observed that miR-320 mimic transfection downregulated cell proliferation rate and miR-320 inhibitor upregulated tumor proliferation rate (Figure 2B). Colony assay demonstrated that transfection of miR-320 mimic significantly decreased colony number, whereas transfection of miR-320 inhibitor increased colony formation ability of BGC-823 cell line (Figure 2C). Matrigel invasion assay showed that overexpression of miR-320 in SGC-7901 cells decreased the number of invading cells. In BGC-823 cells transfected with miR-320 inhibitor, the number of invading cells were remarkably higher than control (Figure 2D). We also validated the function of miR-320 in AGS cell line. As shown in Figure S1, miR-320 inhibited both growth and invasion of AGS cell line.
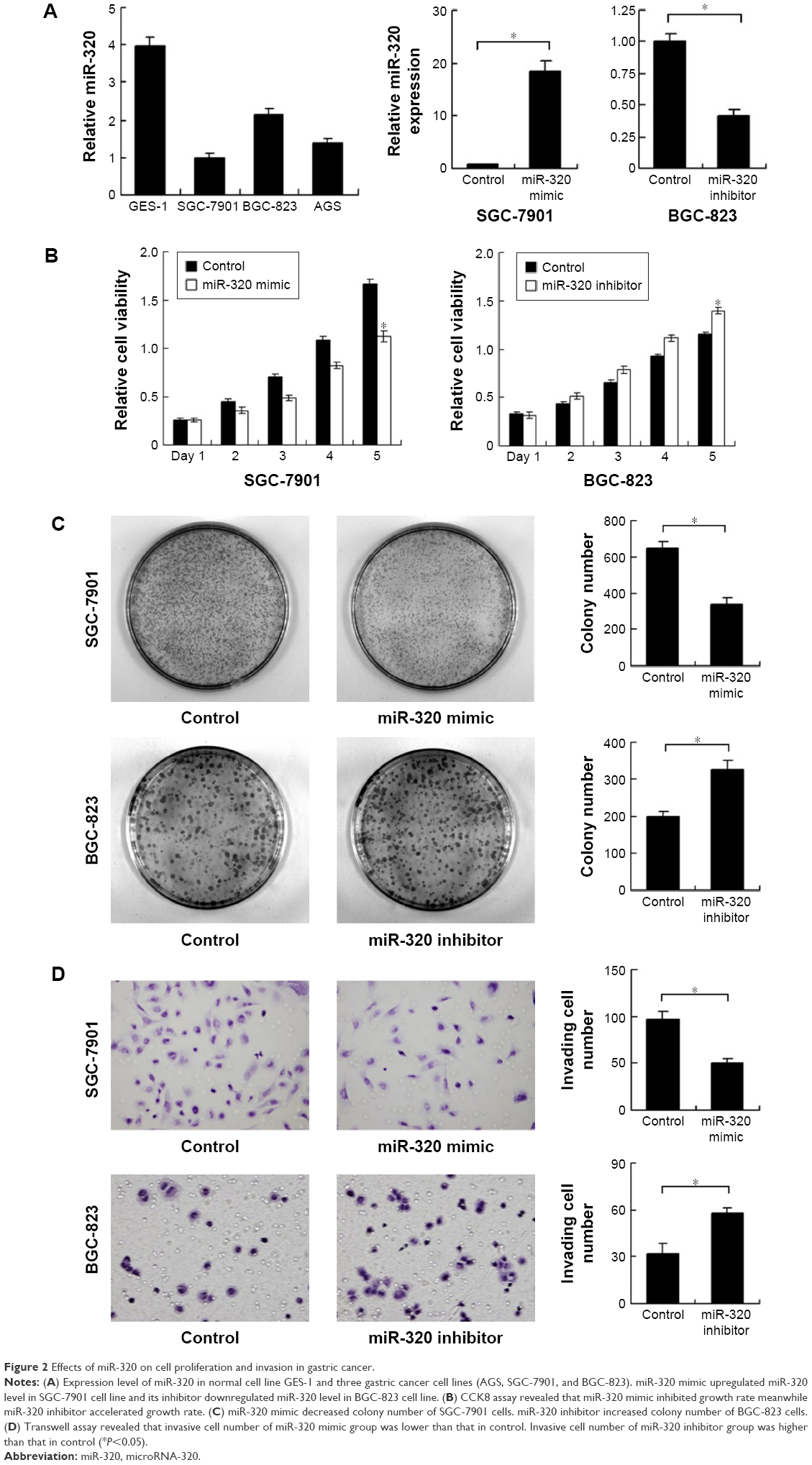 | Figure 2 Effects of miR-320 on cell proliferation and invasion in gastric cancer. |
miR-320 inhibits 5-fluorouracil resistance
To characterize the role of miR-320 on chemoresistance of gastric cancer cells, CCK-8 was used to examine cell survival. As shown in Figure 3A, miR-320 mimic significantly downregulated cell viability in SGC-7901 cells at 48 and 72 h of 5-fluorouracil (5-FU) (5 μg/mL) treatment. Whereas miR-320 inhibitor increased 5-FU resistance by upregulating cell viability. These results demonstrated that miR-320 could inhibit chemoresistance in gastric cancer cells.
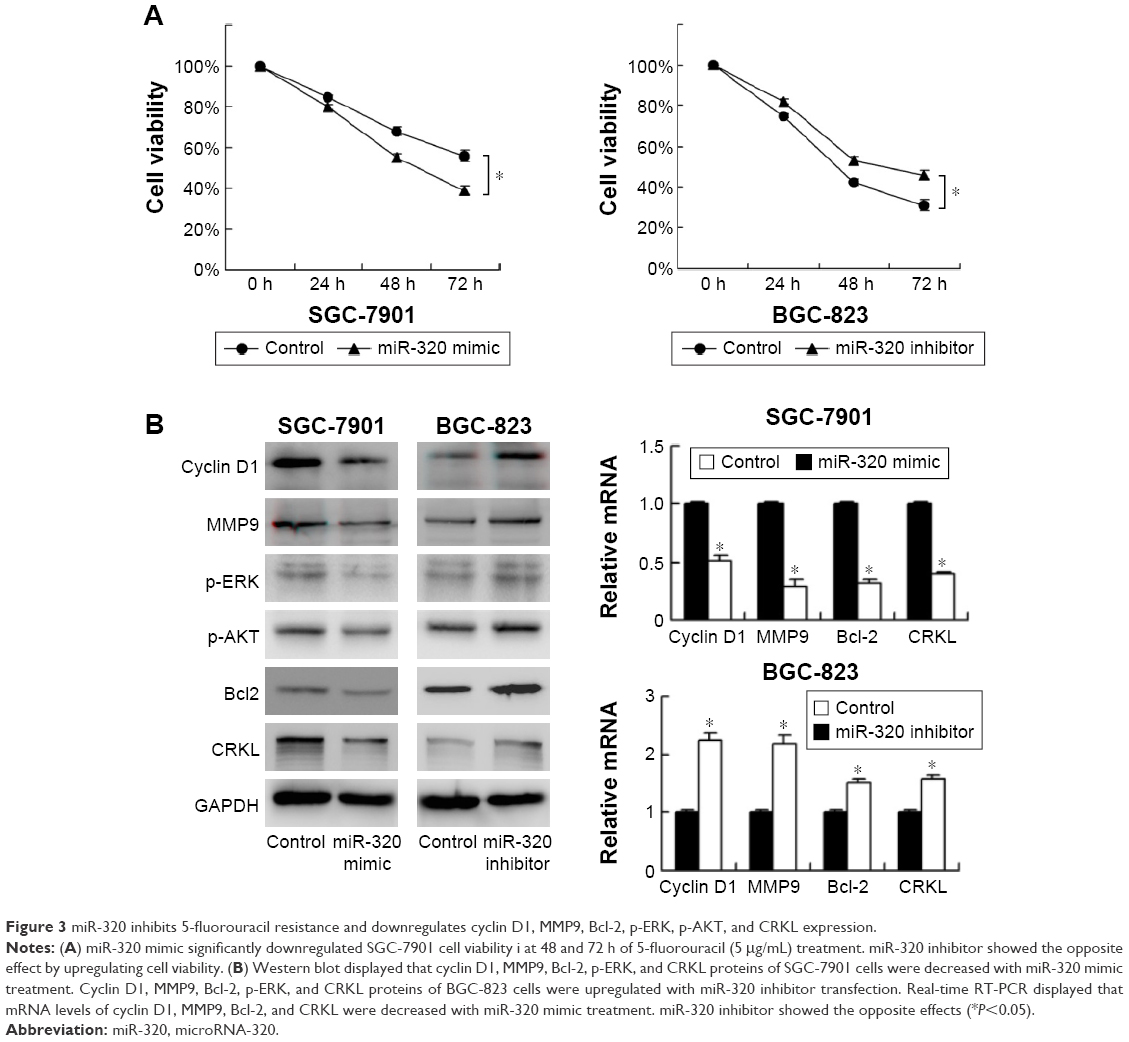 | Figure 3 miR-320 inhibits 5-fluorouracil resistance and downregulates cyclin D1, MMP9, Bcl-2, p-ERK, p-AKT, and CRKL expression. |
miR-320 regulates expression of cyclin D1, MMP9, Bcl-2, p-ERK, and p-AKT expression
To investigate the molecular mechanism underlying the biological effects of miR-320, we checked the change of several related proteins. Transfection of miR-320 mimic decreased cyclin D1, MMP9, and Bcl-2 mRNA and protein in SGC-7901 cells. miR-320 inhibitor in BGC-823 cells increased the expression of cyclin D1, MMP9, and Bcl-2 (Figure 3B). We also checked MAPK pathways and discovered that miR-320 mimic downregulated p-ERK and p-AKT expression, whereas miR-320 inhibitor upregulated the levels of p-ERK and p-AKT (Figure 3B).
miR-320 downregulates CRKL in gastric cancer cells
We explored possible targets of miR-320. TargetScan 7.0 showed that CRKL, a member of the human Crk adapter protein family that serves as an oncogene in many cancers, was in the target list. miR-320 mimic significantly inhibited mRNA and protein expression of CRKL, and miR-320 inhibitor remarkably upregulated the mRNA and protein expression of CRKL (Figure 3B).
CRKL is a miR-320 target
To conform if miR-320 targets CRKL, we used luciferase reporter assay with transfection of reporter plasmid. Wild-type (CCAGCUU) and mutant (CCAAAUU) CRKL 3′-UTR binding sites of miR-320 were cloned into a reporter vector. miR-320 mimic suppressed the reporter activity in SGC-7901 cells transfected with wild-type vector (Figure 4A). No significant change in activity was observed in cells transfected with mutant site vector. These data showed that miR-320 binds to CRKL 3′-UTR and results in its downregulation. In addition, we examined the relationship between miR-320 and CRKL mRNA in gastric cancer tissues. As demonstrated in Figure 1C, there was a negative linear correlation between miR-320 and CRKL mRNA (linear regression, P<0.05).
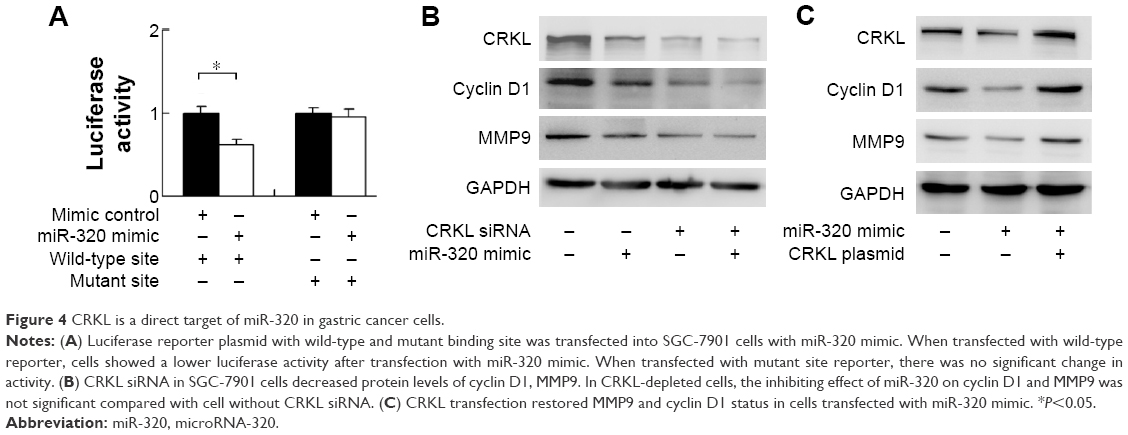 | Figure 4 CRKL is a direct target of miR-320 in gastric cancer cells. |
miR-320 regulates cyclin D1, MMP9, and Bcl-2 through regulation of CRKL
To confirm the involvement of CRKL in the biological effects of miR-320, we transfected SGC-7901 cells with CRKL siRNA and then tested the role of miR-320 in CRKL-depleted cells. As shown in Figure 4B, miR-320 mimic failed to downregulate cyclin D1 and MMP9 in CRKL-depleted cells. CRKL plasmid transfection was also carried out in SGC-7901 cells with miR-320 mimic. Its transfection restored MMP9 and cyclin D1 status, which is shown in Figure 4C. Furthermore, the biological function of CRKL was validated in SGC-7901 cells by plasmid transfection. As shown in Figure S1B, CRKL plasmid facilitated cell growth and invasion. Together, these results indicate that CRKL plays a key role in miR-320-induced inhibition of cell growth and invasion.
Discussion
Growing evidence demonstrate involvement of miRNA during development of various cancers including lung, prostate,9 breast,10 ovarian,11 cervical, and colon cancers.12 miR-320 was reported to be downregulated in human prostate cancer tissues, which inhibited tumorigenesis in vitro and in vivo.13 A report showed that miR-320 inhibited β-catenin expression by targeting the 3′-UTR of β-catenin mRNA, which significantly suppressed stem cell-like properties of cancer cells.6 miR-320 was reported to inhibit angiogenesis driven by vascular endothelial cells in oral cancer by silencing neuropilin 1.14 miR-320 reduced growth rate of osteosarcoma by targeting fatty acid synthase.15 miR-320 also functions as a tumor suppressor in glioma.8 Nevertheless, expression pattern and roles of miR-320 in human gastric cancer remain unexplored. In this study, we found that miR-320 was downregulated in gastric cancer tissues and correlated with advanced stage, which was in accord with previous reports, indicating miR-320 as a potential tumor-inhibiting miRNA in gastric cancers.
Then, we checked its biological effects in gastric cancer cells by CCK8, colony formation, and matrigel invasion assay. The results showed that miR-320 mimic decreased proliferation rate and inhibited colony formation ability. miR-320 weakened cell invasion. These data suggested that miR-320 inhibited malignant progression of gastric cancer. To understand the underlying mechanism, we examined a series of growth and invasion-related proteins. We noticed that cyclin D1 and MMP9 were decreased after treatment with miR-320 mimic and increased after treatment with miR-320 inhibitor. Many reports showed that cyclin D–Cdk4/6 complex phosphorylated Rb to inactivate its function as a transcriptional repressor and release E2F transcription factors, enabling the expression of genes required for G1–S transition and DNA synthesis.16–19 MMP9, which can degrade collagen IV, the major ECM component of the basement membranes, has been regarded as a critical factor of cell invasion and metastasis in a wide range of malignances.20–22 Furthermore, miR-320 negatively modulated ERK activity, which was regarded as the upstream signaling of cyclin D1 and MMP9 proteins.23,24 Collectively, these data indicated that miR-320 inhibited cell growth and invasion possibly through ERK inhibition in gastric cancer.
We also checked the influence of miR-320 on chemoresistance and found that miR-320 significantly reduced drug resistance. In accordance, we found that Bcl-2, an important antiapoptotic protein, was reduced by miR-320 transfection. Many studies have demonstrated that Bcl-2 mediated multidrug resistance of gastric cancer.25 Bcl-2 was also a downstream target of AKT signaling, which was inhibited by miR-320. Together, these results indicated that miR-320 reduced chemoresistance of gastric cancer, possibly through downregulation of Bcl-2 mediated by AKT inhibition.
We predicted potential targets of miR-320 using TargetScan software and CRKL was on the list. CRKL belongs to the human Crk adapter protein family26,27 and is best known as a key substrate and effector of the BCR-ABL oncogenic tyrosine kinase in chronic myelogenous leukemia. Previous data showed that phosphor-CRKL is upregulated in a lot of malignancies including lung cancer, colon cancer, breast cancer, ovarian cancer, and skin cancer.28–31 Our results demonstrated that miR-320 mimic remarkably decreased CRKL expression, whereas miR-320 inhibitor significantly increased CRKL levels. More importantly, we found a negative relationship in gastric cancer tissues between miR-320 mRNA and CRKL mRNA. In addition, luciferase reporter assays revealed that miR-320 could directly bind to the 3′-UTR of CRKL, demonstrating that CRKL is a direct target of miR-320. CRKL is an activator of ERK and AKT signaling pathway, which mediate upregulation of MMP9 and cyclin D1.32,33 Furthermore, our results showed that CRKL depletion abolished the role of miR-320 on its downstream protein cyclin D1 and MMP9. CRKL plasmid transfection restored MMP9 and cyclin D1 levels downregulated by miR-320. Generally speaking, these data demonstrate that miR-320 inhibits cell growth and invasion in gastric cancer by targeting CRKL.
In conclusion, our study demonstrated that miR-320 is downregulated in gastric cancer. Its downregulation correlated with aggressive biological behaviors. miR-320 regulates CRKL directly via binding with its 3′-UTR, making miR-320 a potent tumor suppressor in gastric cancer.
Acknowledgment
This study was supported by National Natural Science Foundation of China (No 81302022) to QD.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | ||
Shrestha S, Hsu SD, Huang WY, et al. A systematic review of microRNA expression profiling studies in human gastric cancer. Cancer Med. 2014;3(4):878–888. | ||
Wang JL, Hu Y, Kong X, et al. Candidate microRNA biomarkers in human gastric cancer: a systematic review and validation study. PLoS One. 2013;8(9):e73683. | ||
Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834–838. | ||
Takamizawa J, Konishi H, Yanagisawa K, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64(11):3753–3756. | ||
Hsieh IS, Chang KC, Tsai YT, et al. MicroRNA-320 suppresses the stem cell-like characteristics of prostate cancer cells by downregulating the Wnt/beta-catenin signaling pathway. Carcinogenesis. 2013;34(3):530–538. | ||
Wan LY, Deng J, Xiang XJ, et al. miR-320 enhances the sensitivity of human colon cancer cells to chemoradiotherapy in vitro by targeting FOXM1. Biochem Biophys Res Commun. 2015;457(2):125–132. | ||
Sun JY, Xiao WZ, Wang F, et al. MicroRNA-320 inhibits cell proliferation in glioma by targeting E2F1. Mol Med Rep. 2015;12(2):2355–2359. | ||
Saini S, Majid S, Yamamura S, et al. Regulatory role of mir-203 in prostate cancer progression and metastasis. Clin Cancer Res. 2011;17(16):5287–5298. | ||
Eades G, Yang M, Yao Y, Zhang Y, Zhou Q. miR-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J Biol Chem. 2011;286(47):40725–40733. | ||
Mateescu B, Batista L, Cardon M, et al. miR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat Med. 2011;17(12):1627–1635. | ||
Borralho PM, Simoes AE, Gomes SE, et al. miR-143 overexpression impairs growth of human colon carcinoma xenografts in mice with induction of apoptosis and inhibition of proliferation. PLoS One. 2011;6(8):e23787. | ||
Sato S, Katsushima K, Shinjo K, et al. Histone deacetylase inhibition in prostate cancer triggers miR-320-Mediated suppression of the androgen receptor. Cancer Res. 2016;76(14):4192–4204. | ||
Wu YY, Chen YL, Jao YC, Hsieh IS, Chang KC, Hong TM. miR-320 regulates tumor angiogenesis driven by vascular endothelial cells in oral cancer by silencing neuropilin 1. Angiogenesis. 2014;17(1):247–260. | ||
Cheng C, Chen ZQ, Shi XT. MicroRNA-320 inhibits osteosarcoma cells proliferation by directly targeting fatty acid synthase. Tumour Biol. 2014;35(5):4177–4183. | ||
Knudsen KE, Diehl JA, Haiman CA, Knudsen ES. Cyclin D1: polymorphism, aberrant splicing and cancer risk. Oncogene. 2006;25(11):1620–1628. | ||
Ratschiller D, Heighway J, Gugger M, et al. Cyclin D1 overexpression in bronchial epithelia of patients with lung cancer is associated with smoking and predicts survival. J Clin Oncol. 2003;21(11):2085–2093. | ||
Roy PG, Thompson AM. Cyclin D1 and breast cancer. Breast. 2006;15(6):718–727. | ||
Keum JS, Kong G, Yang SC, et al. Cyclin D1 overexpression is an indicator of poor prognosis in resectable non-small cell lung cancer. Br J Cancer. 1999;81(1):127–132. | ||
Iizasa T, Fujisawa T, Suzuki M, et al. Elevated levels of circulating plasma matrix metalloproteinase 9 in non-small cell lung cancer patients. Clin Cancer Res. 1999;5(1):149–153. | ||
Shiraga M, Yano S, Yamamoto A, et al. Organ heterogeneity of host-derived matrix metalloproteinase expression and its involvement in multiple-organ metastasis by lung cancer cell lines. Cancer Res. 2002;62(20):5967–5973. | ||
Ylisirnio S, Hoyhtya M, Turpeenniemi-Hujanen T. Serum matrix metalloproteinases -2, -9 and tissue inhibitors of metalloproteinases -1, -2 in lung cancer – TIMP-1 as a prognostic marker. Anticancer Res. 2000;20(2B):1311–1316. | ||
Dong QZ, Wang Y, Tang ZP, et al. Derlin-1 is overexpressed in non-small cell lung cancer and promotes cancer cell invasion via EGFR-ERK-mediated up-regulation of MMP-2 and MMP-9. Am J Pathol. 2013;182(3):954–964. | ||
Cheng SP, Yin PH, Hsu YC, et al. Leptin enhances migration of human papillary thyroid cancer cells through the PI3K/AKT and MEK/ERK signaling pathways. Oncol Rep. 2011;26(5):1265–1271. | ||
Han Z, Hong L, Han Y, et al. Phospho Akt mediates multidrug resistance of gastric cancer cells through regulation of P-gp, Bcl-2 and Bax. J Exp Clin Cancer Res. 2007;26(2):261–268. | ||
ten Hoeve J, Morris C, Heisterkamp N, Groffen J. Isolation and chromosomal localization of CRKL, a human crk-like gene. Oncogene. 1993;8(9):2469–2474. | ||
Feller SM. Crk family adaptors-signalling complex formation and biological roles. Oncogene. 2001;20(44):6348–6371. | ||
ten Hoeve J, Arlinghaus RB, Guo JQ, Heisterkamp N, Groffen J. Tyrosine phosphorylation of CRKL in Philadelphia+ leukemia. Blood. 1994;84(6):1731–1736. | ||
ten Hoeve J, Kaartinen V, Fioretos T, et al. Cellular interactions of CRKL, and SH2-SH3 adaptor protein. Cancer Res. 1994;54(10):2563–2567. | ||
Senechal K, Halpern J, Sawyers CL. The CRKL adaptor protein transforms fibroblasts and functions in transformation by the BCR-ABL oncogene. J Biol Chem. 1996;271(38):23255–23261. | ||
Hemmeryckx B, van Wijk A, Reichert A, et al. Crkl enhances leukemogenesis in BCR/ABL P190 transgenic mice. Cancer Res. 2001;61(4):1398–1405. | ||
Wang Y, Dong QZ, Fu L, Stoecker M, Wang E, Wang EH. Overexpression of CRKL correlates with poor prognosis and cell proliferation in non-small cell lung cancer. Mol Carcinog. 2013;52(11):890–899. | ||
Ladanyi M. CRKL as a lung cancer oncogene and mediator of acquired resistance to EGFR inhibitors: is it all that it is cracked up to be? Cancer Discov. 2011;1(7):560–561. |
Supplementary material
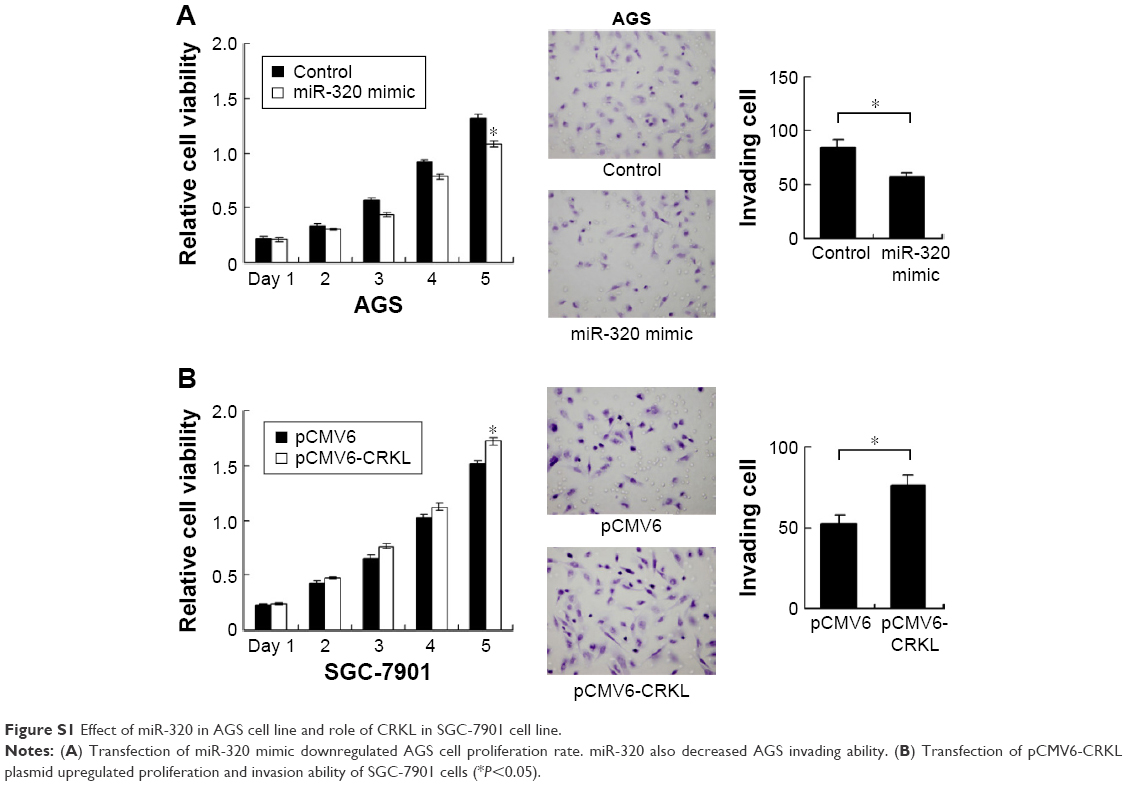 | Figure S1 Effect of miR-320 in AGS cell line and role of CRKL in SGC-7901 cell line. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.