")
Back to Journals » Journal of Inflammation Research » Volume 13
MicroRNA-21 Mediates a Positive Feedback on Angiotensin II-Induced Myofibroblast Transformation
Authors Li D, Mao C, Zhou E, You J, Gao E, Han Z, Fan Y, He Q, Wang C
Received 8 October 2020
Accepted for publication 13 November 2020
Published 27 November 2020 Volume 2020:13 Pages 1007—1020
DOI https://doi.org/10.2147/JIR.S285714
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Dongjiu Li,1,* Chengyu Mao,1,* En Zhou,1,* Jiayin You,2,* Erhe Gao,3 Zhihua Han,1 Yuqi Fan,1 Qing He,1 Changqian Wang1
1Department of Cardiology, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200011, People’s Republic of China; 2Department of Emergency, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200011, People’s Republic of China; 3Center for Translational Medicine, Department of Pharmacology, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, USA
*These authors contributed equally to this work
Correspondence: Changqian Wang Email [email protected]
Objective: Post myocardial infarction (MI) fibrosis has been identified as an important factor in the progression of heart failure. Previous studies have revealed that microRNA-21 (miR-21) plays an important role in the pathogenesis of fibrosis. The purpose of this study was to explore the role of miR-21 in post-MI cardiac fibrosis.
Material and Methods: MI was established in wild-type (WT) and miR-21 knockout (KO) mice. Primary mice cardiac fibroblasts (CFs) were isolated from WT and miR-21 KO mice and were treated with angiotensin II (Ang II) or Sprouty1 (Spry1) siRNA. Histological analysis and echocardiography were used to determine the extent of fibrosis and cardiac function.
Results: Compared with WT mice, miR-21 KO mice displayed smaller fibrotic areas and decreased expression of fibrotic markers and inflammatory cytokines. In parallel, Ang II-induced myofibroblasts transformation was partially inhibited upon miR-21 KO in primary CFs. Mechanistically, we found that the expression of Spry1, a previously reported target of miR-21, was markedly increased in miR-21 KO mice post MI, further inhibiting ERK1/2 activation. In vitro studies showed that Ang II activated ERK1/2/TGF-β/Smad2/3 pathway. Phosphorylated Smad2/3 further enhanced the expression of α-SMA and FAP and may promote the maturation of miR-21, thereby downregulating Spry1. Additionally, these effects of miR-21 KO on fibrosis were reversed by siRNA-mediated knockdown of Spry1.
Conclusion: Our findings suggest that miR-21 promotes post-MI fibrosis by targeting Spry1. Furthermore, it mediates a positive feedback on Ang II, thereby inducing the ERK/TGF-β/Smad pathway. Therefore, targeting the miR-21–Spry1 axis may be a promising therapeutic option for ameliorating post-MI cardiac fibrosis.
Keywords: cardiac fibrosis, myocardial infarction, microRNA-21, Sprouty1, ERK/TGF-β/Smad signaling pathway
Introduction
Myocardial infarction (MI) is the leading cause of death worldwide. It has been reported that approximately 10% of patients with post-MI cardiac fibrosis develop adverse ventricular remodeling, ultimately leading to advanced heart failure.1 Excessive cardiac fibrosis, defined as the expansion of the cardiac interstitium due to excessive deposition of extracellular matrix (ECM) protein, is an important pathological contributor to heart failure.2–4 Although there have been significant developments in pharmacological treatments to improve patient outcomes post MI, an effective therapeutic target for alleviating fibrosis is lacking. Therefore, it is of utmost importance to identify novel targets that can ameliorate post-MI cardiac fibrosis.
MicroRNAs (miRNAs or miRs) are highly conserved single-stranded RNAs (approximately 18–24 nucleotides) that are involved in gene silencing via the degradation of post-transcriptional mRNA or suppression of the expressed protein.5 Several studies have suggested that miRNAs participate in the progression of various cardiovascular diseases, and emerging evidence suggests that they play pivotal roles in the progression of cardiac fibrosis.6,7 Among the several miRNAs identified for cardiac fibrosis, miR-21 has been demonstrated to serve as a biomarker of many fibrotic cardiac disorders, including pressure overload-induced fibrosis in aortic stenosis and atrial fibrillation-induced atrial remodeling.8,9 As early as 2008, Thum, et al10 had found that miR-21 levels were increased selectively in CFs of the failing heart via targeting Sprouty1 (Spry1). In addition, recent studies have reported that miR-21 promotes post-MI cardiac fibrosis by targeting Smad7 and Jagged1.11,12 Both these studies have suggested that miR-21 regulates cardiac fibrosis by manipulating the transforming growth factor-beta (TGF-β) signaling pathway. However, it is known that MI activates the neurohumoral pathways, such as the renin angiotensin system (RAS). Angiotensin II (Ang II), the main effector molecule of RAS, has been reported to activate extracellular signal-regulated kinase (ERK) mitogen-activated protein kinase (MAPK) signaling.13,14 Ang II-induced cardiac fibrosis is partially attributed to the TGF-β/Smad2/3 pathway.13,15–17 In contrast, other studies have reported that TGF-β can induce the upregulation of phosphorylated ERK1/2 (p-ERK1/2) in patients with fibrotic disorders.18,19 These findings suggest that a feedback mechanism may exist between ERK and TGF-β signaling. Recently, Bang et al20 has demonstrated that the expression of the guide strand (miR-21-5p) was higher than that of the passenger strand (miR-21-3p) in CF, the latter of which was enriched in fibroblast-derived exosomes and was transported out of the CF. Based on these evidence and considering that miRNAs usually target a variety of genes, we hypothesized that miR-21 (miR-21-5p) participates in a feedback loop between ERK and TGF-β signaling by targeting other genes post MI.
In the present study, we aimed to test the hypothesis that miR-21 promoted post-MI cardiac fibrosis in a positive feedback manner by targeting Sprouty1 (Spry1). We found that Ang II promoted the phosphorylation of ERK1/2, thereby activating the TGF-β/Smad2/3 pathway, further promoting myofibroblast transformation and stimulating the maturation of miR-21. Spry1, a previously reported target of miR-21,10 could inhibit the activation of ERK1/2 post-MI, thereby providing a feedback for Ang II-induced ERK signaling. Our study suggests that manipulation of the miR-21–Spry1 axis could be a therapeutic target for alleviating post-MI cardiac fibrosis.
Materials and Methods
Animals
The miR-21 cKO mice (B6.129-Mir21a tm1Srcmo), EIIa-Cre mice and male wild-type (WT) mice were purchased from Shanghai Biomodel Organism Science & Technology Development Co., Ltd. B6.129-Mir21a tm1Srcmo mice were crossed with EIIa-Cre mice to obtain heterozygous miR-21 KO mice, the latter of which were crossed to obtain homozygous miR-21 systemic KO mice, in which the male mice were used for the present study (genotyping of miR-21 systemic KO is shown in Supplementary Figure 1). The primers for genotyping were: P1: CAGAATTGCCCAGGCTTTTA; P2: AATCCATGAGGCAAGGTGAC. Mice were fed and bred under standard pathogen-free conditions (temperature: 21°C ± 1°C; humidity: 55–60%) at the Laboratory Animal Resources division of Shanghai Ninth People’s Hospital. All animal studies were approved by the institutional ethics committee of Shanghai Ninth People’s Hospital (HKDL2017300) and were performed in accordance with the international ARRIVE guidelines for animal experiments.21
MI Model
The MI mouse model was generated surgically by Dr. Erhe Gao using a previously described method.22 Briefly, mice were anesthetized with 2% isoflurane inhalation using an isoflurane delivery system (Viking Medical, Medford, NJ) without intubation. A small incision (1.2 cm) was made on the left side of the chest. After dissection and retraction of the pectoral major and minor muscles, the fourth intercostal space was exposed. A small hole was made at the fourth intercostal space using a mosquito clamp to open the pleural membrane and pericardium. Using the clamp to slightly open the intercostal space, the heart was smoothly and gently popped out through the hole via the incision. The left coronary artery was located and ligated at a site ≈ 3mm from its origin using a 6–0 silk suture. The ligation was considered successful when the anterior wall of the left ventricle became pale. After ligation, the heart was immediately placed back into the chest cavity followed by manual evacuation of air and closure of the muscle and skin. The animals were then transferred to a warm pad and maintained at 37°C until they were fully conscious. The sham group underwent the same procedure except that the left coronary artery was not ligated.
Echocardiography
Echocardiography was performed at the second week post MI induction using a 30-MHz ultrasound probe (Vevo3100, Box 66 Toronto, ON, Canada). Two-dimensional and M-mode images of the left ventricle were obtained from six cardiac cycles for analysis. Heart rate, left ventricular end-systolic diameter (LVESD), and left ventricular end-diastolic diameter (LVEDD) were measured. Left ventricular ejection fraction (EF) and fractional shortening (FS) were calculated using the following equations: EF (%) = [(LVEDD3 − LVESD3)/LVEDD3] × 100% and FS (%) = [(LVEDD − LVESD)/LVEDD] × 100%. All parameters were measured by a single experienced echocardiographer who was blinded to the study groups.
Adeno-Associated Virus Serotype 9 (AAV9)-Mediated Gene Delivery to the Heart
AAV9 expressing only green fluorescent protein (GFP) alone or harboring siRNA targeting Spry1 (si-Spry1) or negative control (si-NC) were synthesized and produced by Zorin (Shanghai, China) according to the manufacturer’s instructions. For the experimental protocol, miR-21 KO mice were randomly divided into three groups: miR-21 KO (injected with AAV9 expressing GFP alone, n = 6), AAV9-si-Spry1 (n = 8), and AAV9-NC (n = 6) groups. One hundred microliters of each AAV9 solution (2 × 1011 vector genomes) in phosphate-buffered saline (PBS) was loaded into a 1-mL syringe attached to a 29G needle. The AAV9 solution was injected into the tail vein as described previously.23 Two weeks after injection, the efficiency of AAV9 infection in the myocardium was evaluated and the MI was induced.
Masson’s Trichrome Staining and Immunohistochemistry
Fixation was conducted for 24 h in PBS containing formalin (10%). The cardiac tissues were dehydrated with alcohol and then embedded with paraffin (4%). Hematoxylin and eosin (H&E) and Masson’s trichrome staining were performed on 5-µm sliced sections of the infarcted tissues. The extent of cardiac fibrosis was quantified by detecting the collagen content fraction using the Image-Pro Plus software.
Isolation and Culturing of CFs from Neonatal Mice
Primary CFs were isolated from neonatal mice (1–2-day-old) using a previously described method.24 Briefly, neonatal mice were quickly rinsed in 75% ethanol and decapitated. The heart was removed, atrium was excised, and the ventricles were minced and washed with PBS at 4°C to remove excess blood. The heart was then digested with 0.25% trypsin. The cell suspension was subjected to centrifugation, and the pellet was resuspended in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), streptomycin (100 µg/mL) and penicillin (100 U/mL) at 37°C under 5% CO2. The resuspension was plated onto a culture flask and incubated for 90 min, after which CFs attached to the bottom of the flask. The supernatant was removed and the media was changed. The cells were subcultured upon reaching 90% confluency. Cells from passages 2–4 were used for subsequent experiments.
Cell Transfection
CFs in the exponential growth phase were seeded in 6-well plates (2 × 105 cells/well), followed by incubation for 24 h. Transfection was performed using Lipofectamine 3000 (Invitrogen) following the manufacturer’s instructions. Briefly, analogs and Lipofectamine 3000 were mixed with 200 µL of serum-free Opti-MEM (Gibco) for 20 min at room temperature to form transfection complexes. Then, the CFs were incubated with the transfection complexes in Opti-MEM for 4–6 h. The medium was subsequently replaced with fresh DMEM containing 10% FBS to continue culturing of the cells. The transfection efficiencies were measured via fluorescence assays and were found to be ≥75%. mRNA assays were performed after 48 h and proteomic analysis was performed after 72 h of incubation.
Silencing the Expression of Spry1
si-Spry1 was used to silence the endogenous expression of Spry1. We obtained synthesized siRNA and its corresponding negative control from Zorin Shanghai Co., Ltd. The sequences of these siRNAs are presented: Spry1 siRNA: GCAGGUGUAGAAACUCCAATT; negative control: UUGGAGUUUCUACACCUGCAG. Spry1 siRNA and negative control were transfected into CFs using Lipofectamine 3000. The transfection efficiencies were ≥75%.
Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
Total RNA, including miRNA, was extracted from cultured CFs and cardiac tissues using the TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. RNA concentration was assessed using NanoDrop 2000 (Wilmington, DE, USA). Total RNA (1 µg) was reverse transcribed into cDNA using the Color Reverse Transcription Kit (EZBioscience, USA) according to the manufacturer’s instructions. The mRNA levels of α-SMA, Spry1, and collagen Iα were measured using PrimerScriptTM RT reagent Kit (TaKaRa, Japan; Catalog number: RR037A) using α-tubulin as an internal control. Based on a recent study by Bang et al,20 we selected miR-21-5p for the present study. The level of miR-21 was quantified via qRT-PCR using the Bulge-LoopTM miRNA qRT-PCR Starter Kit (RiboBio, Guangzhou, China; Catalog number: C10211-1). The primers for miR-21 and U6 small nuclear RNA were obtained from RiboBio Company. The sequences are covered by a patent (the catalog number of miR-21 and U6 primers are MQPS0002616-1-100 and MQPS0000002-1-100, respectively). All qRT-PCR analyses were performed on the ABI 7500 System (Applied Biosystems). The method used for calculating PCR data is 2-ΔΔCt method. Primers sequences in qRT-PCR are presented in Table 1.
 |
Table 1 Sequences of Primer Sequences Used in Quantitative Reverse Transcription-Polymerase Chain Reaction |
Western Blotting
Total protein was extracted from frozen ventricular tissues and CFs using the RIPA lysis buffer (Beyotime, China). A total of 15 µg of protein was separated using 12.5% SDS–PAGE gels. Electrophoresis was performed at 60–100 V for 2 h. After electrophoresis, the proteins were transferred onto 0.45-µm PVDF membranes (Millipore, USA) at 300 mA for 1 h. Then, the membranes were blocked with rapid blocking buffer (Shanghai EpiZyme Biotechnology Company, China) for 20 min at room temperature, followed by overnight incubation with primary antibodies at 4°C. All primary antibodies were diluted 1:1000 in 5% BSA. The membranes were washed three times with TBST for 8 min each time and were then incubated for 1 h in secondary horseradish peroxidase (HRP)-conjugated antibodies (goat anti-rabbit IgG (H&L, USA, Abcam, ab81053)) diluted 1:5000 in 5% BSA. The protein blots were visualized using the ECL reagent kit (Millipore, USA). The intensity of the bands was quantified using the Image J software (ImageJ v1.48). The antibodies used in this study are shown in Table 2.
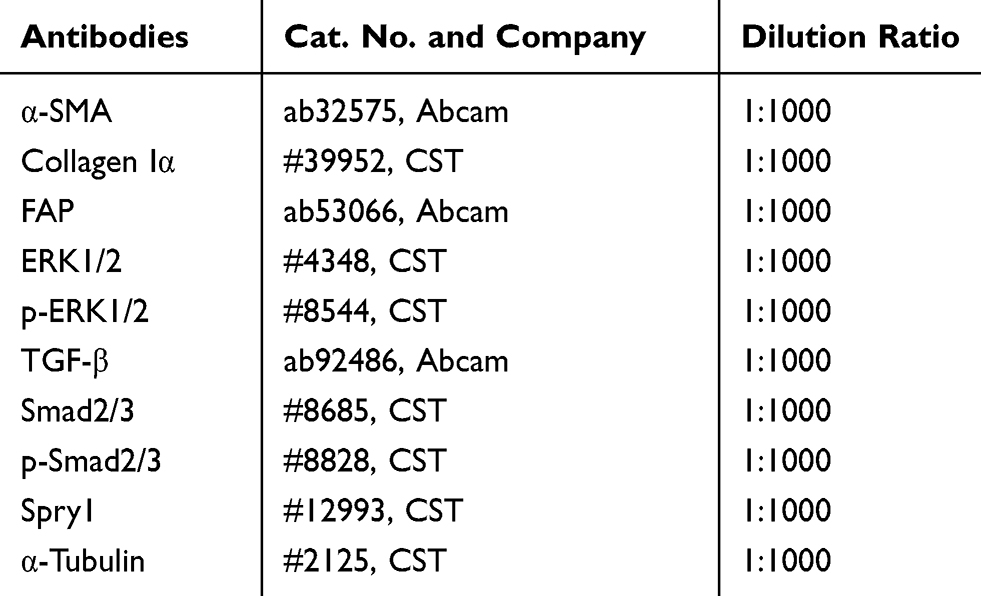 |
Table 2 Antibodies Used for Western Blot Analysis |
Immunofluorescence Analysis
CFs were cultured in 12-well plates. After 96 h, the cells were fixed in 4% paraformaldehyde for 10 min at room temperature, followed by washing three times with PBS for 5 min each. The cells were permeabilized in 0.2% Triton X-100 for 10 min at room temperature and then washed three times with PBS as indicated above. The cells were blocked with 5% BSA for 1 h at room temperature and then incubated with primary antibodies overnight at 4°C. The primary antibodies were diluted 1:100 in 5% BSA. The next day, the cells were washed three times with PBS for 5 min each and then incubated with the secondary antibody (1:1000 in 5% BSA) for 1 h at room temperature. The cells were washed three times with PBS for 5 min each. Nuclei were stained using 0.01% 4′,6-diamidino-2-phenylindole (DAPI) for 1 min. Finally, the cells were washed again and fluorescence was measured. Fluorescent images were acquired using a microscope (Leica Microsystems, Mannheim, Germany) under 200× magnification.
Statistical Analysis
Results are expressed as mean ± SD. Comparisons of continuous variables between groups were performed using two-tailed Student’s t-test or one-way ANOVA followed by the least significant difference t-test and Dunnett’s t-test (SPSS 19.0). All data were tested for normality and homogeneity of variance before comparisons between groups were performed. Statistical analyses were performed using GraphPad Prism 6.0h Software (GraphPad Inc, San Diego, CA, USA). Values of P < 0.05 were considered statistically significant.
Results
miR-21 KO Attenuated Post-MI Cardiac Fibrosis
A total of 67 mice were used, of which 22 were sham group and 45 underwent MI. Three mice died immediately after MI, probably due to excessive bleeding or pneumothorax. The acute mortality rate was about 6.7%. During follow up, 8 mice died during 4–9 days, probably due to cardiac rupture and heart failure. All mice of the sham groups survived.
To investigate the expression profile of miR-21 post MI as well as to determine its impact on cardiac fibrosis, we first examined the expression of miR-21 in the left ventricles of hearts at 2 weeks post-MI induction using qRT-PCR. As shown in Figure 1A, miR-21 expression was significantly upregulated in the myocardium of mice MI models. To further elucidate the association and causal relationship between miR-21 and post-MI cardiac fibrosis, the expression levels of TGF-β, collagen-Iα (Col-Iα), alpha-smooth muscle actin (α-SMA), and fibroblast activation protein (FAP), key markers of the fibrotic process, as well as inflammatory cytokines were assessed and Masson’s trichrome staining was performed. As expected, there were significant upregulation of TGF-β, α-SMA, FAP and collagen Iα expression (Figure 1B–E), inflammatory cytokines (Figure 1F) as well as increased cardiac replacement and interstitial fibrosis (Figure 1G–H) in the infarct zone at 2 weeks post MI. However, miR-21 KO mice that underwent MI induction exhibited reduced fibrotic area in the infarct zone (ie, replacement fibrosis), reduced interstitial fibrosis in the infarct border zone (Figure 1G), and reduced expression of the aforementioned fibrotic markers and inflammatory cytokines (Figure 1B–F) compared with the WT mice. Additionally, the immunofluorescence and Western blotting analysis of α-SMA and FAP in mice myocardium (Figure 1I–L) further confirmed these findings. Furthermore, echocardiography showed that the deterioration of cardiac function 2 weeks after MI in WT mice, as indicated by decreased EF and FS and increased LVEDD, was partially improved in miR-21 KO mice underwent MI (Figure 1M–N).
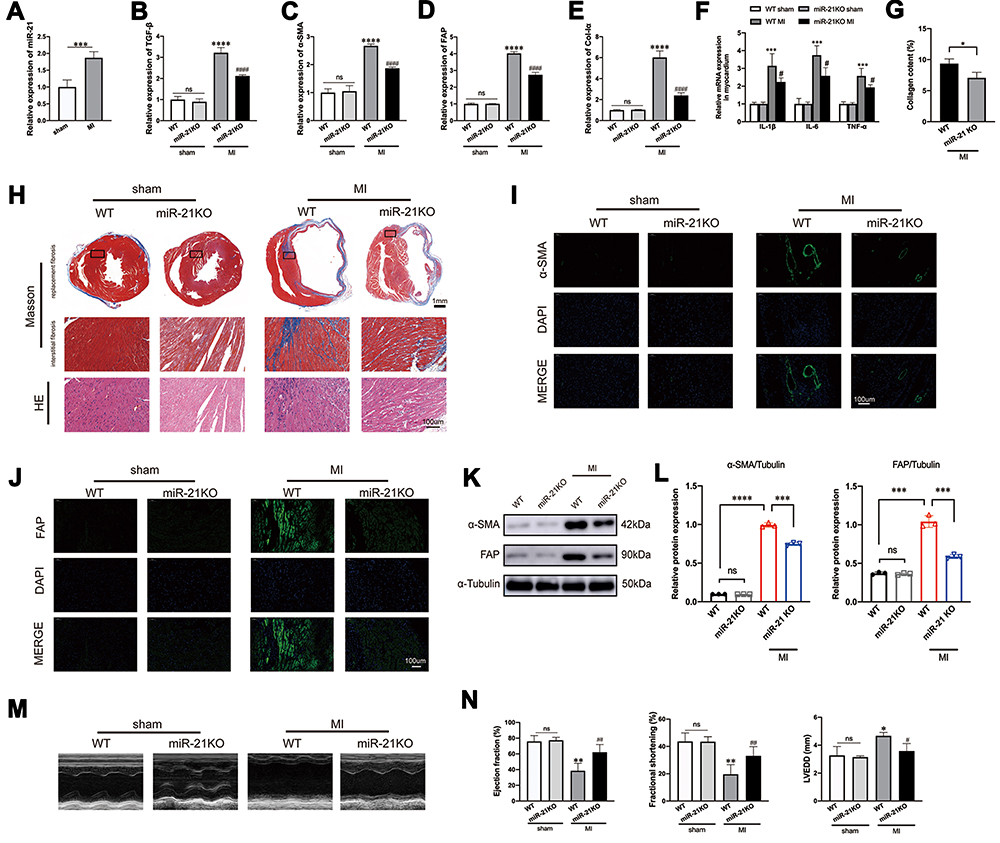 |
Figure 1 miR-21 promotes post-myocardial infarction (MI) cardiac fibrosis and negatively impacted cardiac function. (A) Expression profile of miR-21 at 2 weeks post-MI; ***P < 0.001, n = 5 per group. (B–E) Knockout (KO) of miR-21 decreases the expression of TGF-β, α-SMA, FAP and Col-Iα at 2 weeks post-MI; ****P<0.0001 compared with WT sham, ####P<0.001 compared with wild-type (WT) MI, n = 5 per group. (F) KO of miR-21 decreases the expression of inflammatory cytokines at 2 weeks post-MI; ***P < 0.001 compared with WT sham, #P < 0.05 compared with WT MI, n = 5 per group. (G) Quantitative analysis of collagen content between WT and miR-21 KO mice at 2 weeks post-MI based on Masson’s trichrome staining; *P < 0.05, n = 3 per group. (H) Representative Masson’s trichrome and H&E staining of the cross section of the heart. The representative illustration indicates that miR-21 KO attenuates both replacement fibrosis and interstitial fibrosis in the infarct border zone. (I and J) Representative immunofluorescence staining of α-SMA and FAP in the heart; scale bars = 100 µm. (K and L) Western blotting analysis of α-SMA and FAP in WT and miR-21 KO mice myocardium. Left panel (K), representative blots; right panel (L), quantitative analysis; ns = not significant, ***P < 0.001 and ****P < 0.0001, n = 3 per group. (M and N) Representative (M) and quantitative analysis of cardiac functions at 2 weeks post-MI (N); ns = not significant, *P < 0.05 and **P < 0.01 compared with WT sham, #P < 0.05 and ##P < 0.01 compared with WT MI, n = 4 per group. |
Taken together, these findings highlight the important role of miR-21 in exacerbating post-MI cardiac fibrosis and worsening cardiac function.
Ang II-Induced Myofibroblast Transformation Was Partially Inhibited by miR-21 KO
It is known that RAS is activated during MI and that Ang II is the main effector that drives cardiac fibrosis.25 A previous study10 has shown that miR-21 is predominantly expressed in CFs rather than in cardiomyocytes. Therefore, to explore the impact of miR-21 on cardiac fibrosis in vitro, CFs isolated from both WT and miR-21 KO neonatal mice were treated with Ang II (100 nmol/l), a commonly used stimulant to induce cardiac myofibroblast transformation.26 Western blots (Figure 2A–C) and immunofluorescence images (Figure 2D) showed that the expression levels of α-SMA and FAP in WT CFs were significantly upregulated at 24 h after Ang II treatment. On the other hand, CFs isolated from neonatal miR-21 KO mice showed reduced expression of α-SMA and FAP after Ang II treatment (Figure 2A–D). These results demonstrate that miR-21 can promote Ang II-induced myofibroblasts transformation in CFs.
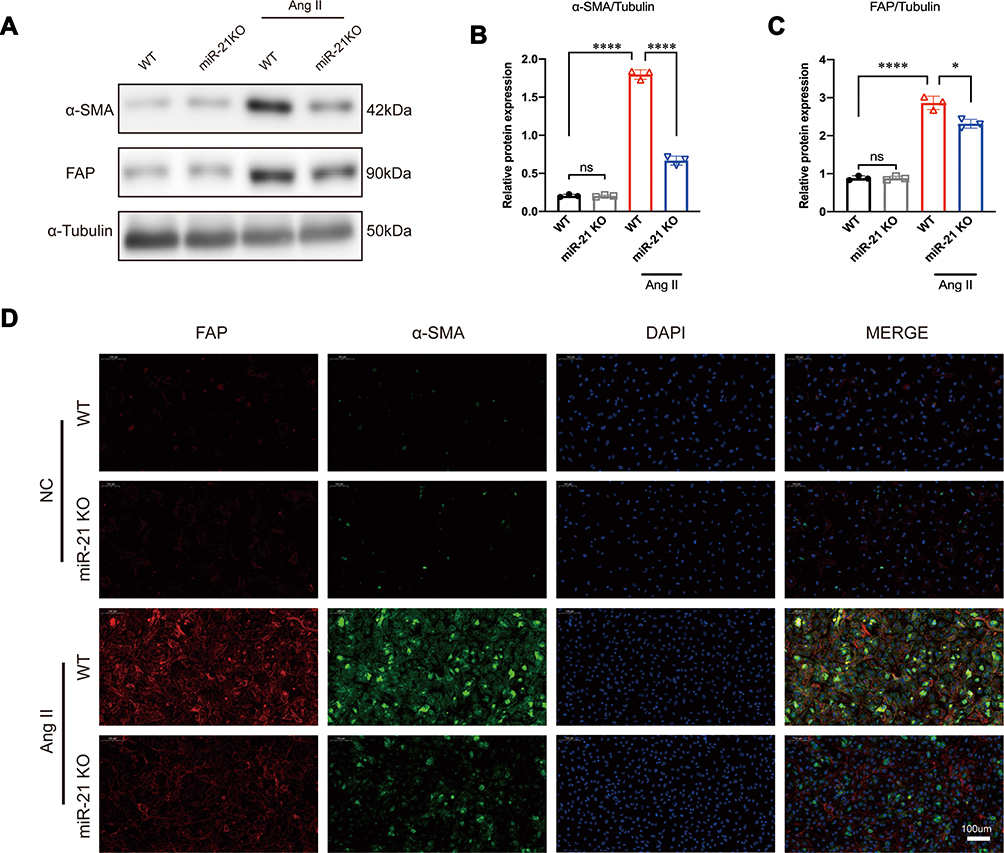 |
Figure 2 miR-21 promotes angiotensin II (Ang II)-induced myofibroblast transformation in vitro. (A–C) Western blotting analysis of myofibroblast markers in wild-type (WT) and miR-21 knockout (KO) primary cardiac fibroblasts (CFs) treated with or without Ang II. Left panel (A), representative blots; right panel (B and C), quantitative analysis; *P < 0.05 and ****P < 0.0001, n = 3 per group. (D) Representative immunofluorescence staining image of α-SMA and FAP in WT and miR-21 KO primary CFs treated with or without Ang II. Scale bar = 100 µm. |
Ang II-Induced ERK/TGF-β/Smad Signaling Could Be Partially Inhibited by miR-21 KO
It is well known that Ang II can activate the MAPK signaling pathway.13,14 c-Jun N-terminal kinase and ERK1/2 (also known as p42/44), key mediators of the MAPK cascade, have been reported to induce the expression of TGF-β in various fibrotic diseases.14,27 Other studies have reported that TGF-β can induce the upregulation of phosphorylated ERK1/2 (p-ERK1/2) in patients with fibrotic disorders.18,19 These results suggest a positive feedback mechanism between ERK1/2 and TGF-β signaling. Based on our aforementioned results that blocking miR-21 could inhibit Ang II-induced myofibroblast transformation, we further hypothesized that the positive feedback between ERK1/2 and TGF-β signaling is mediated by miR-21. To test this hypothesis, CFs were isolated from both WT and miR-21 KO neonatal mice and were treated with Ang II as described above. We found that in WT CFs, Ang II treatment significantly induced the expression of p-ERK1/2, TGF-β, and p-Smad2/3 (Figure 3A–E). In contrast, miR-21 KO mice CFs treated with Ang II showed reduced expression of p-ERK1/2, TGF-β, and p-Smad2/3 compared with WT counterparts (Figure 3A–E). These results indicate that Ang II-induced ERK1/2 and TGF-β signaling are partially regulated by miR-21.
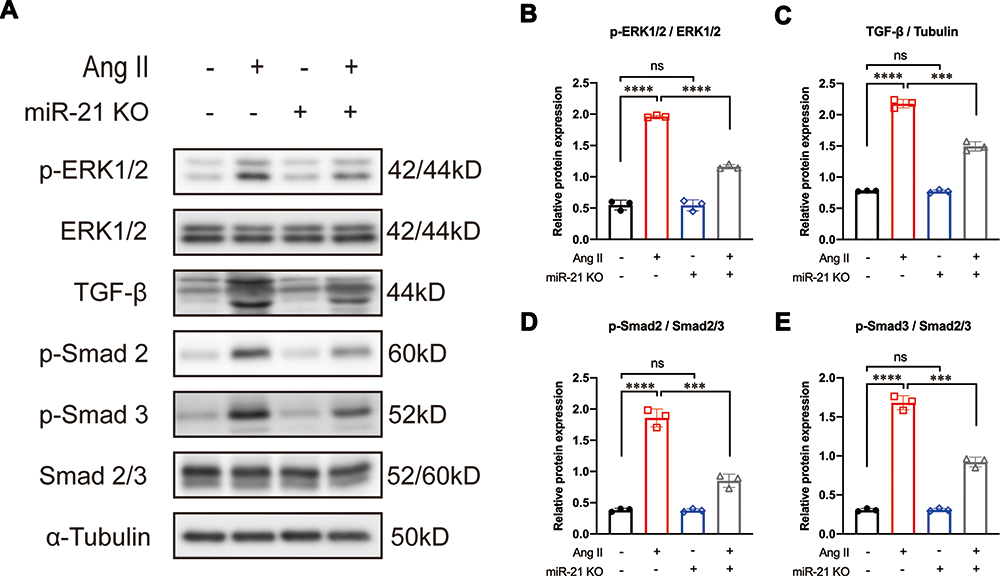 |
Figure 3 Knockout of miR-21 partially reverses angiotensin II (Ang II)-induced ERK/TGF-β/Smad activation. (A) Representative and (B–E) quantitative analysis of Western blots of miR-21 knockout (KO) on Ang II-induced ERK/TGF-β/Smad signaling in primary cardiac fibroblasts (CFs). Ang II induces the activation of ERK/TGF-β/Smad signaling in primary CFs. This effect could be reversed by the KO of miR-21. ***P < 0.001, ****P < 0.0001, n = 3 per group. |
Spry1 is a Target of miR-21 That Can Inhibit ERK Signaling
Previous studies identified Spry1 as one of the target genes of miR-21 that participated in pressure overload-induced cardiac fibrosis and temporomandibular joint osteoarthritis (TMJOA) process.10,28 Some other studies have also demonstrated that Spry1 as a potent inhibitor of the Ras/MEK/ERK pathway.29,30 Therefore, we further speculated that the positive feedback occurring between ERK and TGF-β signaling was mediated via the miR-21–Spry1 axis. To tests this hypothesis, we first explored the expression profile of Spry1 in miR-21 KO mice post MI. As expected, the qRT-PCR data showed that the expression of Spry1 was markedly increased in post-MI miR-21 KO mice compared with WT mice (Figure 4A). To explore the impact of Spry1 on ERK signaling, Spry1 was knocked down via AAV9 harboring siRNA administered via tail vein injection 14 days before the MI procedure. Two weeks after injection the heart was harvested to confirm that GFP was expressed in the myocardium (Supplementary Figure 2). As shown in Figure 4B–D, the protein expression level of Spry1 in miR-21 KO mice post-MI was significantly increased compared with that in WT mice, accompanied by decreased expression of p-ERK. Furthermore, inhibition of Spry1 in miR-21 KO mice reversed the effects of Spry1 on ERK signaling. These results suggest that Spry1, as one of the targets of miR-21, can inhibit the activation of ERK signaling post MI.
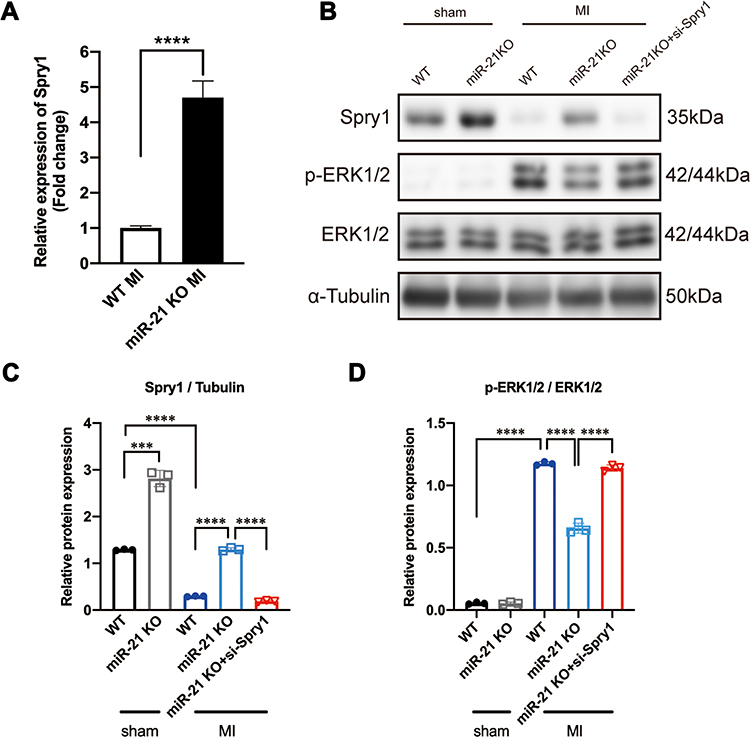 |
Figure 4 Sprouty1 (Spry1), an miR-21 target, inhibits the activation of ERK1/2 post-myocardial infarction (MI). (A) Relative expression of Spry1 between wild-type (WT) and miR-21 knockout (KO) mice at 2 weeks post-MI; ****P < 0.0001, n = 5 per group. (B–D) Representative (B) and quantitative Western blots (C and D) of Spry1, p-ERK, and ERK in WT and miR-21 KO mice myocardium; miR-21 KO mice that underwent MI induction exhibited increased expression of Spry1 and decreased expression of p-ERK1/2. Inhibition of Spry1 promotes the phosphorylation of ERK1/2. ***P < 0.001, ****P < 0.0001, n = 3 per group. |
Spry1 Inhibition Reversed the Effects of miR-21 KO on Ang II-Induced ERK/TGF-β Signaling and Myofibroblast Transformation
To investigate the impact of the miR-21–Spry1 axis on ERK/TGF-β signaling in CFs, si-Spry1 was used to transfect CFs isolated from miR-21 KO mice to evaluate whether it could reverse the effects of miR-21 on Ang II-induced downstream signaling. The results (Figure 5A–H) showed that the inhibitory effects of miR-21 KO on Ang II-induced ERK and TGF-β signaling as well as on α-SMA and FAP expression were abrogated upon Spry1 knockdown. These results confirm the role of the miR-21–Spry1 axis in regulating Ang II-induced ERK/TGF-β signaling in CFs.
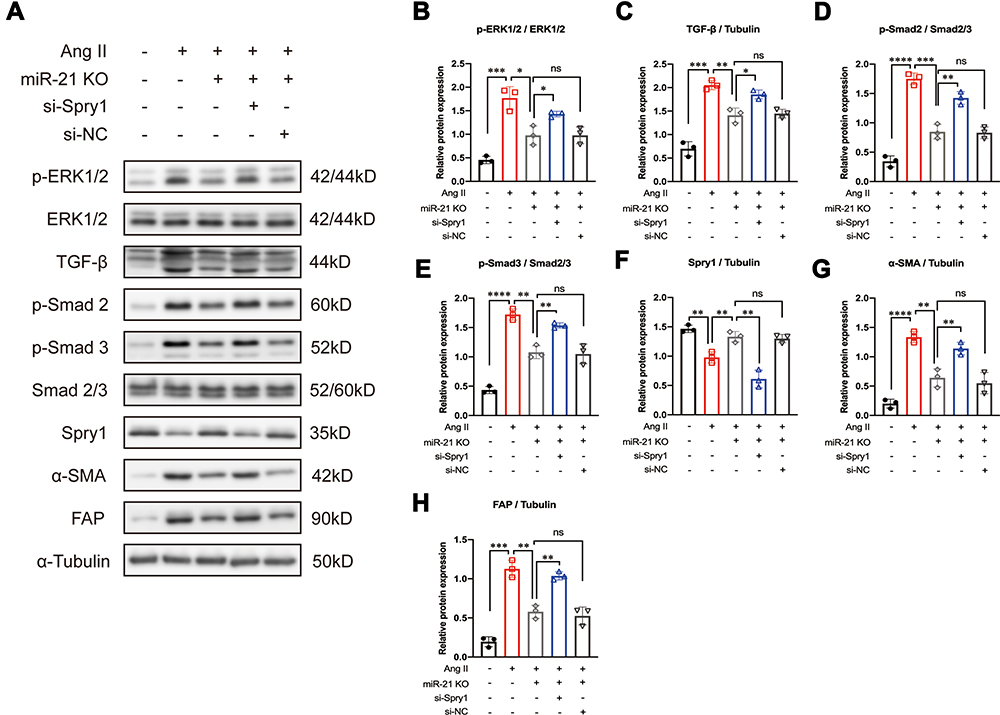 |
Figure 5 Inhibition of Sprouty1 (Spry1) reverses the effect of miR-21 knockout (KO) on angiotensin II (Ang II)-induced ERK/TGF-β/Smad activation in primary cardiac fibroblasts (CFs). (A) Representative and (B–H) quantitative analysis of Western blots. Inhibition of Spry1 reverses the effects of miR-21 KO on Ang II-induced ERK/TGF-β/Smad signaling in primary CFs; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n = 3 per group. |
Inhibition of Spry1 Reversed the Effects of miR-21 KO on Post-MI Fibrosis
To further test whether Spry1 knockdown could reverse the effects of miR-21 KO on post-MI cardiac fibrosis, AAV9 harboring Spry1 siRNA was injected into miR-21 KO mice via the tail vein. We found that when miR-21 KO mice were treated with Spry1 siRNA, the protective effects of miR-21 inhibition on post-MI cardiac fibrosis were abrogated, as evidenced by an enlarged fibrotic area (both replacement fibrosis and peri-infarct interstitial fibrosis) (Figure 6A–B). In addition, there were increased expression of fibrotic markers (Figure 6C–F) compared with miR-21 KO mice injected with AAV9-GFP and AAV9-si-NC, respectively. These results further confirm the role of the miR-21–Spry1 axis at the in vivo level of cardiac fibrosis.
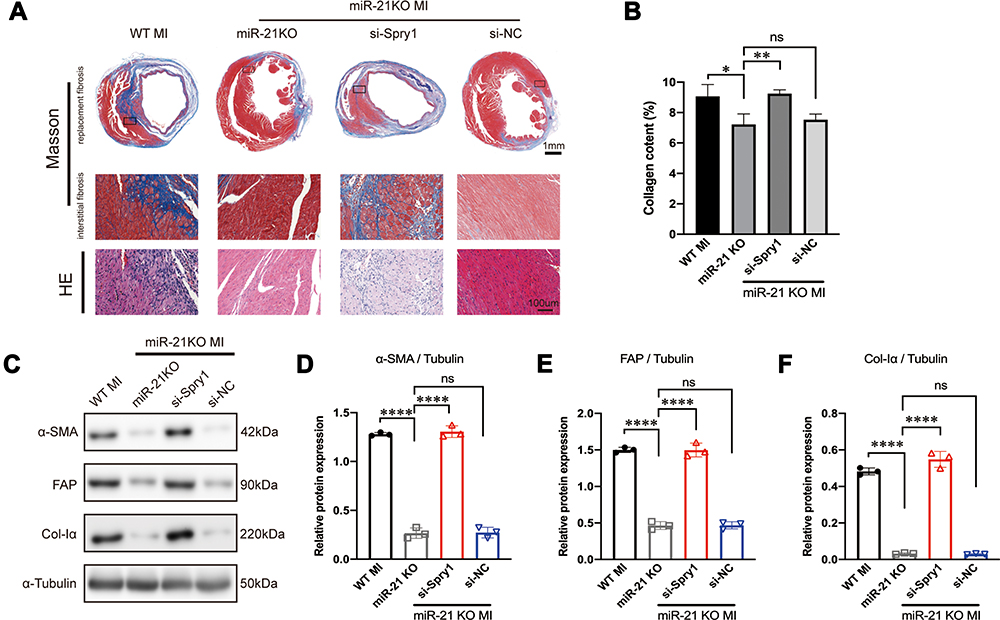 |
Figure 6 Inhibition of Spry1 reverses the effects of miR-21 knockout (KO) on post-myocardial infarction (MI) cardiac fibrosis. (A) Representative Masson’s trichrome and hematoxylin and eosin (H&E) staining of the cross section of the heart. The image shows that the inhibition of Spry1 reverses the effects of miR-21 KO on both replacement fibrosis and interstitial fibrosis post-MI. (B) Quantitative analysis of collagen content (%) between the groups shown in (A); *P < 0.05, **p < 0.01, n = 3 per group. (C) Representative Western blotting and (D–F) quantitative analysis of fibrotic markers in mice myocardium subjected to MI; ****P < 0.0001, n = 3 per group. |
Previous studies have shown that TGF-β/Smad signaling may promote DROSHA-mediated miR-21 maturation.31–34 Therefore, we proposed a positive feedback loop model of Ang II-induced post-MI fibrosis (Figure 7), in which mice underwent MI activate RAS and Ang II, thereby inducing ERK1/2 and TGF-β/Smad2/3 signaling. On the one hand, p-Smad2/3 could promote the transformation of CFs into myofibroblasts, leading to ECM production. In addition, it promoted the maturation and upregulation of miR-21, further enhancing the activation of ERK1/2 by targeting Spry1, an inhibitor of ERK.
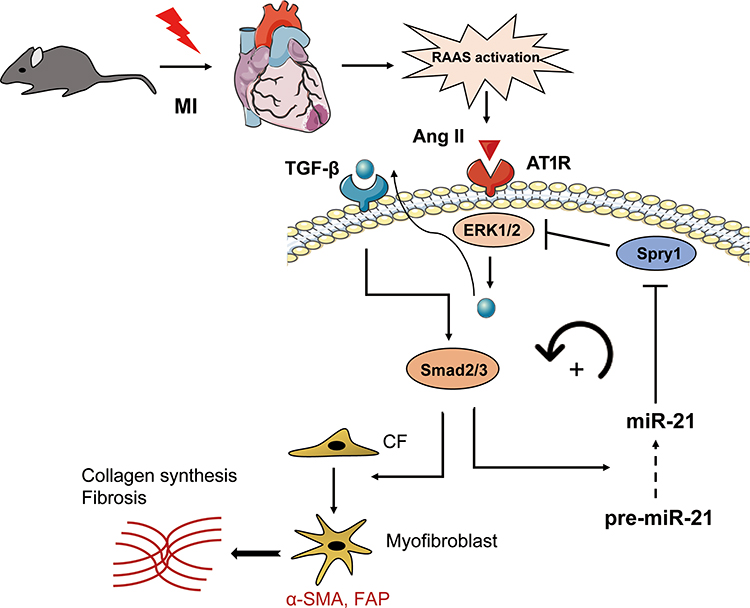 |
Figure 7 Schematic representation of the mechanisms by which miR-21 promotes post-MI cardiac fibrosis. |
Discussion
Several studies have demonstrated the role of miRNAs in pathological fibrosis. Among these fibrosis-related miRNAs, miR-21 has been identified as an important regulator of fibrosis in multiple organs, including the heart.10,33,35,36 Although recent studies have identified that miR-21 promotes post-MI cardiac fibrosis by targeting Smad7 and Jagged1,11,12 these studies only confirmed that TGF-β, the key regulator of fibrosis, and its downstream signaling were partially regulated by miR-21. As far as we know, both circulating and local RAS are rapidly activated post MI. The effects of RAS on fibroblast activation may be mediated, at least in part, via TGF-β signaling.25 Ang II, the primary effector of RAS, exerts its profibrotic activity in infarct myofibroblasts mainly via the type I (AT1) receptor.37 Previous studies have suggested that Ang II activates ERK signaling,13,14 while others have reported that TGF-β induces ERK activation in patients with fibrotic disorders.18,19 Therefore, we hypothesized that a feedback loop occurs between Ang II/ERK and TGF-β signaling. In the present study, we reported that miR-21 mediates a positive feedback between Ang II-induced ERK and TGF-β signaling by targeting Spry1 post-MI.
As we know, there are two strands of miR-21, in which miR-21-5p is the guide strand and miR-21-3p is the passenger strand. Bang et al20 has demonstrated that the expression of the guide strand (miR-21-5p) was higher than that of the passenger strand (miR-21-3p) in CF. The study further demonstrated that miR-21-3p was enriched in fibroblast-derived exosomes and was transported out of the CF, further mediating cardiomyocyte hypertrophy, whereas miR-21-5p remained inside the CF. Therefore, we speculate that miR-21-5p mediates biological and pathological functions mainly inside the CF. Hence we chose miR-21-5p (abbreviated as miR-21) for the present study. We first established a mice model of post-MI cardiac fibrosis using WT and miR-21 KO mice to verify the roles of miR-21 in post-MI fibrosis. Consistent with a previous study,12 we demonstrated that miR-21 was significantly upregulated in the infarcted region 14 days post-MI. Moreover, TGF-β, collagen Iα, and α-SMA, all key markers in the development of fibrosis, as well as inflammatory cytokines, were upregulated. FAP, a previously identified cell surface glycoprotein that is expressed only in activated fibroblasts,38,39 was also detected in the infarcted zone post-MI. In vivo KO of miR-21 attenuated fibrosis post-MI, as evidenced by Masson’s trichrome staining and the production of fibrotic markers and inflammatory cytokines, and improved cardiac function. Additionally, in vitro cultivation of primary CFs demonstrated that miR-21 promotes myofibroblast transformation triggered by Ang II. These results suggest that miR-21 promotes post-MI cardiac fibrosis in CFs.
Previous studies have reported that Ang II can stimulate the production and release of TGF-β from CFs by activating the MAPK pathway.13,40,41 Ang II-induced cardiac fibrosis is partially attributed to the TGF-β/Smad2/3 pathway.13,15–17 In the present study, to evaluate the role of miR-21 in Ang II-induced ERK/TGF-β/Smad signaling we performed in vitro isolation of WT and miR-21 KO primary CFs. As expected, CFs treated with Ang II exhibited the increased expression of p-ERK1/2, TGF-β, Smad2, and Smad3, whereas KO of miR-21 in CFs attenuated Ang II-induced signaling. These results suggest that miR-21 partially promotes fibrosis by regulating the Ang II/ERK/TGF-β/Smad pathway.
Previous studies have shown that biogenesis of miR-21 is regulated by TGF-β/Smad signaling in smooth muscle cell.31,32 Another study has found that TGF-β can induce miR-21 expression post MI.11 Based on our aforementioned results, we speculated that miR-21 and one of its targets participate in the feedback between ERK and TGF-β signaling and identified Spry1, a previously reported target of miR-21 in pressure overload-induced cardiac fibrosis and temporomandibular joint osteoarthritis process,10,28 which is a potent inhibitor of the Ras/MEK/ERK signaling pathway.29,30 Numerous studies have shown that Spry1 promotes fibroblast apoptosis by deactivating the ERK/MAPK pathway.10,42–44 Therefore, we explored the role of the miR-21–Spry1 axis in the Ang II/ERK/TGF-β/Smad pathway. Our results showed that miR-21 KO mice subjected to the MI procedure exhibited increased mRNA expression of Spry1 compared with the WT MI mice. Western blotting further confirmed that the upregulation of Spry1 was accompanied by decreased phosphorylation of ERK1/2. In addition, inhibition of Spry1 by AAV9 siRNA in miR-21 KO MI mice promoted the activation of ERK signaling. Moreover, we noted that the expression of Spry1 in miR-21 KO mice post MI was decreased compared with that in miR-21 sham mice. We cannot exclude the possibility that Spry1 was targeted by other miRNAs post MI. In conclusion, we suggest that miR-21 promotes Ang II/ERK signaling by targeting Spry1 post MI.
To further confirm the role of the miR-21–Spry1 axis in Ang II/ERK/TGF-β signaling and myofibroblast transformation, we performed an in vitro rescue study. Inhibition of Spry1 in miR-21 KO CFs reversed the effects of miR-21 KO on ERK/TGF-β signaling and promoted myofibroblast transformation. Moreover, studies have shown that TGF-β/Smad signaling is important for DROSHA-mediated miRNA-21 maturation.31,32 Our data confirmed that Ang II-induced TGF-β/Smad2/3 activation was regulated by miR-21; however, the miR-21–Spry1 axis could inversely regulate Ang II-induced ERK signaling. Therefore, we speculate that the Ang II-mediated activation of TGF-β/Smad2/3 in this study may regulate DROSHA protein levels and ultimately control miR-21 levels in activated CFs. However, this possibility needs to be thoroughly investigated in future studies.
To further verify the role of miR-21/Spry1 in post-MI cardiac fibrosis, we performed an in vivo rescue study, in which inhibition of Spry1 partially reversed the effects of miR-21 on cardiac fibrosis and expression of fibrotic markers. These results further confirm the role of the miR-21–Spry1 axis at an in vivo level.
We proposed a pathway model to elucidate the potential role of miR-21 in post-MI cardiac fibrosis. In brief, acute MI activates RAS, thereby promoting Ang II expression, which further activates ERK/MAPK signaling. Activated ERK1/2 (ie, p-ERK1/2) subsequently promotes TGF-β/Smad2/3 activation, which may further promotes the maturation of miR-21. By targeting Spry1, miR-21 exerts a positive feedback on Ang II-induced ERK activation. Our study is the first to evaluate the role of miR-21 and its target Spry1 in MI and subsequent cardiac fibrosis. In addition, we further suggested that miR-21 and Spry1 mediated a positive feedback loop between Angiotensin II/ERK and TGF-β/Smad signaling, which is also novel. Therefore, manipulating the miR-21–Spry1 axis may block this positive feedback loop, thereby providing a promising therapeutic target for alleviating post-MI cardiac fibrosis and remodeling.
Our study has some limitations. First, although our results suggest that miR-21 plays a key role in regulating Ang II-induced post-MI cardiac fibrosis, it is important to note that miR-21 has many downstream targets in addition to Spry1. Therefore, we cannot exclude the fact that miR-21 promotes Ang II-induced cardiac fibrosis by targeting other genes. Second, because previous studies have indicated that the TGF-β/Smad2/3 pathway promotes the maturation of miR-21 in smooth muscle cells, we have speculated that a similar mechanism exists in CFs. Therefore, further investigations are warranted to elucidate the positive feedback mechanism of miR-21 in promoting Ang II-induced cardiac fibrosis.
Conclusions
In summary, the present study explored the correlation and causal relationship of miR-21 with post-MI cardiac fibrosis in vivo and determined the positive feedback of miR-21 on the Ang II-induced ERK/TGF-β/Smad pathway. Both the in vivo and in vitro results demonstrate that miR-21 promotes cardiac fibrosis by targeting Spry1. Our findings may provide new insights into the mechanisms underlying post-MI cardiac fibrosis and may help in identifying novel therapeutic targets for ameliorating post-MI cardiac fibrosis.
Data Sharing Statement
The data used to support the findings of this study are available from the corresponding author upon request.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81870264).
Disclosure
The authors declare that they have no conflicting interests.
References
1. McLaughlin S, McNeill B, Podrebarac J, et al. Injectable human recombinant collagen matrices limit adverse remodeling and improve cardiac function after myocardial infarction. Nat Commun. 2019;10(1):4866. doi:10.1038/s41467-019-12748-8
2. Davis J, Molkentin JD. Myofibroblasts: trust your heart and let fate decide. J Mol Cell Cardiol. 2014;70:9–18. doi:10.1016/j.yjmcc.2013.10.019
3. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71(4):549–574.
4. Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. 2009;105(12):1164–1176. doi:10.1161/CIRCRESAHA.109.209809
5. Lorenzen J, Kumarswamy R, Dangwal S, Thum T. MicroRNAs in diabetes and diabetes-associated complications. RNA Biol. 2012;9(6):820–827. doi:10.4161/rna.20162
6. van Rooij E, Olson EN. MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J Clin Invest. 2007;117(9):2369–2376. doi:10.1172/JCI33099
7. Thum T. Noncoding RNAs and myocardial fibrosis. Nat Rev Cardiol. 2014;11(11):655–663. doi:10.1038/nrcardio.2014.125
8. He X, Zhang K, Gao X, et al. Rapid atrial pacing induces myocardial fibrosis by down-regulating Smad7 via microRNA-21 in rabbit. Heart Vessels. 2016;31(10):1696–1708. doi:10.1007/s00380-016-0808-z
9. Villar AV, Garcia R, Merino D, et al. Myocardial and circulating levels of microRNA-21 reflect left ventricular fibrosis in aortic stenosis patients. Int J Cardiol. 2013;167(6):2875–2881. doi:10.1016/j.ijcard.2012.07.021
10. Thum T, Gross C, Fiedler J, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456(7224):980–984. doi:10.1038/nature07511
11. Zhou XL, Xu H, Liu ZB, Wu QC, Zhu RR, Liu JC. miR-21 promotes cardiac fibroblast-to-myofibroblast transformation and myocardial fibrosis by targeting Jagged1. J Cell Mol Med. 2018;22(8):3816–3824. doi:10.1111/jcmm.13654
12. Yuan J, Chen H, Ge D, et al. Mir-21 promotes cardiac fibrosis after myocardial infarction via targeting Smad7. Cell Physiol Biochem. 2017;42(6):2207–2219. doi:10.1159/000479995
13. Pan XC, Liu Y, Cen YY, et al. Dual role of triptolide in interrupting the NLRP3 inflammasome pathway to attenuate cardiac fibrosis. Int J Mol Sci. 2019;20(2):360. doi:10.3390/ijms20020360
14. Li L, Fan D, Wang C, et al. Angiotensin II increases periostin expression via Ras/p38 MAPK/CREB and ERK1/2/TGF-beta1 pathways in cardiac fibroblasts. Cardiovasc Res. 2011;91(1):80–89. doi:10.1093/cvr/cvr067
15. Ding J, Tang Q, Luo B, et al. Klotho inhibits angiotensin II-induced cardiac hypertrophy, fibrosis, and dysfunction in mice through suppression of transforming growth factor-beta1 signaling pathway. Eur J Pharmacol. 2019;859:172549. doi:10.1016/j.ejphar.2019.172549
16. Ge W, Zhang W, Gao R, Li B, Zhu H, Wang J. IMM-H007 improves heart function via reducing cardiac fibrosis. Eur J Pharmacol. 2019;857:172442. doi:10.1016/j.ejphar.2019.172442
17. Chen RR, Fan XH, Chen G, et al. Irisin attenuates angiotensin II-induced cardiac fibrosis via Nrf2 mediated inhibition of ROS/TGFbeta1/Smad2/3 signaling axis. Chem Biol Interact. 2019;302:11–21. doi:10.1016/j.cbi.2019.01.031
18. Li J, Feng M, Sun R, et al. Andrographolide ameliorates bleomycin-induced pulmonary fi brosis by suppressing cell proliferation and myofibroblast differentiation of fibroblasts via the TGF-beta1-mediated Smad-dependent and -independent pathways. Toxicol Lett. 2020;321:103–113. doi:10.1016/j.toxlet.2019.11.003
19. Wang D, Yan Z, Bu L, et al. Protective effect of peptide DR8 on bleomycin-induced pulmonary fibrosis by regulating the TGF-beta/MAPK signaling pathway and oxidative stress. Toxicol Appl Pharmacol. 2019;382:114703. doi:10.1016/j.taap.2019.114703
20. Bang C, Batkai S, Dangwal S, et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124(5):2136–2146. doi:10.1172/JCI70577
21. Percie Du Sert N, Hurst V, Ahluwalia A, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol. 2020;18(7):e3000410. doi:10.1371/journal.pbio.3000410
22. Gao E, Lei YH, Shang X, et al. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res. 2010;107(12):1445–1453. doi:10.1161/CIRCRESAHA.110.223925
23. Carroll KJ, Makarewich CA, McAnally J, et al. A mouse model for adult cardiac-specific gene deletion with CRISPR/Cas9. Proc Natl Acad Sci U S A. 2016;113(2):338–343. doi:10.1073/pnas.1523918113
24. Fu Q, Lu Z, Fu X, Ma S, Lu X. MicroRNA 27b promotes cardiac fibrosis by targeting the FBW7/Snail pathway. Aging (Albany NY). 2019;11(24):11865–11879. doi:10.18632/aging.102465
25. Frangogiannis NG. Pathophysiology of myocardial infarction. Compr Physiol. 2015;5(4):1841–1875.
26. Schnee JM, Hsueh WA, Angiotensin II. adhesion, and cardiac fibrosis. Cardiovasc Res. 2000;46(2):264–268. doi:10.1016/S0008-6363(00)00044-4
27. Aoki H, Ohnishi H, Hama K, et al. Autocrine loop between TGF-beta1 and IL-1beta through Smad3- and ERK-dependent pathways in rat pancreatic stellate cells. Am J Physiol Cell Physiol. 2006;290(4):C1100–8. doi:10.1152/ajpcell.00465.2005
28. Ma S, Zhang A, Li X, et al. MiR-21-5p regulates extracellular matrix degradation and angiogenesis in TMJOA by targeting Spry1. Arthritis Res Ther. 2020;22(1):99. doi:10.1186/s13075-020-2145-y
29. Hanafusa H, Torii S, Yasunaga T, Nishida E. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat Cell Biol. 2002;4(11):850–858. doi:10.1038/ncb867
30. Casci T, Vinos J, Freeman M. Sprouty, an intracellular inhibitor of Ras signaling. Cell. 1999;96(5):655–665. doi:10.1016/S0092-8674(00)80576-0
31. Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454(7200):56–61. doi:10.1038/nature07086
32. Blahna MT, Hata A. Smad-mediated regulation of microRNA biosynthesis. FEBS Lett. 2012;586(14):1906–1912. doi:10.1016/j.febslet.2012.01.041
33. Zhong X, Chung AC, Chen HY, Meng XM, Lan HY. Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J Am Soc Nephrol. 2011;22(9):1668–1681. doi:10.1681/ASN.2010111168
34. Verma SK, Garikipati VNS, Krishnamurthy P, et al. Interleukin-10 inhibits bone marrow fibroblast progenitor cell-mediated cardiac fibrosis in pressure-overloaded myocardium. Circulation. 2017;136(10):940–953. doi:10.1161/CIRCULATIONAHA.117.027889
35. Zhao J, Tang N, Wu K, et al. MiR-21 simultaneously regulates ERK1 signaling in HSC activation and hepatocyte EMT in hepatic fibrosis. PLoS One. 2014;9(10):e108005. doi:10.1371/journal.pone.0108005
36. Liu G, Friggeri A, Yang Y, et al. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med. 2010;207(8):1589–1597. doi:10.1084/jem.20100035
37. Sun Y, Weber KT. Infarct scar: a dynamic tissue. Cardiovasc Res. 2000;46(2):250–256. doi:10.1016/S0008-6363(00)00032-8
38. Aghajanian H, Kimura T, Rurik JG, et al. Targeting cardiac fibrosis with engineered T cells. Nature. 2019;573(7774):430–433. doi:10.1038/s41586-019-1546-z
39. Tillmanns J, Hoffmann D, Habbaba Y, et al. Fibroblast activation protein alpha expression identifies activated fibroblasts after myocardial infarction. J Mol Cell Cardiol. 2015;87:194–203. doi:10.1016/j.yjmcc.2015.08.016
40. Autelitano DJ, Ridings R, Pipolo L, Thomas WG. Adrenomedullin inhibits angiotensin AT1A receptor expression and function in cardiac fibroblasts. Regul Pept. 2003;112(1–3):131–137. doi:10.1016/S0167-0115(03)00031-4
41. Artlett CM. The role of the NLRP3 inflammasome in fibrosis. Open Rheumatol J. 2012;6:80–86. doi:10.2174/1874312901206010080
42. Adam O, Lohfelm B, Thum T, et al. Role of miR-21 in the pathogenesis of atrial fibrosis. Basic Res Cardiol. 2012;107(5):278. doi:10.1007/s00395-012-0278-0
43. Qiao G, Xia D, Cheng Z, Zhang G. Role of Sprouty1 (Spry1) in the pathogenesis of atrial fibrosis. Pathol Res Pract. 2018;214(2):308–313. doi:10.1016/j.prp.2017.04.021
44. Wang W, Zhang K, Li X, et al. Doxycycline attenuates chronic intermittent hypoxia-induced atrial fibrosis in rats. Cardiovasc Ther. 2018;36(3):e12321. doi:10.1111/1755-5922.12321
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.