")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Special Issue on diverse applications in Nano-Theranostics
Microglial AGE-albumin is critical for neuronal death in Parkinson's disease: a possible implication for theranostics
Authors Bayarsaikhan E, Bayarsaikhan D, Lee J, Son M, Oh S, Moon J, Park H, Roshini A, Kim S, Song B, Jo S, Byun K, Lee B
Received 25 August 2015
Accepted for publication 17 December 2015
Published 23 August 2016 Volume 2015:10(Special Issue on diverse applications in Nano-Theranostics) Pages 281—292
DOI https://doi.org/10.2147/IJN.S95077
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Enkhjargal Bayarsaikhan,1,2,* Delger Bayarsaikhan,1,* Jaesuk Lee,1 Myeongjoo Son,1,3 Seyeon Oh,1 Jeongsik Moon,1 Hye-Jeong Park,1 Arivazhagan Roshini,1 Seung U Kim,4 Byoung-Joon Song,5 Seung-Mook Jo,6 Kyunghee Byun,1,3 Bonghee Lee1,3
1Center for Regenerative Medicine, Lee Gil Ya Cancer and Diabetes Institute, Gachon University, Incheon, Republic of Korea; 2Department of General Laboratory, National Cancer Center of Mongolia, Ulaanbaatar, Mongolia; 3Department of Anatomy and Cell Biology, Graduate School of Medicine, Gachon University, Incheon, Republic of Korea; 4Department of Medicine, University of British Columbia, Vancouver, Canada; 5Laboratory of Membrane Biochemistry and Biophysics, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, MD, USA; 6Department of Emergency Medical Services, Eulji University, Seongnam-si, Gyeonggi-do, Republic of Korea
*These authors contributed equally to this work
Abstract: Advanced glycation end products (AGEs) are known to play an important role in the pathogenesis of neurodegenerative diseases, including Parkinson’s disease (PD), by inducing protein aggregation and cross-link, formation of Lewy body, and neuronal death. In this study, we observed that AGE-albumin, the most abundant AGE product in the human PD brain, is synthesized in activated microglial cells and accumulates in the extracellular space. AGE-albumin synthesis in human-activated microglial cells is distinctly inhibited by ascorbic acid and cytochalasin treatment. Accumulated AGE-albumin upregulates the receptor to AGE, leading to apoptosis of human primary dopamine (DA) neurons. In animal experiments, we observed reduced DA neuronal cell death by treatment with soluble receptor to AGE. Our study provides evidence that activated microglial cells are one of the main contributors in AGE-albumin accumulation, deleterious to DA neurons in human and animal PD brains. Finally, activated microglial AGE-albumin could be used as a diagnostic and therapeutic biomarker with high sensitivity for neurodegenerative disorders, including PD.
Keywords: theragnostic, microglia, AGE-albumin, neuronal death, receptor of AGE, Parkinson’s disease
Introduction
Parkinson’s disease (PD) is a common neurodegenerative disorder due to loss of brain dopamine (DA) neurons in humans.1–4 The symptoms of human PD occur after 60%–80% of DA neurons in brain are damaged by various causes, including genetic or sporadic with toxic chemicals and drugs.5–9 In genetic PD, several mutations centered on α-synuclein are reported and are responsible for protein misfolding and aggregation and mitochondrial dysfunction.10,11 However, the majority of neuronal cell deaths are associated with exposure to environmental toxins such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and rotenone, which are widely used as herbicide parquet, insecticide, and fish poison.12–15
Many studies have shown that activated microglial cells are key contributors to a variety of neurodegenerative disease progression, including PD and Alzheimer’s disease (AD).16–19 We recently reported that human microglial cells synthesize and secrete advanced glycation end product-albumin (AGE-albumin) in AD, and AGE-albumin is one of the possible inducers of neuronal death through an increase in the receptor to AGE (RAGE) in humans and animals.20–23 The pathophysiology of PD brain damage is also highly associated with inflammation through microglial activation. Accumulated AGE-albumin from activated microglial cells continuously induces neuronal RAGE-albumin, resulting in DA neuronal death.
RAGE is a member of the immunoglobulin family. There is a single transmembrane-spanning domain and a very short (50 amino acids) cytoplasmic domain that is highly charged.24 The cytoplasmic domain of RAGE is actively required for RAGE-dependent signaling. AGE–RAGE binding stimulates activation of diverse signaling cascades – mitogen-activated protein kinases and phosphoinositol-3 kinase/Akt to Jak/stat pathways, as examples – and finally leads to apoptotic pathway.25
Thus, we aimed at assessing whether AGE-albumin from activated microglial cells is a key inducer of dopaminergic neuronal degeneration in human and mouse PD and whether AGE-albumin accumulation could be used as a diagnostic and therapeutic biomarker with high sensitivity for the same.
Materials and methods
Cell culture
An immortalized human microglial cell line (HMO6) was used for in vitro studies. HMO6 cells were grown in Dulbecco’s Modified Eagle’s Medium (Thermo Fisher Scientific, Waltham, MA, USA) containing a high glucose concentration supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) and 20 mg/mL gentamycin (Sigma-Aldrich Co., St Louis, MO, USA). These cells were maintained at 37°C with 5% CO2 and humidified 95% air. HMO6 cells were exposed to rotenone (Rot; Sigma-Aldrich Co.) at concentrations from 1 to 10 nM during four times of passage. For inhibition studies, HMO6 cells were first exposed to 500 nM cytochalasin D (CD; Sigma-Aldrich Co.) for 20 minutes and 500 μM ascorbic acid (Acs; Sigma-Aldrich Co.) for 30 minutes, followed by fresh medium with rotenone treatment. All procedures including the use of human cells and tissues were approved by the Institutional review board of LCDI, Gachon University.
Human DA neuron
Human Lund human mesencephalic (LUHMES) cells were kindly provided with the courtesy donation from Dr Marcel Leist of Kontanz University, Germany.26 LUHMES cells were grown at 37°C in a humidified 95% air and 5% CO2 atmosphere. For this cell culture, plastic cell culture dishes, flasks, and multiwell plates were precoated with 50 mg/mL poly-L-orthinine (Sigma-Aldrich Co.) and 1 μg/mL fibronectin (Sigma-Aldrich Co.). Before differentiation, LUHMES cells were maintained in proliferation medium (advanced DMEM/F12 medium; Thermo Fisher Scientific) supplemented with N-2 (Thermo Fisher Scientific), 2 mM L-glutamine (Thermo Fisher Scientific), and 40 ng/mL recombinant basic fibroblast growth factor (R&D Systems, Inc., Minneapolis, MN, USA). Cells were splatted from one dish into ten new dishes when confluence reached 80%. For differentiation, 8×106 LUHMES were seeded into a T175 flask (Thermo Fisher Scientific) in proliferation medium. After 24 hours of attachment, the proliferation medium was replaced with differentiation medium (consisting of advanced DMEM/F12, N-2 supplement, 2 mM L-glutamine, 1 mM dibutyryl 3′,5′-cyclic adenosine monophosphate; Sigma-Aldrich Co.), 1 μg/mL tetracycline (Sigma-Aldrich Co.), and 2 ng/mL recombinant human glial cell-derived neurotrophic factor (R&D Systems, Inc.), and predifferentiation was completed 48 hours later; cells were then seeded into poly-L-orthinine/fibronectin precoated multiwell plates at a density of 1.5×105 cells/cm2. After additional 72 hours of incubation, cultivation of DA neuronal differentiation was completed.
Human brain tissue
Brain tissues of normal (n=5) and PD patients (n=5), including substantia nigra (SN) part, were received from the Department of Structural Pathology, Institute of Nephrology, Nigata University, Japan. Lee Gil Ya Cancer and Diabetes Institute’s EC/IRB does not require patient’s consent because brain samples were already approved from Niigata University Hospital for research purpose.
Rotenone-exposed animals
Animal experiments were conducted in the Center of Animal Care and Use of Lee Gil Ya Cancer and Diabetes Institute of Gachon University, with C57BL/6N mice (20–22 g). Eight-week-old male mice were randomly divided and housed with five mice per cage under a temperature-controlled environment with a 12-hour light–dark cycle, with free access to food and water. All the animal protocols described in this study were approved by the Center of Animal Care and Use animal center ethical board. We followed the guidelines of Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) and IRB approval number was LCDI-2011-0041.
To establish a suitable PD model, 30 mg/kg of rotenone (Sigma-Aldrich Co.) suspended in 0.5% carboxymethyl cellulose was administered orally once a day for 2 months. Rotenone treatment concentration and time were chosen based on a previous report.27
Soluble RAGE or pyridoxamine injection by stereotaxic manipulation and tissue preparation
On day 30 of rotenone oral injection, 30 animals were randomly divided into the following three groups: rotenone, rotenone/soluble RAGE (sRAGE) (BioVendor, Brno, Czech Republic), and rotenone/pyridoxamine (PM) (Sigma-Aldrich Co.) injection groups, with ten mice in each group. Animals were anesthetized with ketamine HCl (0.75 mg/kg body weight) and xylazine (1 mg/kg body weight) prior to surgical procedures. Injection was performed by unilateral administration of sRAGE or PM into the right SN (posteriorly 0.4 mm, laterally 1.8 mm, and ventrally 3.5 mm from Bregma). sRAGE or PM was injected using a 30 G Hamilton syringe attached to an automated microinjector (KD Scientific, Holliston, MA, USA). Three microliters of 10 μM of sRAGE and PM were injected slowly at the rate of 1 μL/min using an automatic microinjector. Then the syringe was removed slowly, and the surgical wounds were sutured with wound clips, followed by topical treatment with antibiotics. To assess the effect of drug on the neurons, perfusion was performed at intervals of 1 week and 1 month.
Perfusion was performed under anesthesia through the heart with 50 mL phosphate-buffered saline (PBS; Thermo Fisher Scientific) followed by 50 mL cold fixative consisting of 4% paraformaldehyde (PFA; Sigma-Aldrich Co.). After perfusion, the brain was removed and post fixed for 5 hours in 4% PFA and then overnight in 20% sucrose. Cryoprotected brain blocks were cut into 10 μm slices on a cryostat.
Immunostaining
Paraffin-embedded brain tissue sections of normal and PD patients were deparaffinized at room temperature in xylene two times for 10 minutes, followed by dehydration with a graded series of ethanol (95% ethanol for 15 minutes, 70% ethanol for 1 minute, and 50% ethanol for 1 minute). Heat-induced antigen retrieval step was performed in Tris-ethylenediaminetetraacetic acid (EDTA) buffer pH 9.0 (Sigma-Aldrich Co.) by boiling at 100°C. Tissues were rinsed with cold distilled water and Tris-PBS. Nonspecific binding of antibodies was blocked using 3% normal serum. Frozen sections of mouse brain were washed five times with PBS and incubated with protein-specific antibodies (Table 1).
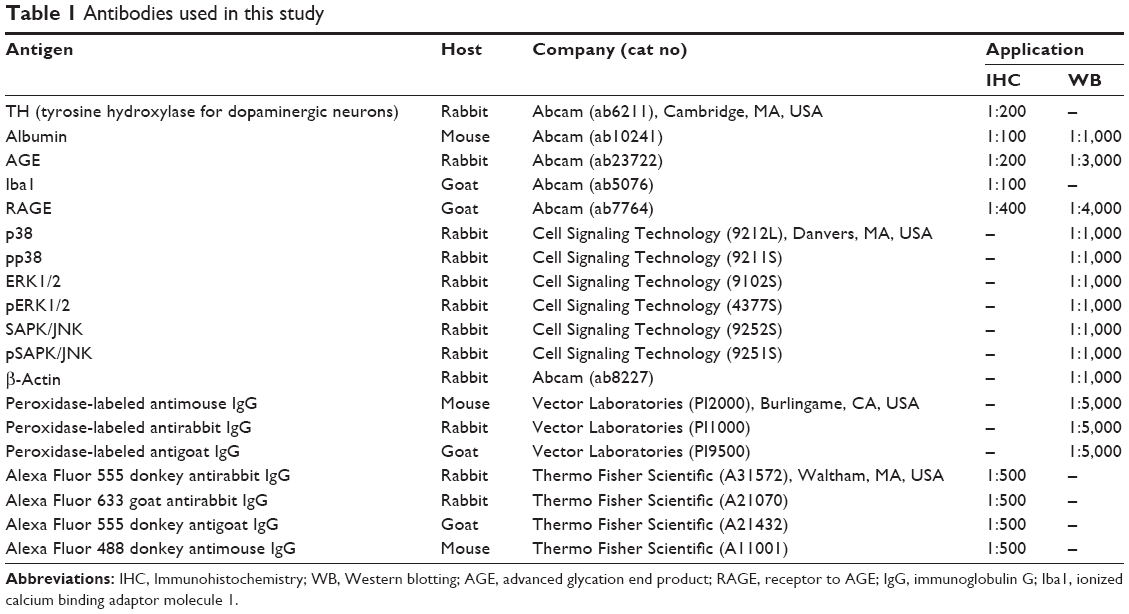 | Table 1 Antibodies used in this study |
HMO6 cells were grown in chamber slides, rinsed in PBS, and fixed in 100% methyl alcohol for 20 minutes, followed by washing three times with PBS. Nonspecific binding of antibodies was blocked by normal goat, rabbit, or horse serum (Vector Laboratories, Burlingame, CA, USA). After overnight incubation of primary antibodies at 4°C, samples were washed with PBS, followed by secondary antibody incubation step at room temperature for 1 hour. For counterstaining of nucleus, samples were incubated with 4′,6-diamino-2-phenylindole (1 μg/mL; Sigma-Aldrich Co.) for 20 seconds. After washing with PBS, coverslips were mounted on glass slides using Vectashield mounting media (Vector Laboratories) and analyzed using an LSM 710 confocal microscope (Carl Zeiss Meditec AG, Jena, Germany).
Cresyl violet staining
Frozen sections of mouse brain were dried at room temperature for 5 minutes and washed five times with PBS every 10 minutes, followed by incubation in graded ethanol (95% ethanol for 15 minutes, 70% ethanol for 1 minute, and 50% ethanol for 1 minute). After washing with distilled water, brain tissues were stained in 0.5% cresyl violet acetate (Sigma-Aldrich Co.) solution for 2 minutes and passed through distilled water (1 minute), 50% ethanol (1 minute), 70% ethanol (2 minutes), 95% ethanol (two times for 2 minutes), 100% ethanol (1 minute), and finally cleansed in xylene (5 minutes). Stained slides were mounted using DPX mounting medium for histological analysis (Sigma-Aldrich Co.).
Western blotting
Cell lysates were prepared with lysis buffer containing 7 M urea, 2 M thiourea, 4% CHAPS, and 5 mM DTT (Amresco, Solon, OH, USA), and protease inhibitor was added (Hoffman-La Roche Ltd., Basel, Switzerland) followed by sonication. The lysates were centrifuged at 14,000× g for 20 minutes at 4°C. Total protein concentration was measured by QUBIT (Thermo Fisher Scientific) according to the manufacturer’s method. Equal amounts (30 μg) of proteins were separated in 12% polyacrylamide gels (Thermo Fisher Scientific) and transferred to a nitrocellulose membrane (EMD Millipore, Billerica, MA, USA). Proteins were detected with protein-specific antibodies. ECL (Sigma-Aldrich Co.) detection reagent was used to visualize the immunoreactive proteins on the membrane.
Coimmunoprecipitation
Cell lysates were prepared with lysis buffer containing 20 mM Tris HCL, 10% glycerol, 1% Triton X-100, 2 mM EDTA, and 137 mM NaCl (Amresco), and protease inhibitor (Hoffman-La Roche Ltd.) was added, followed by sonication. The lysates were centrifuged at 14,000× g for 20 minutes at 4°C. The collected supernatant was incubated overnight with antibody at 4°C under rotation. Immunoprecipitates were collected by protein G agarose beads (Thermo Fisher Scientific). Nonspecific binding of agarose beads was blocked by 5% bovine serum albumin. One hundred microliters of bead were incubated with samples for 4 hours at 4°C under rotation, followed by centrifugation, and the supernatant was removed. Beads were washed with lysis buffer, and the immunoprecipitates were resuspended in sodium dodecyl sulfate sample buffer and then heated at 70°C for 10 minutes. Equal amounts (10 μL) of protein were separated in 12% polyacrylamide gels (Thermo Fisher Scientific) and transferred to a nitrocellulose membrane (EMD Millipore). Proteins were detected with protein-specific antibodies.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay
On the first day of the incubation, the cells were seeded into 96-well plates (5×104 cells/well). After attachment of cells for 24 hours, the medium was changed with differentiation medium, and after an additional 72 hours, the cells were treated with AGE-albumin (50 mM; Sigma-Aldrich Co.) for 12 hours. Cotreated cells were incubated with sRAGE for 1 hour before AGE-albumin (50 mM) treatment and incubated for 12 hours. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich Co.) assay was manipulated for evaluation of DA neuronal cell death. Yellow MTT compound was converted by living cells to form purple formazan crystals, which is soluble in dimethyl sulfoxide (Sigma-Aldrich Co.); 0.5 mg/mL MTT was added to each well, followed by incubation for 2 hours, and dimethyl sulfoxide was added. Intensity of purple staining in culture medium presented the number of live cells proportionally and was measured by spectrophotometer at 540 and 570 nm.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling assay
Frozen sections of mouse brain were washed five times with PBS for 10 minutes and permeabilization was performed by freshly prepared 0.1% Triton X-100 in 0.1% citrate buffer on ice for 2 minutes. After rinsing the slides twice with PBS, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) reaction mixture (Hoffman-La Roche Ltd.) was added on samples, followed by incubation for 60 minutes at 37°C in a humidified atmosphere in the dark. Slides were rinsed three times in PBS and then incubated with 4′,6-diamino-2-phenilindole (1 μg/mL; Sigma-Aldrich Co.) for 20 seconds; coverslips were mounted on glass slides using Vectashield mounting media (Vector Laboratories) and analyzed using an LSM 710 confocal microscope (Carl Zeiss Meditec AG).
Enzyme-linked immunosorbent assay
A 96-well plate was coated with 1 μg/mL of albumin antibody in 100 mM carbonate/bicarbonate buffer (pH 9.6) at 4°C overnight. After washing the plate twice with PBS, remaining protein-binding sites were blocked by addition of 5% skim milk (Sigma-Aldrich Co.) at 4°C overnight. After washing in PBS, the sample was added, followed by incubation for 90 minutes at 37°C. The plate was rinsed in PBS and 1 μg/mL of AGE antibody was added, followed by incubation for 2 hours at room temperature. The plate was washed with PBS and incubated for 2 hours at room temperature with horse radish peroxidase-conjugated secondary antibody (Vector Laboratories). TMB (3,3′,5,5′-tetramethylbenzidine) solution (Sigma-Aldrich Co.) was added, followed by incubation for 30 minutes, and mixed with equal volume of stopping solution (2 N, H2SO4; Sigma-Aldrich Co.), and the optical density was read at 450 nm.
Densitometry and statistical analysis
The densitometry intensity of each immunoreactive band was determined using Image-Pro gel digitizing software (Media Cybernetics, Inc., Bathesda, MD, USA). All data shown in this study represent results from at least three independent experiments. Statistical analyses were performed using the Student’s t-test, and P<0.05 was considered significant (*P<0.05, **P<0.001, and ***P<0.0001).
Results
AGE-albumin was colocalized with activated microglial cells in substantia nigra of human and mouse PD brains
We first investigated the distribution of AGE-albumin in the brains of human PD and normal individuals to study the mechanisms by which AGE-albumin was increased and how it promoted dopaminergic neuronal cell death. We observed that activated microglial cells synthesize and secrete AGE-albumin in human PD patient midbrain (Figure 1). To investigate AGE-albumin distribution in midbrain area, we compared human normal and PD patient brain sections by triple-labeled immunostaining (Figure 1A–C). In SN area of PD midbrain, activated microglial cells were increased in the population compared to normal human brain. By contrast, other brain areas including cerebral cortex showed no accumulation of activated microglial cells (Figure 1D–F). To further demonstrate in animal PD models, a rotenone-treated PD mouse model was used. By immunohistochemical staining, interestingly, most AGE-albumin was colocalized with activated microglial cells in SN of PD model mouse midbrain, same as human brains (Figure 2A–C). Fluorescence intensity analysis indicated that AGE-albumin was highly expressed in PD animal brains in Iba1-positive microglial cells of rotenone-treated PD mouse brain (Figure 2B and C). By contrast, other brain areas including cerebral cortex showed no accumulation of activated microglial cells in PD animal models (Figure 2D–F).
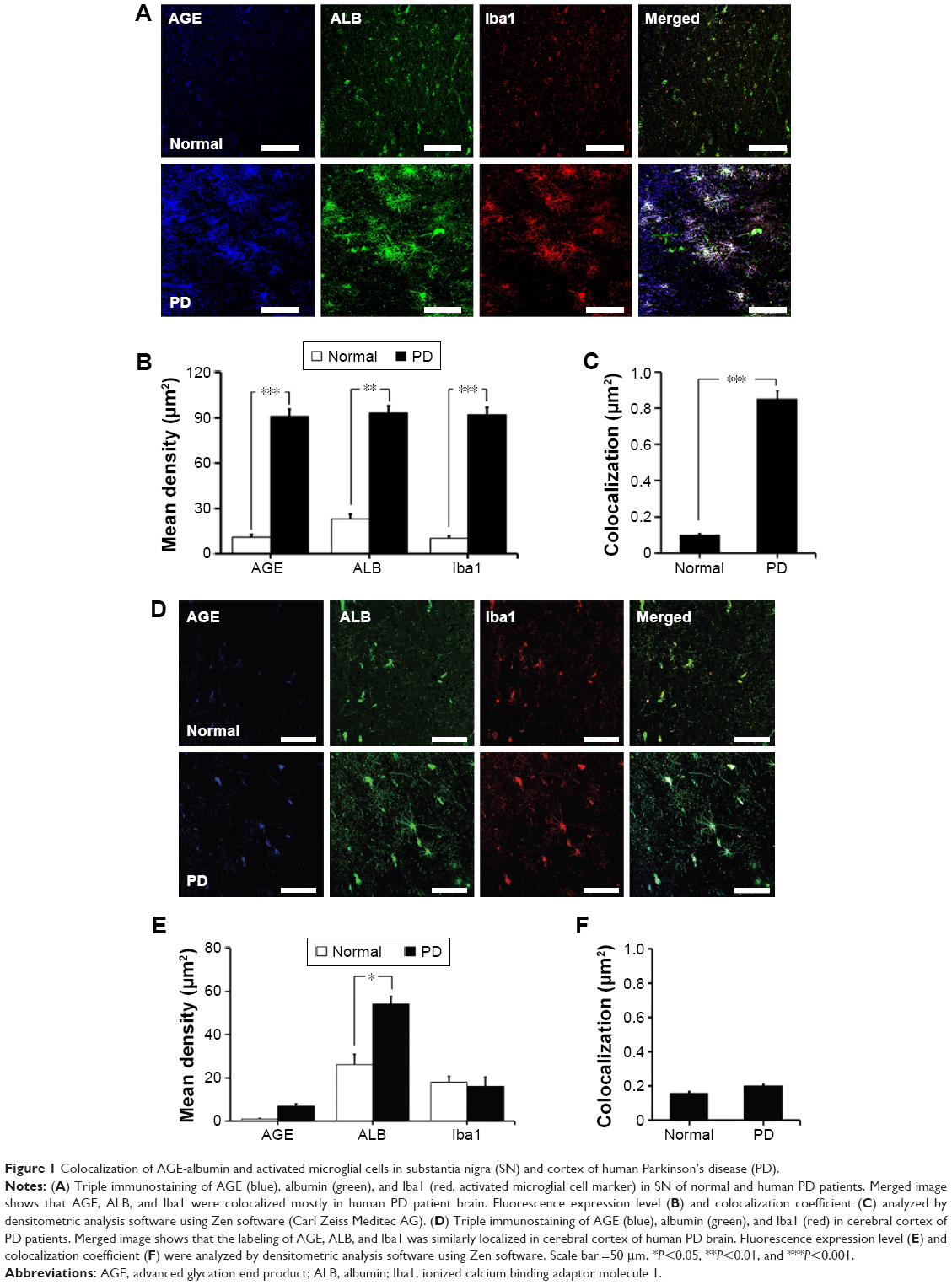 | Figure 1 Colocalization of AGE-albumin and activated microglial cells in substantia nigra (SN) and cortex of human Parkinson’s disease (PD). |
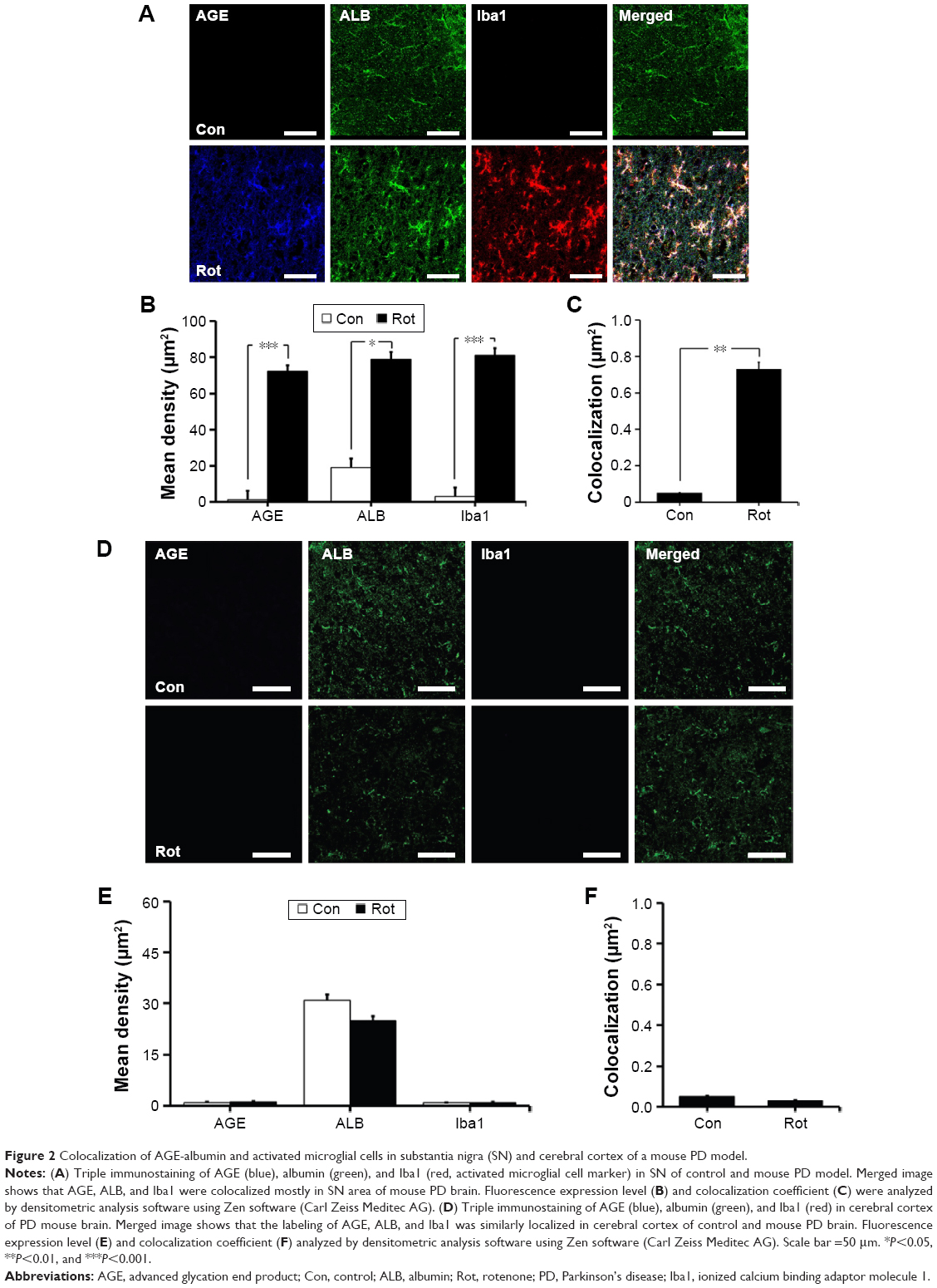 | Figure 2 Colocalization of AGE-albumin and activated microglial cells in substantia nigra (SN) and cerebral cortex of a mouse PD model. |
Activated microglial cells synthesize and secrete AGE-albumin
Our previous study demonstrated that only AGE-albumin has microglia-specific expression in pathological conditions such as AD.20–22 To determine the AGE-albumin synthesis and secretion in activated microglial cells, we evaluated whether AGE-albumin synthesis and secretion are elevated by α-synuclein (Figure 3A).
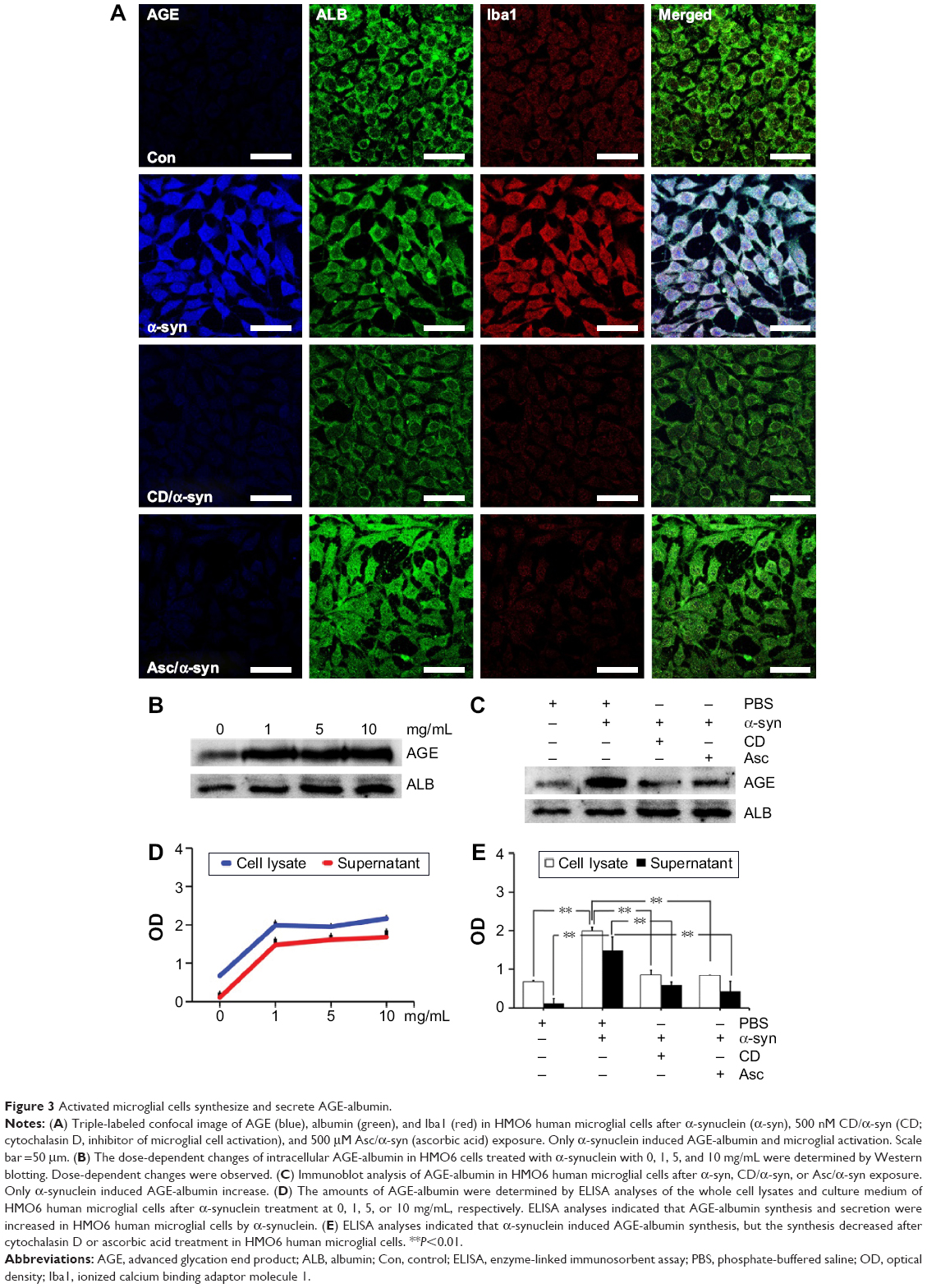 | Figure 3 Activated microglial cells synthesize and secrete AGE-albumin. |
After α-synuclein exposure, human microglial cells showed significant elevation of AGE-albumin. Thus, we induced DA neuronal cell damage by high-dose rotenone exposure, and microglial cells were exposed to culture medium for analysis of activation. After inhibition of human microglial cell activation by CD or antioxidant – ascorbic acid, AGE-albumin synthesis and secretion showed a dramatic decrease (Figure 3A). In addition, intracellular AGE-albumin in HMO6 cells showed a dose-dependent increase in response to α-synuclein exposure (Figure 3B). Immunoblot analysis of AGE-albumin in HMO6 human microglial cells after exposure to α-syn, CD/α-syn, or Asc/α-syn showed similar results with immunohistochemistry (Figure 3C). Enzyme-linked immunosorbent assay (ELISA) analyses indicated that AGE-albumin synthesis and secretion were increased dose dependently in HMO6 human microglial cells by α-synuclein (Figure 3D). Immunoblot analyses indicated that α-synuclein induced AGE-albumin synthesis, but the synthesis decreased after CD or ascorbic acid treatment in HMO6 human microglial cells (Figure 3E).
AGE-albumin induced human dopaminergic neuronal death by RAGE and MAPK pathway
Immunostaining data showed that RAGE was increased in human dopaminergic neurons after AGE-albumin treatment but decreased dramatically after cotreatment with sRAGE (Figure 4A). To test whether dopaminergic neuronal death was induced via RAGE-MAPK pathway, immunoblot analysis of RAGE and MAPK pathways was performed. RAGE was increased after AGE-albumin treatment but decreased dramatically after cotreatment with sRAGE (Figure 4B). Phospho-p38, pERK1/2, pSAPK/JNK, and Bax were significantly increased after AGE-albumin treatment but decreased dramatically after cotreatment with sRAGE (Figure 4B). MTT analysis (Figure 4D) also showed that human dopaminergic neuronal survival was reduced by AGE-albumin treatment, but cell survival was recovered by sRAGE and AGE-albumin cotreatment (Figure 4C).
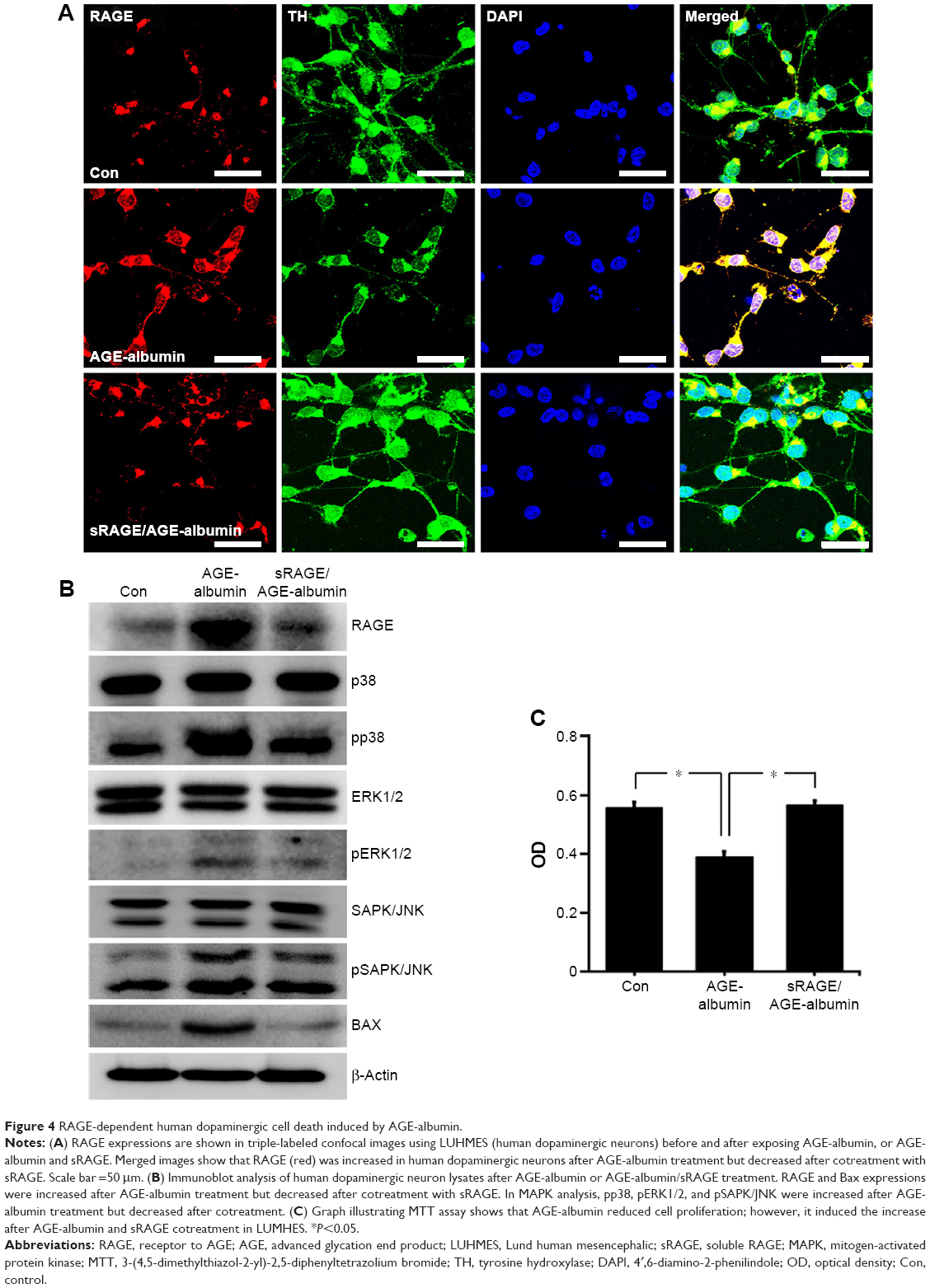 | Figure 4 RAGE-dependent human dopaminergic cell death induced by AGE-albumin. |
AGE-albumin inhibition protects against DA neuronal cell death
To determine whether AGE-albumin is critical in PD animal models, we analyzed the neuronal cell population after oral injection of rotenone for 2 months. Cresyl violet staining of control and PD mouse brains revealed that the neuronal population was reduced in rotenone-treated animals; however, the population increased in rotenone with sRAGE-treated or rotenone with PM-treated groups (Figure 5A and B).
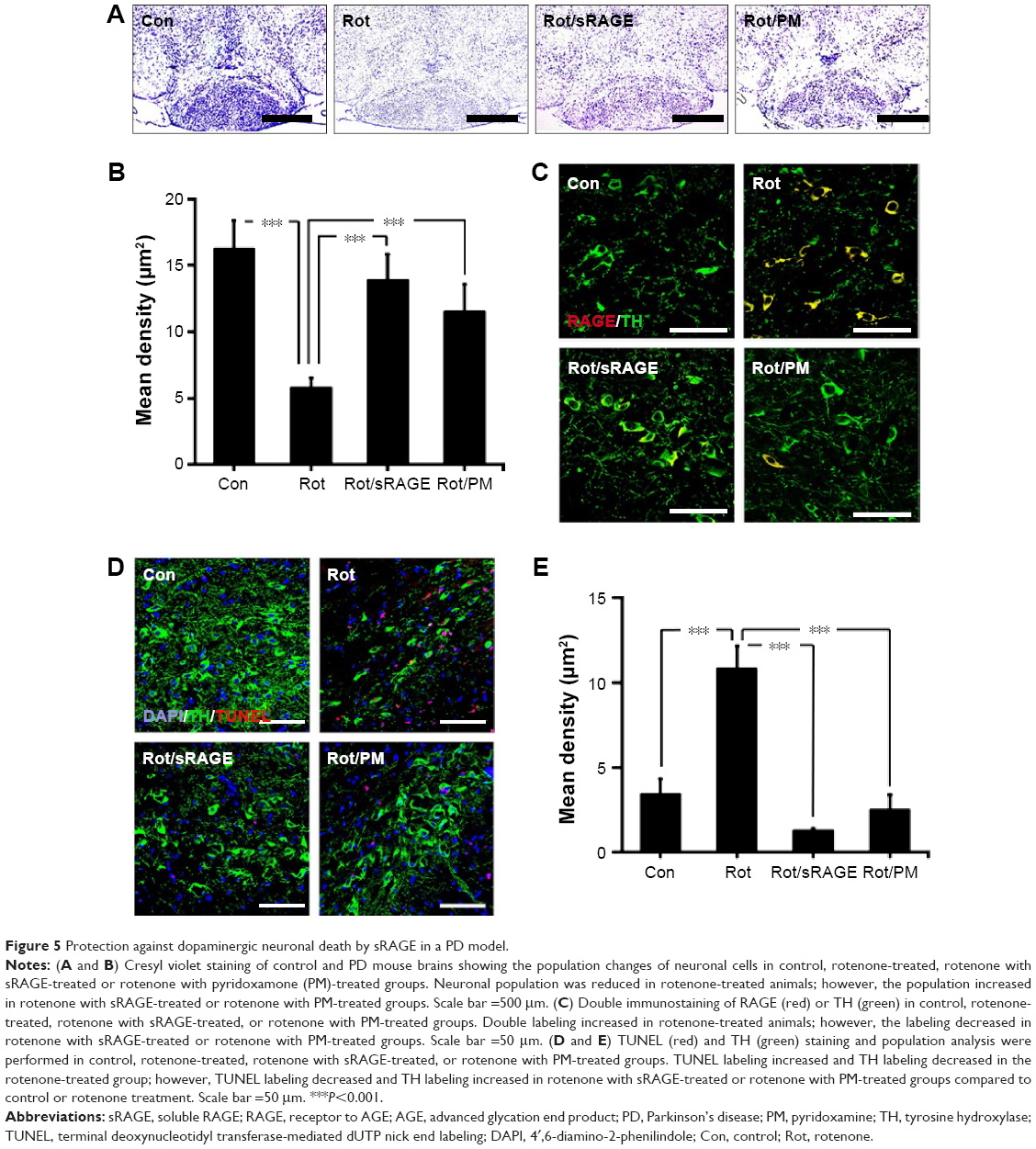 | Figure 5 Protection against dopaminergic neuronal death by sRAGE in a PD model. |
To determine whether activated microglial AGE-albumin is critical in dopaminergic neuronal death via RAGE increase in PD animals, double immunostaining of RAGE and tyrosine hydroxylase (TH) was performed. Double labeling of RAGE (red) and TH (green) showed that double labeling increased in rotenone-treated animals but decreased in rotenone with sRAGE-treated or rotenone with PM-treated groups (Figure 5C).
TUNEL (red) and TH (green) staining and population analysis were performed in control, rotenone-treated, rotenone with sRAGE-treated, or rotenone with PM-treated groups. TUNEL labeling increased and TH labeling decreased in the rotenone-treated group; however, TUNEL labeling decreased and TH labeling increased in rotenone with sRAGE-treated or rotenone with PM-treated group (Figure 5D and E).
Discussion
Recent postmortem studies on neurodegenerative diseases have commonly indicated that AGE levels were elevated and accumulated in damaged brain areas.28–31 They also reported the implication of AGEs on neurodegeneration by promoting formation of pathological protein aggregations; however, the origins of AGEs have not been clearly reported. In this study, we observed that activated microglial cells and AGE-albumin were colocalized in SN area of human PD patient’s brain and animal PD models. These results of the triple labelings were similar to those of previous reports in that the most abundant glycated product is AGE-albumin in AD and alcoholic brains.21,23
In experiments with human microglial cells and a PD mouse model, we found that activated microglial cells synthesize and secrete AGE-albumin and its accumulation is continuously deleterious to DA neurons. Immunostaining, ELISA, and immunoblot data showed that human microglial cells are responsible for AGE-albumin synthesis and secretion. In a mouse PD model, activated microglial cells were accumulated in SN area and mostly double-labeled AGE-albumin. These results are also similar with those for AD and alcoholic brains.21,23
Microglial activation can also be inhibited by CD, which we used for validation of AGE-albumin produced after activation.25,32,33 To further confirm whether AGE-albumin synthesis and secretion are related to microglial activation, we performed inhibition study with ascorbic acid and CD.
In our results, ELISA and immunoblotting analysis showed that ascorbic acid or CD treatment in human microglial cells led to reduced synthesis and secretion of AGE-albumin from activated microglial cells. These results are also similar with those for AD and alcoholic brains.21–23
Next, we investigated whether AGE-albumin is key for inducing dopaminergic neuronal death in LUMHES cells. Immunohistochemical and immunoblot analyses using PD animal models demonstrated that AGE-albumin upregulates RAGE expression in human dopaminergic neurons and induced cell death by MTT analysis. AGE-albumin-mediated dopaminergic neuronal death was inhibited by sRAGE treatment. AGE-RAGE-related neuronal death has been reported widely in many neurodegenerative diseases.34–37 In our immunohistochemistry result, the dopaminergic neuronal population was also increased after cotreatment of rotenone with sRAGE, indicating that sRAGE successfully binds with accumulated AGE-albumin and protected against its pathologic effect.
AGE-albumin increased the level of Bax, pp38, pERK1/2, and pSAPK/JNK of MAPK pathway in human dopaminergic neurons. Our data show that AGE-albumin induced DA neuronal cell death recovered by sRAGE. Expression of Bax, pp38, pERK1/2, and pSAPK/JNK was decreased again after sRAGE treatment. Based on these results, accumulated microglial AGE-albumin has a key role on dopaminergic neuronal death in PD models. AGEs are known as major structural cross-linkers, which cause formation of Lewy bodies in human dopaminergic neurons.
In summary, our current data show that AGE-albumin, the most abundant form of brain AGEs, is synthesized in activated microglial cells and secreted and accumulated into extracellular space. The rate of AGE-albumin synthesis is markedly increased by dose-dependent α-synuclein treatment. In addition, AGE-albumin promotes RAGE-pp38-, pERK1/2-, and pSAPK/JNK-Bax-mediated apoptosis in human dopaminergic neurons of PD.
Conclusion
In conclusion, our data suggest that activated microglial cells are one of the main contributors in AGE-albumin accumulation, and targeting AGE-albumin could provide useful therapeutic and diagnostic biomarkers with high sensitivity at the microscopic level.
Acknowledgments
The authors appreciate the courtesy donation of LUHMES cell line from Dr Marcel Leist of Kontanz University, Germany. This work was supported by the Gachon University research fund (Nos 2014-5105 and 2014-5102) and a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (grant number: HI13C2098).
Disclosure
The authors report no conflicts of interest in this work.
References
Poirier J, Kogan S, Gauthier S. Environment, genetics and idiopathic Parkinson’s disease. Can J Neurol Sci. 1991;18(1):70–76. | ||
De Rijk MC, Tzourio C, Breteler MM, et al. Prevalence of parkinsonism and Parkinson’s disease in Europe: the EUROPARKINSON Collaborative Study. European Community concerted action on the epidemiology of Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1997;62(1):10–15. | ||
Calabrese VP. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;69(2):223–224. | ||
Przedborski S. Pathogenesis of nigral cell death in Parkinson’s disease. Parkinsonism Relat Disord. 2005;11 Suppl 1:S3–S7. | ||
Albin RL, Dauer WT. Parkinson syndrome. Heterogeneity of etiology; heterogeneity of pathogenesis? Neurology. 2012;79(3):202–203. | ||
Przedborski S. Inflammation and Parkinson’s disease pathogenesis. Mov Disord. 2010;25 Suppl 1:S55–S57. | ||
Vives-Bauza C, Przedborski S. PINK1 points Parkin to mitochondria. Autophagy. 2010;6(5):674–675. | ||
Lang AE, Obeso JA. Challenges in Parkinson’s disease: restoration of the nigrostriatal dopamine system is not enough. Lancet Neurol. 2004;3(5):309–316. | ||
Michel PP, Hirsch EC, Agid Y. Parkinson disease: mechanisms of cell death. Rev Neurol. 2002;158 Spec no 1:S24–S32. | ||
Bognar C, Baldovic M, Benetin J, Kadasi L, Zatkova A. Analysis of Leucine-rich repeat kinase 2 (LRRK2) and Parkinson protein 2 (parkin, PARK2) genes mutations in Slovak Parkinson disease patients. Gen Physiol Biophys. 2013;32(1):55–66. | ||
Kitada T, Mizuno Y. Molecular pathogenesis of familial Parkinson’s disease. Nihon Rinsho. 2000;58(10):2016–2021. | ||
Hirsch EC, Hoglinger G, Rousselet E, et al. Animal models of Parkinson’s disease in rodents induced by toxins: an update. J Neural Transm Suppl. 2003;(65):89–100. | ||
Thany SH, Reynier P, Lenaers G. Neurotoxicite des pesticides Quel impact sur les maladies neurodegeneratives? [Neurotoxicity of pesticides: its relationship with neurodegenerative diseases]. Med Sci. 2013;29(3):273–278. French. | ||
Binienda ZK, Sarkar S, Mohammed-Saeed L, et al. Chronic exposure to rotenone, a dopaminergic toxin, results in peripheral neuropathy associated with dopaminergic damage. Neurosci Lett. 2013;541:233–237. | ||
Iancu R, Mohapel P, Brundin P, Paul G. Behavioral characterization of a unilateral 6-OHDA-lesion model of Parkinson’s disease in mice. Behav Brain Res. 2005;162(1):1–10. | ||
Hirsch EC, Hunot S, Damier P, Faucheux B. Glial cells and inflammation in Parkinson’s disease: a role in neurodegeneration? Ann Neurol. 1998;44(3 Suppl 1):S115–S120. | ||
Luber-Narod J, Rogers J. Immune system associated antigens expressed by cells of the human central nervous system. Neurosci Lett. 1988;94(1–2):17–22. | ||
Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6(4):193–201. | ||
Hirsch EC, Hunot S, Hartmann A. Neuroinflammatory processes in Parkinson’s disease. Parkinsonism Relat Disord. 2005;11 Suppl 1:S9–S15. | ||
Ahn SM, Byun K, Cho K, et al. Human microglial cells synthesize albumin in brain. PLoS One. 2008;3(7):e2829. | ||
Byun K, Bayarsaikhan E, Kim D, et al. Induction of neuronal death by microglial AGE-albumin: implications for Alzheimer’s disease. PLoS One. 2012;7(5):e37917. | ||
Byun K, Bayarsaikhan E, Kim D, et al. Activated microglial cells synthesize and secrete AGE-albumin. Anat Cell Biol. 2012;45(1):47–52. | ||
Byun K, Bayarsaikhan D, Bayarsaikhan E, et al. Microglial AGE-albumin is critical in promoting alcohol-induced neurodegeneration in rats and humans. PLoS One. 2014;9(8):e104699. | ||
Neeper M, Schmidt AM, Brett J, et al. Cloning and expression of a cell surface receptor for advanced glycosylation endproducts of proteins. J Biol Chem. 1992;267:4998–5004. | ||
Yan SF, Ramasamy R, Schmidt AM. The RAGE axis: a fundamental mechanism signaling danger to the vulnerable vasculature. Circ Res. 2010;106:842–853. | ||
Scholz D, Poltl D, Genewsky A, et al. Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J Neurochem. 2011;119(5):957–971. | ||
Inden M, Kitamura Y, Abe M, Tamaki A, Takata K, Taniguchi T. Parkinsonian rotenone mouse model: reevaluation of long-term administration of rotenone in C57BL/6 mice. Biol Pharm Bull. 2011;34(1):92–96. | ||
Munch G, Luth HJ, Wong A, et al. Crosslinking of alpha-synuclein by advanced glycation endproducts – an early pathophysiological step in Lewy body formation? J Chem Neuroanat. 2000;20(3–4):253–257. | ||
Castellani R, Smith MA, Richey PL, Perry G. Glycoxidation and oxidative stress in Parkinson disease and diffuse Lewy body disease. Brain Res. 1996;737(1–2):195–200. | ||
Shaikh S, Nicholson LF. Advanced glycation end products induce in vitro cross-linking of alpha-synuclein and accelerate the process of intracellular inclusion body formation. J Neurosci Res. 2008;86(9):2071–2082. | ||
Munch G, Westcott B, Menini T, Gugliucci A. Advanced glycation endproducts and their pathogenic roles in neurological disorders. Amino Acids. 2012;42(4):1221–1236. | ||
Zhang W, Wang T, Pei Z, et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 2005;19(6):533–542. | ||
Shaikh SB, Nicholson LF. Effects of chronic low dose rotenone treatment on human microglial cells. Mol Neurodegener. 2009;4:55. | ||
Bucciarelli LG, Wendt T, Rong L, et al. RAGE is a multiligand receptor of the immunoglobulin superfamily: implications for homeostasis and chronic disease. Cell Mol Life Sci 2002;59(7):1117–1128. | ||
Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108(7):949–955. | ||
Walker DG, Lue LF. Investigations with cultured human microglia on pathogenic mechanisms of Alzheimer’s disease and other neurodegenerative diseases. J Neurosci Res. 2005;81(3):412–425. | ||
Rondeau P, Bourdon E. The glycation of albumin: structural and functional impacts. Biochimie. 2011;93(4):645–658. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.