")
Back to Journals » International Journal of General Medicine » Volume 14
Microarray Expression Profile and Analysis of Circular RNA Regulatory Network in Pulmonary Thromboembolism
Authors Peng D, Hou ZL, Zhang HX, Zhang S, Zhang SM, Lin RY, Xing ZC, Yuan Y, Yang KY, Wang JX
Received 6 February 2021
Accepted for publication 18 March 2021
Published 9 April 2021 Volume 2021:14 Pages 1239—1249
DOI https://doi.org/10.2147/IJGM.S304199
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Dan Peng, Zi-Liang Hou, Hong-Xia Zhang, Shuai Zhang, Shu-Ming Zhang, Rui-Yan Lin, Zhen-Chuan Xing, Yuan Yuan, Kai-Yuan Yang, Jin-Xiang Wang
Department of Respiratory and Critical Care Medicine, Beijing Luhe Hospital, Capital Medical University, Beijing, People’s Republic of China
Correspondence: Jin-Xiang Wang
Department of Respiratory and Critical Care Medicine, Beijing Luhe Hospital, Capital Medical University, Xinhuananlu No. 82, Tongzhou District, Beijing, 101100, People’s Republic of China
Email [email protected]
Background: Pulmonary thromboembolism (PTE) is a common disease which may be a serious condition and has high mortality. Recently, it has been shown that circRNAs play an important role in the development of various diseases, including thromboembolic disease. However, circRNAs expression profiling is not clear in PTE, this study aims to identify the circRNAs expressed in PTE and to elucidate their possible role in pathophysiology of PTE.
Methods: A total of 5 patients with CTPA-confirmed PTE and 5 healthy controls were recruited for the present study. The circRNAs expression profile was analyzed by microarray.
Results: In total, 256 differentially expressed circRNAs (up 142, down114) and 1162 mRNA (up 446, down 716) were summarized by analyzing the circRNAs microarray data. The top 3 up-regulated and 3 down-regulated circRNAs were validated by Real-Time Polymerase Chain Reaction (qRT-PCR). Two differentially expressed circRNAs (hsa_circ_0000891, hsa_circ_0043506) were selected for further analysis. Finally, we construct a circRNA-miRNA-mRNA ceRNA network with a bioinformatic prediction tool. Pathway analysis shows that the enriched mRNAs targets take part in Protein processing in endoplasmic reticulum, Systemic lupus erythematosus, Endocytosis, Spliceosome, HTLV-I infection and Ubiquitin mediated proteolysis.
Conclusion: Our findings indicated that aberrantly expressed circRNAs (hsa_circ_0000891, hsa_circ_0043506) may be involved in the development of PTE.
Keywords: circRNA, gene, pulmonary thromboembolism, circRNA-miRNA-mRNA network, functional enrichment
Introduction
Pulmonary thromboembolism (PTE) is a common and potentially lethal disease characterized by acute onset, serious complications, and high mortality. In the past 40 years, the mortality rate has been hovering around 15%, and the sudden death rate is as high as 11%. Up to now, computed tomography pulmonary angiography (CTPA) has been considered the gold-standard diagnostic modality in patients with suspected pulmonary embolism. However, its application in practical practice is limited for some specific patients with renal failure or allergic reactions to contrast agent.1 D-dimer is well established in the diagnostic procedure of PTE because of its long half-life, but its clinical benefit is limited due to low specificity.2 There is an urgent need to find new biomarkers with high sensitivity and specificity to improve the diagnostic efficiency and further to explore the pathogenesis of PTE.
With the popularity of high-throughput sequencing, more and more big data of genomics are being captured. Network Topology analysis has greater advantages over traditional reductionist approach high clinical heterogeneity characterizing CV patients. A multi-omics panel of network biomarkers based on big data may superior to traditional population-based risk prediction algorithms in order to identify high-risk subjects as well as patients with different diagnosis, prognosis, and response to specific drug treatments.3 The molecular mechanism of PTE is currently elusive. Some pieces of evidence indicate that non-coding RNAs may be involved in the development of PTE. Currently, the vast majority of research is focused on microRNAs (miRNAs). It is notable that miRNA expression is related to platelets, endothelial progenitor cells (EPCs) and coagulation factors.4 Recently, another class of non-coding RNA molecules, circRNAs, has drawn increasing interests for their regulatory roles in cancer and cardiovascular (CV) diseases. CircRNAs are a kind of endogenous noncoding RNAs that regulate gene expression through acting as competitive endogenous RNAs (ceRNAs) and modulating gene.5,6 In addition, circRNAs are highly abundant in human cells, its covalently closed continuous loop structure confers them the resistance to RNases and more stable (half-life of about 48 hours, endowing them clear advantages as novel diagnostic markers in comparison to linear RNA (eg, mRNAs, 10 hours).7–9 Many piece of evidence indicated that circRNAs may play important roles in CV diseases (HF, CHD and AMI).10 To date, the expression pattern and roles of circRNAs in PTE remain elusive. We assume that it may serve as a new potential plasma biomarker and provide new ideas for the pathogenesis of PTE. To explore the issues, we firstly investigated the expression profile of circRNAs in PTE using the microarray analysis. We identified a large number of differentially expressed circRNAs. Also, a circRNA-miRNA-mRNA network was established to identify the potential regulatory role of circRNAs in PTE.
Materials and Methods
Study Populations
This study was carried out ethically in conformity to the World Medical Association Declaration of Helsinki and approved by the Ethics Committee of Beijing Luhe Hospital, Capital Medical University (2018-LHKY-004-01). All patients provided written informed consent. Peripheral blood of 5 PTE patients and 5 healthy controls (HC) was collected from June 2018 through October 2019. All PTEs were diagnosed by CTPA. The subjects had no history of severe trauma, staying in bed after surgery within 2 weeks, autoimmune diseases, long-term hormone therapy, central venous catheterization, acute myocardial infarction, atrial fibrillation, new cerebral infarction and cancers.
Blood Sample Collection and Processing
Venous blood samples were drawn from all patients within the first 24 hours after admission. This sample was centrifuged at 3000 rpm at 4°C for 10 minutes. The layer of white cells was collected and placed in a 15 mL centrifuge tube containing 10 mL of 10% PBS. The centrifuge tube was centrifuged in a cooling centrifuge at 4°C for 10 minutes at 3000 rpm. Finally, the buffy coat layer just above the line dividing two fractions of the supernate and the red blood cells was then extracted and transferred to an RNase/DNase-free tubes. One milliliter Trizol was added and mixed well with cell and then stored at −80°C.
CircRNA, miRNA and mRNA Microarray Analysis
This experiment was carried out in strict accordance with the Thermo Fisher official standard. TRIzol method was used to extract RNA from samples. The concentration and purity of total RNA were assessed using a NanoDrop Spectrophotometer (NanoDrop Technologies). RNA integrity and DNA contamination were checked by denaturing agarose gel electrophoresis. Total RNA concentration was measured using Qubit 3.0 Fluorometer. The total RNA samples were enriched for polyA RNA and then reverse-transcribed to synthesize double-stranded cDNA. The cDNA was purified, quantified, fragmented and fluorescently labeled with dNTP (Cy3dCTP). The labeled cDNAs were hybridized onto Clariom D Human Assay Microarrays. After hybridization, the GeneChip arrays were washed and stained using the Affymetrix GeneChip Fluidics Station 450 and then scanned using the GeneChip Scanner 3000 7G. Fluorescent scan images were preserved. Affymetrix GeneChip Operating Software (GCOS) was used to generate signal intensities for each transcript and create CEL data files.
Validation of Candidate circRNAs Using Quantitative Reverse Transcription Real-Time Polymerase Chain Reaction (qRT-PCR)
Based on the results of the microarray analysis, we selected the top 3 up-regulated and 3 down-regulated circRNAs for further validation using qRT-PCR. Total RNA was extracted using the TRNzol isolation kit according to the manufacturer’s specification. The concentration and purity of total RNA were determined by UV absorption at 260:280, using NanoDrop ND-2000 Spectrophotometer. Reverse transcription of circRNAs into cDNA was performed through a PrimeScript RT reagent kit with gDNA Eraser. All primers were synthesized by Premedical Technology Corporation (Beijing, China) (Table 1) and qRT-PCR was performed on the ABI Prism 7900HT Sequence Detection System (Applied Biosystems). The real-time PCR reactions were performed for targeted circRNAs and the internal reference (U6). Each sample was analyzed in triplicate. In order to obtain relative circRNAs expression data, the relative gene expression data were analyzed by the 2 −(∆∆Ct) method.
 |
Table 1 Specific circRNA Primers for Quantitative of RT-PCR Analysis |
Prediction of miRNAs and mRNAs for Candidate circRNAs and Bioinformatics Analysis
CircRNAs contain miRNA response elements (MREs). We predicted miRNA binding sites (MREs) with the Circular RNA Interactome (https://circinteractome.nia.nih.gov/). TargetScan (http://www.targetscan.org/) was used to predict target miRNAs of the candidate circRNAs. The interactions of miRNAs and mRNAs were established by TargetScan11 (http://www.targetscan.org) and miRDB12 (http://www.mirdb.org). The predicted miRNAs and mRNAs were compared with detected miRNAs and mRNAs by the microarray assay, respectively. We identified mRNAs overlapping between the predicted target genes of the circRNAs and detected mRNAs. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were used to analyze the functional roles of the circRNAs-targeted genes. Go analysis and KEGG pathway were acquired using DAVID online tools (http://david.abcc.ncifcrf.gov/). The target genes of the circRNAs were analyzed by GO and KEGG enrichment using R packages Cluster profiler.
Construction of circRNA-miRNA-mRNA Interaction Networks
CircRNAs regulate miRNA and downstream gene expression through a competing endogenous RNA (ceRNA) mechanism.8 A circRNA-miRNA-mRNA regulatory network was further assessed according to interaction information derived from TargetScan and miRDB. Finally, the interaction network of circRNAs, miRNAs, and mRNAs was built using the Cytoscape platform.
Statistical Analysis
The ‘limma’ data packet in R software was applied to analyze the differentially expressed circRNAs (DECs), differentially expressed miRNAs (DEMis) and differentially expressed mRNAs (DEMs) between PTE samples and healthy control samples. Hierarchical clustering and principal component analysis (PCA) were performed to visualize distinguishable gene expression patterns among the samples. The DECs were evaluated using t test, with statistical significance set at a P < 0.05 and |logFC |≥1, and the value of DEMis was P < 0.05 and |logFC |≥0.5, while the value of DEMs was P < 0.05, |logFC| ≥1.5 was statistically significant. Student’s t-test was used to analyze qRT-PCR data. Statistical analysis was performed using SPSS Statistics 19 software package (IBM, Chicago, IL, USA), and P < 0.05 was considered statistically significant.
Results
Characteristics of Study Populations
A total of 5 patients with CTPA-confirmed PTE were recruited for the present study, and 5 healthy controls were recruited as the control group. The demographic and clinical characteristics of study populations are shown in Supplementary Table S1.
Profiles of Differentially Expressed circRNAs, miRNAs and mRNAs
The Clariom D Human Array was used to analyze circRNAs miRNA and mRNAs expression in peripheral blood of subjects. We detected the expression of 12960 circRNAs, 777 miRNAs and 20665 mRNAs. The limma R package was applied to screen differentially expressed circRNAs, miRNAs and mRNA. A total of 256 differentially expressed circRNAs (up 142, down 114), 11 differentially expressed miRNAs (up 3, down 8) and 1162 mRNAs (up 446, down 716) were summarized by analyzing the circRNAs microarray data from 5 pairs of PTE and control group. Hierarchical clustering showed obvious discrimination between the two groups (Figure 1A–C). Volcano plots were performed to show the distinguishable circRNAs miRNAs and mRNAs expression (Figure 1D–F). In the total 256 circRNAs, the top 3 up-regulated and top 3 down-regulated circRNAs selected according to the P value are listed in Table 2.
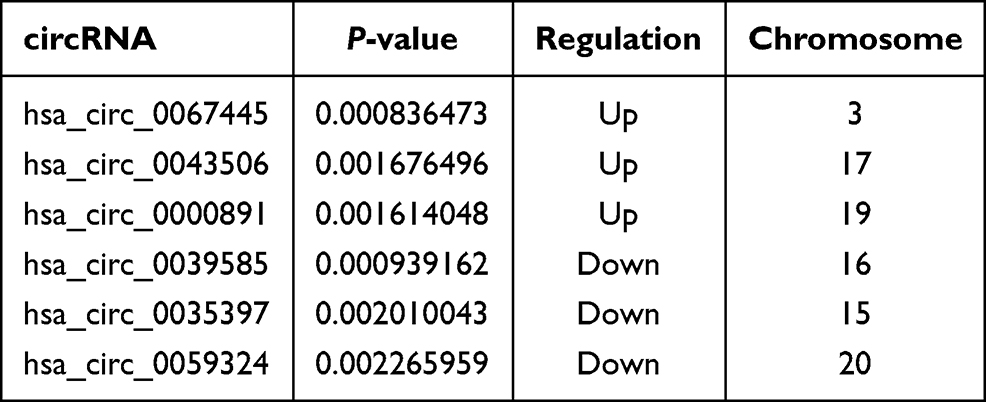 |
Table 2 The Top 3 Up-Regulated and Down-Regulated circRNAs |
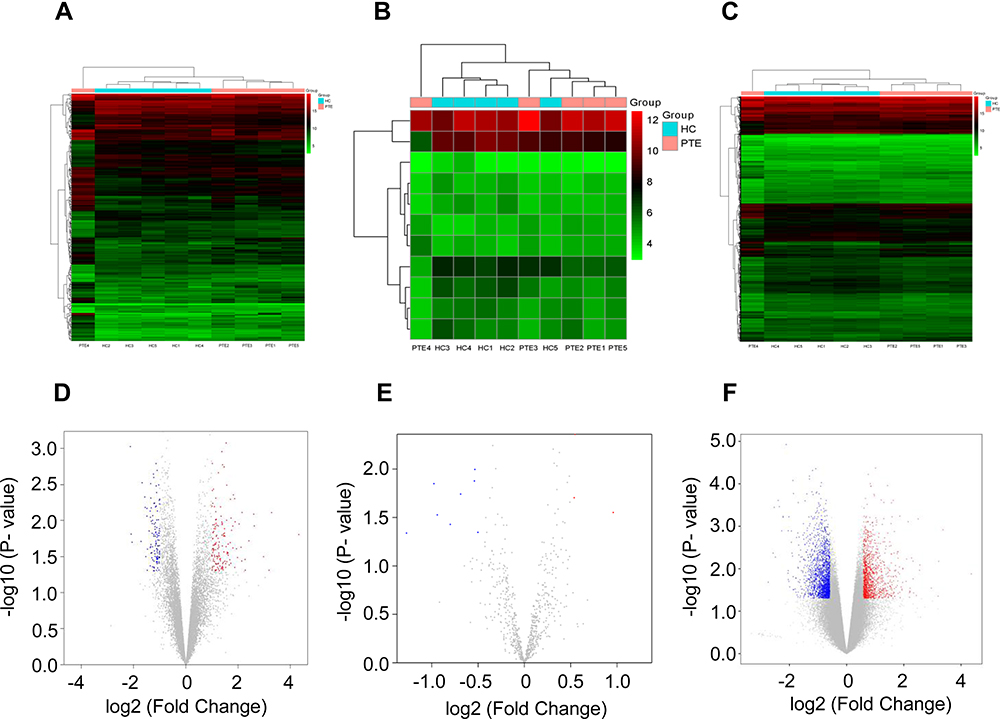 |
Figure 1 Microarray analysis of differential expression of circRNAs, miRNAs and mRNAs in two groups. Hierarchical clustering analysis of differentially expressed circRNAs (|logFC| ≥2.0, P < 0.05) (A), miRNAs (|logFC| ≥0.5, P < 0.05) (B) and mRNAs (|logFC| ≥1.5, P < 0.05) (C) between PTE group and HC group; each group included five individuals. “Red” indicates high relative expression, and “Blue” indicates low relative expression. Volcano plot showing the differentially expressed circRNAs (D), miRNAs (E) and mRNAs (F) in the two groups, with “Red” and “Green” points representing the up-regulated and down-regulated transcripts, respectively. Abbreviations: PTE, pulmonary thromboembolism; HC, healthy control. |
Gene Ontology (GO) was classified into three categories, including biological process (BP), molecular function (MF), and cellular component (CC). We analyzed the functional roles of the differentially expressed mRNAs using GO analysis. For the down-regulated mRNAs (Figure 2A), the top 10 enriched GO terms in the BP, CC, and MF category were identified. The main enriched BP categories involved in regulation of immune response, T cell costimulation, and positive regulation of natural killer cell mediated cytotoxicity. The CC involved in nucleoplasm, spliceosomal complex, and catalytic step 2 spliceosome. While the MF category revealed that they mainly involved in nucleic acid binding, protein binding, and poly (A) RNA binding. On the other hand, the GO enrichment analysis on the up-regulated mRNAs (Figure 2B) showed that the BP was primarily enriched in nucleosome assembly, innate immune response in mucosa, and antibacterial humoral response. In the CC category, the main enriched categories were nucleosome, nuclear nucleosome, and extracellular exosome. In the MF category, the main enriched categories were protein heterodimerization activity, chromatin DNA binding, and protein binding.
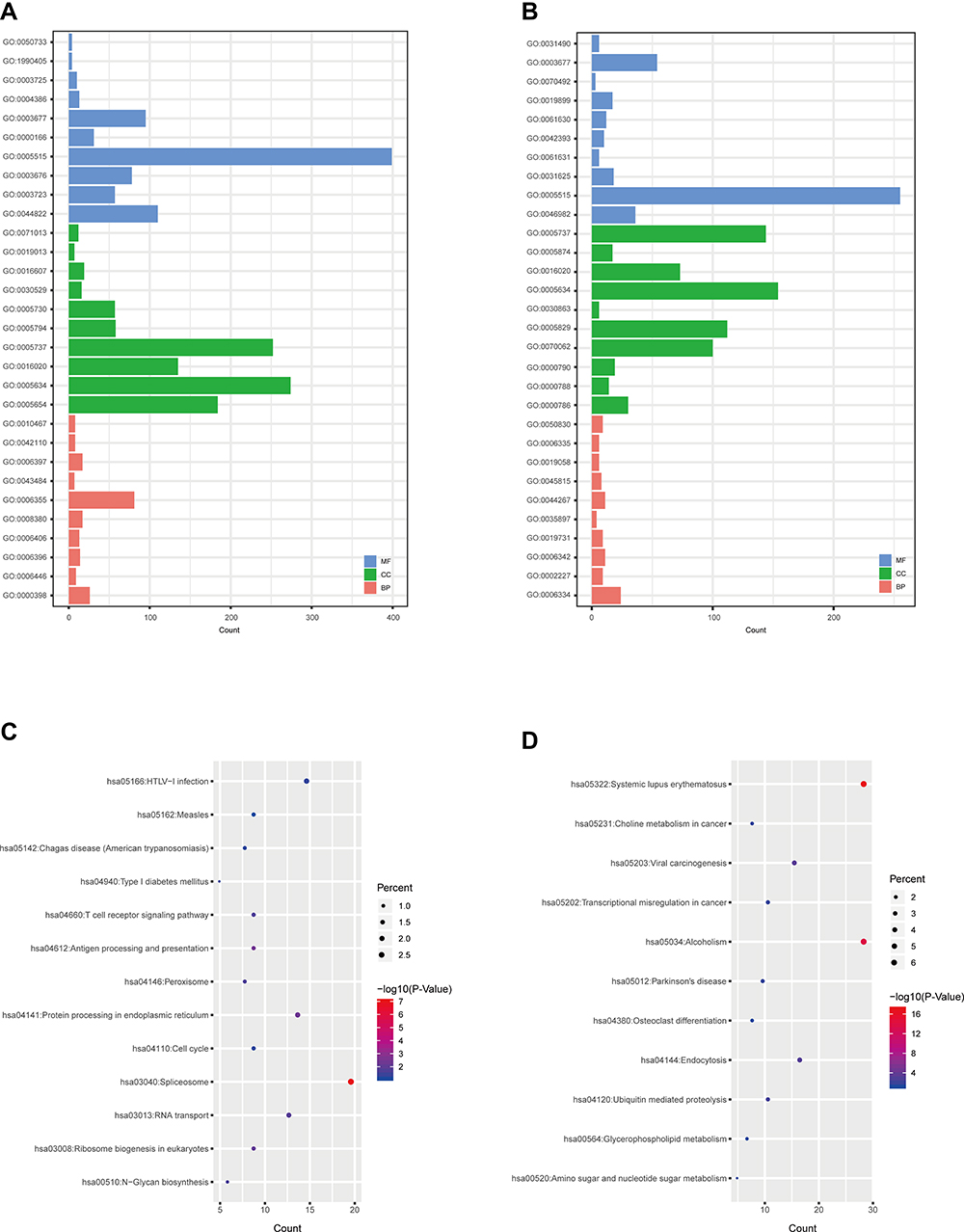 |
Figure 2 GO and KEGG analyses of differentially expressed mRNAs. GO annotation of down-regulated (A) and up-regulated (B) mRNAs. KEGG pathway enrichment analyses for down-regulated (C) and up-regulated (D) mRNAs. Abbreviations: GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; BP, biological process; CC, cellular component; MF, molecular function. |
In the KEGG pathway analysis, the 8 KEGG pathways were also identified for the down-regulated mRNAs (Figure 2C). The related pathways sorted by GeneRatio included T cell receptor signaling pathway, Antigen processing and presentation, Natural killer cell mediated cytotoxicity, Measles, Hematopoietic cell lineage, HTLV-I infection, Protein processing in endoplasmic reticulum, and Chagas disease (American trypanosomiasis). Moreover, there were 4 KEGG pathways on the up-regulated mRNAs (Figure 2D) involving in Systemic lupus erythematosus, Alcoholism, Viral carcinogenesis, and Glycerophospholipid metabolism.
Principal Component Analysis (PCA) for the Differentially Expressed circRNA Candidates and mRNAs
In order to confirm the validity of microarray assay, we used principal component analysis (PCA) to analyze the differentially expressed circRNAs, and the results showed that the PTE group and the HC group could be clearly distinguished (Figure 3A). On the other hand, the PCA plot of the differentially expressed mRNAs from microarray data indicated that the PTE group was also separated from the HC group (Figure 3B). Therefore, we believe that differentially expressed circRNAs and mRNAs can distinguish between the PTE group and the HC group.
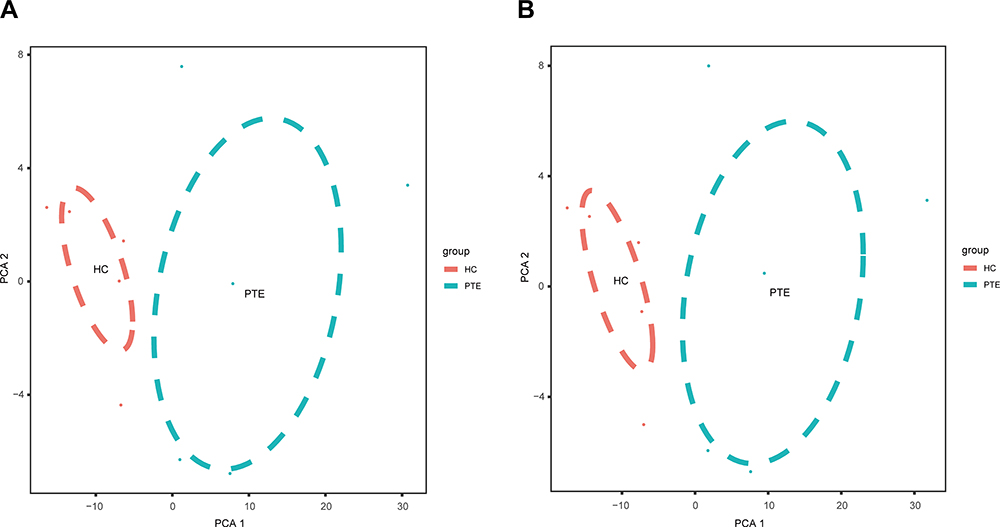 |
Figure 3 The principle component analysis (PCA) on the microarray data. (A) PCA was showed on the differentially expressed circRNAs. (B) PCA plot of the differentially expressed mRNAs. Abbreviations: PTE, pulmonary thromboembolism; HC, healthy control. |
Validation of Differentially Expressed circRNA Candidates Using qRT-PCR
In order to confirm the microarray results, we selected the top 3 up-regulated circRNAs and the top 3 down-regulated circRNAs for qRT-PCR validation. The three up-regulated circRNAs were as follows: hsa_circ_0067445, hsa_circ_0000891, and hsa_circ_0043506. The three down-regulated circRNAs were as follows: hsa_circ_0039585, hsa_circ_0035397, and hsa_circ_0059324. The result of RT-qPCR revealed that two of the up-regulated circRNAs and one of the down-regulated circRNAs exhibited changes in their expression levels that were identical with the results of the microarray. The relative expression of has_circ_0043506 and has_circ_0000891 was confirmed to be up-regulated in PTE group compared with that in the HC group (Figure 4A and B). The relative expression of hsa_circ_0059324 in PTE had down-regulated trend compared with that in HC, but not statistically significant (Figure 4C). So we chose has_circ_0043506 and has_circ_0000891 for further analysis.
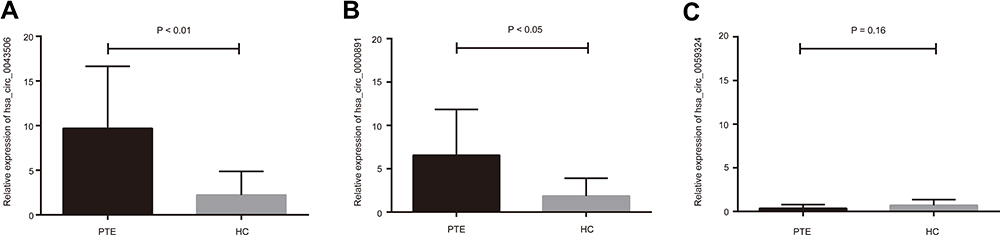 |
Figure 4 Validation of candidate circRNAs using qRT-PCR. (A and B) The relative expression of has_circ_0043506 and has_circ_0000891 was confirmed to be up-regulated. (C) The relative expression of hsa_circ_0059324 had a down trend, but not statistically significant. Abbreviations: qRT-PCR, real time-quantitative-polymerase chain reaction; PTE, pulmonary thromboembolism; HC, healthy control. |
Prediction of circRNAs Targeted Genes and Bioinformatics Analysis for the ceRNA Network
The 2 significantly up-regulated circRNAs (hsa_circ_0000891, hsa_circ_0043506) were selected for further study. A total of 65 target miRNAs were predicted according to the complementary matching sequence by CircInteractome for the two circRNAs. Of which, 61 target miRNAs for hsa_circ_0043506 and 4 target miRNAs for hsa_circ_0000891. Among the 65 miRNAs, 17 miRNAs were simultaneously expressed on the microarray. Table 3 demonstrates the two validated circRNAs and their miRNA binding sites. The 16 miRNA-targeted genes are predicted with TargetScan and miRDB, and compared with the microarray results. In the end, we got 514 down-regulated genes and 264 up-regulated genes. In order to explore the function of two up-regulated circRNAs, we performed GO and pathway enrichment analyses for the predicted target genes. The GO analysis showed that the top five enriched GO terms were negative regulation of telomere capping, negative regulation of protein phosphorylation, regulation of translational initiation, regulation of protein ubiquitination and response to endoplasmic reticulum stress in BP (Figure 5A). The KEGG analysis indicated that the total six enriched in Protein processing in endoplasmic reticulum, systemic lupus erythematosus, endocytosis, spliceosome, HTLV-I infection and Ubiquitin mediated proteolysis (Figure 5B). At the same time, we performed GO and KEGG analyses for the predicted target genes of differently expressed miRNA (Figure S1). Interestingly, GO and pathway enrichment for the predicted target genes of miRNA and circRNA share many of the same pathways. On the basis of the miRNA and mRNA targets for the 2 circRNAs (hsa_circ_0000891, hsa_circ_0043506), a ceRNA network of circRNA-miRNA-mRNA interactions was established with Cytoscape software (Figure 6). Finally, we hope to draw the putative regulations on these targets in the pathogenesis of pulmonary embolism.
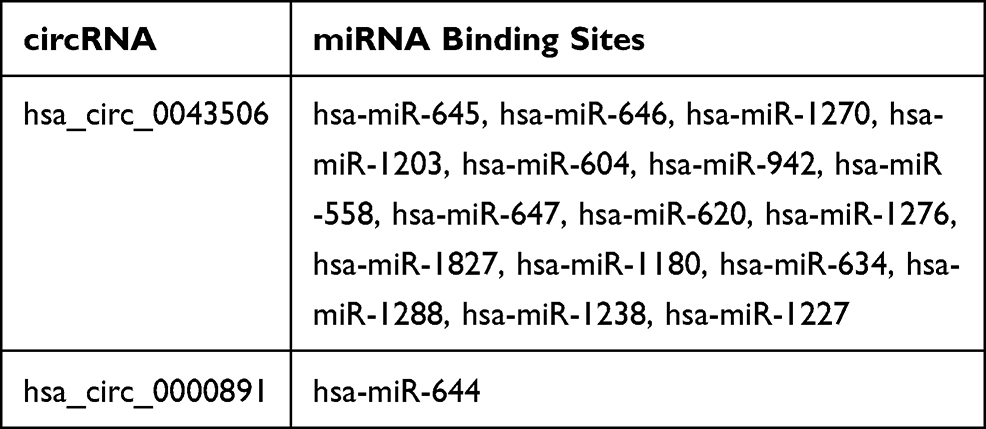 |
Table 3 Potential Target Genes of the Specific circRNAs |
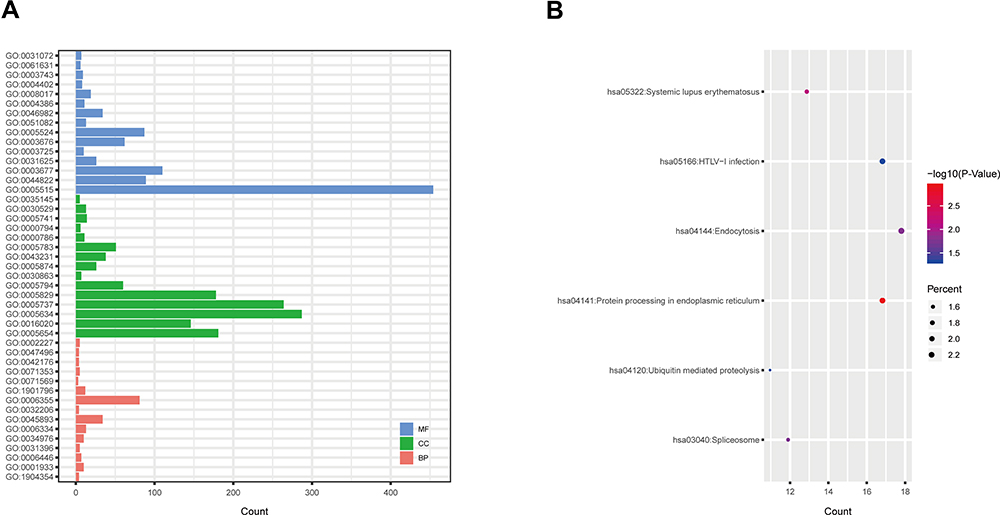 |
Figure 5 GO and KEGG analyses were performed for the predicted target genes of differentially expressed circRNA. (A) GO enrichment for targeted mRNAs. (B) KEGG pathway enrichment for targeted mRNAs. Abbreviations: GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes. |
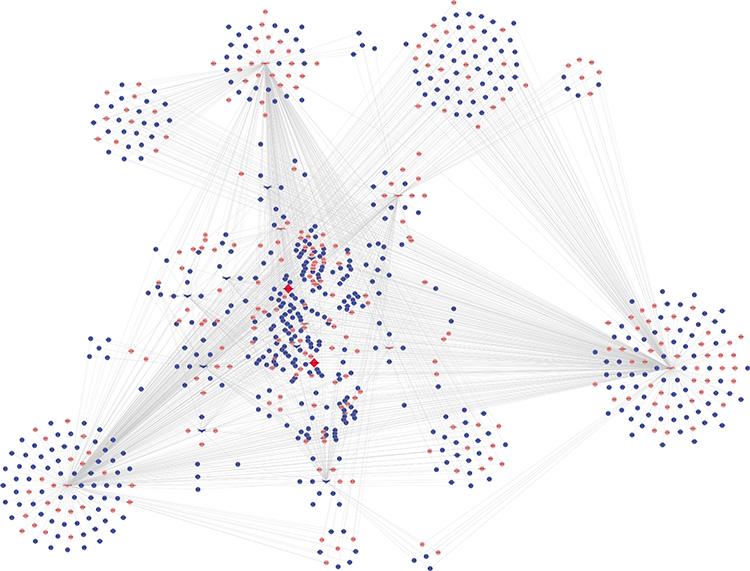 |
Figure 6 A ceRNA network of circRNA-miRNA-mRNA interactions. Diamond, circRNA; V, miRNA; round, mRNA; red, up-regulated; blue, down-regulated. |
Discussion
PTE is a clinical and pathophysiological syndrome of the pulmonary circulation and respiratory insufficiency, which lack of a reliable screening tool. The high morbidity, misdiagnosis and mortality rate make PTE a worldwide health problem. PTE is the third leading cause of death in the United States, after cancer and myocardial infarction.13 In fact, the molecular mechanism of the PTE is still unclear. Noncoding RNA may be determinant for various pathological and physiological processes of thrombotic events, including PTE. In the present study, the circRNAs expression pattern was found to be different between the PTE and control group. And then, the final outcome were used to construct a circRNA-miRNA-mRNA regulatory network. Therefore, we speculated that the special circRNAs might be involved in the pathogenesis of PTE.
As a result of the rapid development of high-throughput technologies including microarray and high-throughput sequencing. Increasing studies have shown that circRNAs can affect a variety of disease processes, including cancer,8 cardiovascular disease,14 and nervous system diseases.15 CircRNAs consist of no 5′ or 3′ free terminus, which formed a naturally closed loop structure to avoid the degradation of RNase enzyme. Mounting evidence has proved that the function of circRNAs could act as a miRNA ‘sponge’ and inhibit miRNAs’ function.16 In the present study, a total of 256 differentially expressed circRNAs were identified including 142 up-regulated circRNAs and 114 down-regulated circRNAs. To verify the circRNAs sequencing data, we selected three up-regulated and three down-regulated identified circRNAs for the analysis of RT-qPCR. The data confirmed that hsa_circ_0000891 and hsa_circ_0043506 was up-regulated in peripheral blood of PTE by RT-qPCR. At present, there is no significant evidence to prove that hsa_circ_0000891 and hsa_circ_0043506 is associated with PTE. Many studies have indicated that miR-942 is closely involved in the development of various disease including chronic thromboembolic pulmonary hypertension,17 cancer,18,19 Liver Fibrosis,20 Nephrotic Syndrome,21 infections.22 In a study of 512 patients with nephrotic syndrome, pulmonary embolism and/or renal vein thrombosis (RVT) were found in 35% of them, and pulmonary embolism was more common.23 Tumor aggravates high blood coagulation state. PTE is more common in patients with malignant tumors than in the general population, and has a high fatality rate, disability rate and clinical missed diagnosis rate.24–26 Based on miRNAs sponge mechanism and the above studies, we speculated that the target miRNAs of hsa_circ_0043506 (miR-942) might be involved in the pathogenesis of PTE, although there is no direct evidence that tumor pathways are associated with PE.
CAPN2, a target gene of miR-942, was also detected by microarray analysis, which enriched in pathways in protein processing in endoplasmic reticulum. CAPN2 adjust endothelial integrity which involve in membrane fusion, cytoskeletal remodeling, cell differentiation and proliferation.27–29 Activated CAPN2 cleaves eNOS and cytoskeletal proteins and then induces apoptosis.30 Mice with knockout of CAPN2 are lethal defects in vasculogenesis with characteristic rounding of the endothelial cells.31 Furthermore, inhibiting CAPN2 reduces spreading, actin filament networks and formation of focal adhesions of endothelial cells.32 The previous research indicates that CAPN2 inhibition significantly reduced endothelial nitric oxide (NO) synthase (NOS-3)-mediated NO production.33 Specific CAPN2 facilitates the NOS-3 activity in porcine pulmonary arterial endothelial cells.34 In the pathological mechanism of PTE, pulmonary endothelial remodeling has been emphasized. Thrombus that is trapped in pulmonary vessels can destroy vascular endothelial cells, which stimulate the release of inflammatory mediators.35 In PTE mice model, NO in the peripheral blood was reduced while eNOS was up-regulated in pulmonary endothelial cells. A decrease in the activity of NO results further affects other regulatory networks, such as inhibition EPCs differentiating into mature vascular endothelial cells and repair of the vascular endothelium.36 Combining these findings with the results of our present study, we speculate that hsa_circ_0043506-miR-942-CAPN2 may play an important role in the pathophysiology of PTE. However, further studies are needed to verify our conclusions.
Gene expression profiles of a target gene in PTE can help us understand its biological function of the differentially expressed circRNAs. We did GO enrichment analysis, revealing the biological process, cellular component, and molecular function of the target genes about hsa_circ_0043506 and hsa_circ_0000891. GO enrichment analysis of target genes of candidate circRNAs showed that the enriched GO terms referred mainly to regulation of translational initiation, telomere capping, protein ubiquitination, transcription and response to endoplasmic reticulum stress so on. We speculate that the observations be connected with PTE, but further work is needed to confirm this. On the other hand, KEGG analysis illustrated that there were 6 pathways related to the 2 up-regulated circRNAs, including Protein processing in endoplasmic reticulum, systemic lupus erythematosus, endocytosis, spliceosome, HTLV-I infection and ubiquitin mediated proteolysis. Factor V and factor VIII were regulated in Protein processing in endoplasmic reticulum. Systemic lupus erythematosus (SLE) is a systemic inflammatory disease. Another pathway (Endocytosis) play an important role in Syndromic multisystem autoimmune disease.37 And the pathway (Ubiquitin mediated proteolysis) play an important role in cancer disease.38,39 Plenty of studies indicated that the most common pulmonary manifestations of autoimmune disease are pulmonary thromboembolism and pulmonary hypertension.40 All these results suggest that the top 2 up-regulated circRNAs may be involved in pathogenesis of PTE.
In conclusion, to the best of our knowledge we are the first to report the circRNAs profile in PTE patients and to propose that hsa_circ_0043506-miR-942-CAPN2 pathways may be key mechanisms in PTE development. However, there are still some shortcomings in this study. First, the sample size was still limited, and we should try our best to recruit larger number of PTE samples to confirm our results. Second, we consider hsa_circ_0043506- miR-942-CAPN2 pathways may participate in the mechanism of PTE development, but other pathways need to be elaborated in further studies. Third, the results here obtained or predicted needs further confirmation by performing a set of cellular and molecular experiments.
Data Sharing Statement
The analyzed data sets generated during the present study are available from the first author on reasonable request.
Acknowledgments
We thank all partners who were involved in this study, especially our colleagues Danyang Zhang.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Grants from Yunhe Project of Tongzhou, Beijing, China [2017B0400386]; Health Development Research Project of Tongzhou, Beijing, China [TFZXPT-20180103].
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Schembri GP, Miller AE, Smart R. Radiation dosimetry and safety issues in the investigation of pulmonary embolism. Semin Nucl Med. 2010;40:442–454. doi:10.1053/j.semnuclmed.2010.07.007
2. Stein PD, Hull RD, Patel KC, et al. D-dimer for the exclusion of acute venous thrombosis and pulmonary embolism: a systematic review. Ann Intern Med. 2004;140:589–602. doi:10.7326/0003-4819-140-8-200404200-00005
3. Benincasa G, Marfella R, Della Mura N, Schiano C, Napoli C. Strengths and opportunities of network medicine in cardiovascular diseases. Circ J. 2020;84:144–152. doi:10.1253/circj.CJ-19-0879
4. Sunderland N, Skroblin P, Barwari T, et al. MicroRNA biomarkers and platelet reactivity: the clot thickens. Circ Res. 2017;120:418–435. doi:10.1161/CIRCRESAHA.116.309303
5. Zhao B, Chen Y, Hu S, et al. Systematic analysis of non-coding RNAs involved in the angora rabbit () hair follicle cycle by RNA sequencing. Front Genet. 2019;10:407. doi:10.3389/fgene.2019.00407
6. Wang Y, Mo Y, Gong Z, et al. Circular RNAs in human cancer. Mol Cancer. 2017;16:25. doi:10.1186/s12943-017-0598-7
7. Salzman J, Gawad C, Wang PL, et al. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 2012;7:e30733. doi:10.1371/journal.pone.0030733
8. Suzuki H, Tsukahara T. A view of pre-mRNA splicing from RNase R resistant RNAs. Int J Mol Sci. 2014;15:9331–9342.
9. Benincasa G, Mansueto G, Napol C. Fluid-based assays and precision medicine of cardiovascular diseases: the ‘hope’ for Pandora’s box? J Clin Pathol. 2019:1–15.
10. Zhou M-Y, Yang J-M, Xiong X-D. The emerging landscape of circular RNA in cardiovascular diseases. J Mol Cell Cardiol. 2018;122:134–139. doi:10.1016/j.yjmcc.2018.08.012
11. Enright AJ, John B, Gaul U, et al. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1. doi:10.1186/gb-2003-5-1-r1
12. Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43:D146–D52. doi:10.1093/nar/gku1104
13. Garg K, Welsh CH, Feyerabend AJ, et al. Pulmonary embolism: diagnosis with spiral CT and ventilation-perfusion scanning–correlation with pulmonary angiographic results or clinical outcome. Radiology. 1998;208:201–208. doi:10.1148/radiology.208.1.9646814
14. Altesha MA, Ni T, Khan A, et al. Circular RNA in cardiovascular disease. J Cell Physiol. 2019;234:5588–5600. doi:10.1002/jcp.27384
15. Ghosal S, Das S, Sen R, et al. Circ2Traits: a comprehensive database for circular RNA potentially associated with disease and traits. Front Genet. 2013;4:283. doi:10.3389/fgene.2013.00283
16. Liu L, Wang J, Khanabdali R, et al. Circular RNAs: isolation, characterization and their potential role in diseases. RNA Biol. 2017;14:1715–1721. doi:10.1080/15476286.2017.1367886
17. Miao R, Wang Y, Wan J, et al. Microarray expression profile of circular RNAs in chronic thromboembolic pulmonary hypertension. Medicine. 2017;96:e7354. doi:10.1097/MD.0000000000007354
18. Patnaik SK, Yendamuri S, Kannisto E, et al. MicroRNA expression profiles of whole blood in lung adenocarcinoma. PLoS One. 2012;7:e46045. doi:10.1371/journal.pone.0046045
19. Ge C, Wu S, Wang W, et al. miR-942 promotes cancer stem cell-like traits in esophageal squamous cell carcinoma through activation of Wnt/β-catenin signalling pathway. Oncotarget. 2015;6:10964–10977. doi:10.18632/oncotarget.3696
20. Tao L, Xue D, Shen D, et al. MicroRNA-942 mediates hepatic stellate cell activation by regulating BAMBI expression in human liver fibrosis. Arch Toxicol. 2018;92:2935–2946. doi:10.1007/s00204-018-2278-9
21. Teng J, Sun F, Yu P-F, et al. Differential microRNA expression in the serum of patients with nephrotic syndrome and clinical correlation analysis. Int J Clin Exp Pathol. 2015;8:7282–7286.
22. Yan Q, Shen C, Qin J, et al. HIV-1 Vpr inhibits kaposi’s sarcoma-associated herpesvirus lytic replication by inducing MicroRNA miR-942-5p and activating NF-κB signaling. J Virol. 2016;90:8739–8753. doi:10.1128/JVI.00797-16
23. Zhang L, Zhang Z, Li S, et al. Pulmonary embolism and renal vein thrombosis in patients with nephrotic syndrome: prospective evaluation of prevalence and risk factors with CT. Radiology. 2014;273:897–906. doi:10.1148/radiol.14140121
24. Cushman M, Tsai AW, White RH, et al. Deep vein thrombosis and pulmonary embolism in two cohorts: the longitudinal investigation of thromboembolism etiology. Am J Med. 2004;117:19–25. doi:10.1016/j.amjmed.2004.01.018
25. Timp JF, Braekkan SK, Versteeg HH, et al. Epidemiology of cancer-associated venous thrombosis. Blood. 2013;122:1712–1723. doi:10.1182/blood-2013-04-460121
26. Van Es N, Bleker SM, Di Nisio M. Cancer-associated unsuspected pulmonary embolism. Thromb Res. 2014;133:S172–S8. doi:10.1016/S0049-3848(14)50028-X
27. Gonscherowski V, Becker BF, Moroder L, et al. Calpains: a physiological regulator of the endothelial barrier? Am J Physiol Heart Circ Physiol. 2006;290:H2035–H2042. doi:10.1152/ajpheart.00772.2004
28. Bialkowska K, Kulkarni S, Du X, et al. Evidence that beta3 integrin-induced Rac activation involves the calpain-dependent formation of integrin clusters that are distinct from the focal complexes and focal adhesions that form as Rac and RhoA become active. J Cell Biol. 2000;151:685–696. doi:10.1083/jcb.151.3.685
29. Goll DE, Thompson VF, Li H, et al. The calpain system. Physiol Rev. 2003;83:731–801. doi:10.1152/physrev.00029.2002
30. Dong Y, Wu Y, Wu M, et al. Activation of protease calpain by oxidized and glycated LDL increases the degradation of endothelial nitric oxide synthase. J Cell Mol Med. 2009;13:2899–2910. doi:10.1111/j.1582-4934.2008.00416.x
31. Arthur JS, Elce JS, Hegadorn C, et al. Disruption of the murine calpain small subunit gene, Capn4: calpain is essential for embryonic development but not for cell growth and division. Mol Cell Biol. 2000;20:4474–4481. doi:10.1128/MCB.20.12.4474-4481.2000
32. Kulkarni S, Saido TC, Suzuki K, et al. Calpain mediates integrin-induced signaling at a point upstream of Rho family members. J Biol Chem. 1999;274:21265–21275. doi:10.1074/jbc.274.30.21265
33. Liu D, Yan Z, Minshall RD, et al. Activation of calpains mediates early lung neutrophilic inflammation in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2012;302:L370–L379. doi:10.1152/ajplung.00349.2011
34. Su Y, Block ER. Role of calpain in hypoxic inhibition of nitric oxide synthase activity in pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2000;278:L1204–L12. doi:10.1152/ajplung.2000.278.6.L1204
35. Wood KE. Major pulmonary embolism: review of a pathophysiologic approach to the golden hour of hemodynamically significant pulmonary embolism. Chest. 2002;121:877–905.
36. Ning H, Liu G, Lin G, et al. Fibroblast growth factor 2 promotes endothelial differentiation of adipose tissue-derived stem cells. J Sex Med. 2009;6:967–979. doi:10.1111/j.1743-6109.2008.01172.x
37. Zhao D, Guo M, Liu B, et al. Frontline Science: tim-3-mediated dysfunctional engulfment of apoptotic cells in SLE. J Leukoc Biol. 2017;102:1313–1322. doi:10.1189/jlb.3HI0117-005RR
38. Davis MI, Pragani R, Fox JT, et al. Small molecule inhibition of the ubiquitin-specific protease USP2 accelerates cyclin D1 degradation and leads to cell cycle arrest in colorectal cancer and mantle cell lymphoma models. J Biol Chem. 2016;291:24628–24640. doi:10.1074/jbc.M116.738567
39. Roberti A, Rizzolio F, Lucchetti C, et al. Ubiquitin-mediated protein degradation and methylation-induced gene silencing cooperate in the inactivation of the INK4/ARF locus in Burkitt lymphoma cell lines. Cell Cycle. 2011;10:127–134. doi:10.4161/cc.10.1.14446
40. Tselios K, Urowitz MB. Cardiovascular and pulmonary manifestations of systemic lupus erythematosus. Curr Rheumatol Rev. 2017;13:206–218. doi:10.2174/1573397113666170704102444
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.