")
Back to Journals » International Journal of Nanomedicine » Volume 13
Micelle-templated, poly(lactic-co-glycolic acid) nanoparticles for hydrophobic drug delivery
Authors Nabar GM , Mahajan KD, Calhoun MA, Duong AD, Souva MS, Xu J , Czeisler C , Puduvalli VK , Otero JJ, Wyslouzil BE, Winter JO
Received 24 May 2017
Accepted for publication 15 July 2017
Published 10 January 2018 Volume 2018:13 Pages 351—366
DOI https://doi.org/10.2147/IJN.S142079
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Gauri M Nabar,1 Kalpesh D Mahajan,1 Mark A Calhoun,2 Anthony D Duong,1 Matthew S Souva,1 Jihong Xu,3,4 Catherine Czeisler,5 Vinay K Puduvalli,3,4 José Javier Otero,5 Barbara E Wyslouzil,1,6 Jessica O Winter1,2
1William G Lowrie Department of Chemical and Biomolecular Engineering, 2Department of Biomedical Engineering, 3Division of Neuro-oncology, College of Medicine, The Ohio State University Comprehensive Cancer Center, 4Dardinger Laboratory for Neuro-oncology and Neurosciences, Department of Neurosurgery, College of Medicine, The Ohio State University Comprehensive Cancer Center, 5Department of Pathology and the Neurological Research Institute, College of Medicine, 6Department of Chemistry and Biochemistry, The Ohio State University, Columbus, OH, USA
Purpose: Poly(lactic-co-glycolic acid) (PLGA) is widely used for drug delivery because of its biocompatibility, ability to solubilize a wide variety of drugs, and tunable degradation. However, achieving sub-100 nm nanoparticles (NPs), as might be desired for delivery via the enhanced permeability and retention effect, is extremely difficult via typical top-down emulsion approaches.
Methods: Here, we present a bottom-up synthesis method yielding PLGA/block copolymer hybrids (ie, “PolyDots”), consisting of hydrophobic PLGA chains entrapped within self-assembling poly(styrene-b-ethylene oxide) (PS-b-PEO) micelles.
Results: PolyDots exhibit average diameters <50 nm and lower polydispersity than conventional PLGA NPs. Drug encapsulation efficiencies of PolyDots match conventional PLGA NPs (ie, ~30%) and are greater than those obtained from PS-b-PEO micelles (ie, ~7%). Increasing the PLGA:PS-b-PEO weight ratio alters the drug release mechanism from chain relaxation to erosion controlled. PolyDots are taken up by model glioma cells via endocytotic mechanisms within 24 hours, providing a potential means for delivery to cytoplasm. PolyDots can be lyophilized with minimal change in morphology and encapsulant functionality, and can be produced at scale using electrospray.
Conclusion: Encapsulation of PLGA within micelles provides a bottom-up route for the synthesis of sub-100 nm PLGA-based nanocarriers with enhanced stability and drug-loading capacity, and tunable drug release, suitable for potential clinical applications.
Keywords: PLGA, nanoparticles, micelles, drug delivery, hydrophobic drug, block copolymer, glioma, electrospray
Plain language summary
Drugs that are not soluble in blood, such as dexamethasone steroids, are difficult to deliver to the body. Nanoparticles (NPs) can serve as carriers to transport these drugs to a desired location, and can also control the timing of drug release. Two common materials used to deliver drugs are micelles and solid polymer NPs. In this paper, a NP that combines the benefits of these two particle types is described. The “PolyDot” consists of polymer chains used to form solid polymer NPs that are encapsulated within micelles. The ratio of these two materials was shown to have only modest impact on PolyDot size, which remained small, but could alter the way that drugs were released. Toward clinical implementation, methods to produce large amounts of PolyDots and to enable their storage are shown. PolyDot uptake by cells was also studied, indicating that drug delivery is possible. These studies show that PolyDots have important advantages for drug delivery, such as tightly controlled size, high drug-loading ability, and controllable drug release.
Introduction
Hydrophobic drugs have poor solubility in blood; thus, alternative delivery schemes are required to deliver these drugs to their targets.1 Although excipients can be used to enhance solubility, such as Cremophor for Paclitaxel delivery to cancer patients,2 these often have toxic side effects. Nanoparticle (NP) carriers offer a crucial alternative to the use of excipients for hydrophobic drug delivery which present minimal increases in toxicity.3,4 To achieve optimal circulation in vivo, carriers must be larger than 10 nm to avoid rapid renal clearance,5 but smaller than ~200 nm to avoid sequestration by macrophages in the reticuloendothelial system.6 In cancer therapy, carriers of <~100 nm are preferred, as they can extravasate through the gap junctions between endothelial cells of the hyperpermeable tumor vasculature.7
Poly(lactic-co-glycolic acid) (PLGA) microparticles and NPs have been widely used in drug delivery because of their biocompatibility, tunable degradation times, and ability to encapsulate a wide variety of drugs.8 PLGA is a “generally regarded as safe” polymer, has been incorporated into FDA-approved drug delivery systems such as the Lupron® depot,9 and has been employed for cardiovascular disease treatment, vaccine delivery, and chemotherapy.10 The most common methods employed to synthesize PLGA NPs are nanoprecipitation11 and emulsion-based processes.12 Using these approaches, particles >200 nm in size can be easily and uniformly produced by optimizing PLGA concentration,13,14 PLGA–solvent interactions,15 continuous- and dispersed-phase viscosities,16 or the rate of mixing and shear stress17 applied for emulsion formation. However, synthesis of sub-100 nm PLGA NPs, as might be required for systemic delivery, has proven particularly challenging.18 For example, some commonly used emulsion approaches19 result in PLGA NPs that appear sub-100 nm via scanning electron microscopy, but that are connected via polymer bridges, thus resulting in a much larger effective diameter. Additionally, drug loading can alter morphology, increasing the NP size to several hundred nanometers.16
As an alternative to PLGA NPs, micelles have been used for hydrophobic drug delivery, resulting in clinically approved products.20,21 Lipid-polymer and block copolymer (BCP)-based micelle carriers are typically formed via bottom-up self-assembly in solvents selective for one of the blocks. Because assembly is controlled in part by the thermodynamics governing the system, micelles can exhibit tightly controlled, reproducible sizes. Self-assembling BCP micelles display enhanced vascular permeability, narrower size distributions, and better release characteristics compared to microparticles.22 However, low drug-loading capacity resulting from poor drug–BCP compatibility can be a limitation in micellar drug delivery systems.23 Also, the lack of strong intermolecular interactions between the drug and the micelle can cause rapid leakage of physically entrapped drugs.24 For example, micelles composed of PLGA-b-poly(ethylene glycol) (PEG) BCPs released physically entrapped doxorubicin in only 3 days.25
Here, we examined the combination of these two popular drug release systems by using micelles to encapsulate high-molecular-weight PLGA polymer chains, yielding PolyDots. This approach is an extension of our previous work using poly(styrene-b-ethylene oxide) (PS-b-PEO) BCPs (molecular weight 9500-b-18000 Da) to encapsulate hydrophobic NPs.26–29 PS has been previously used for hydrophobic drug delivery30 and may promote cellular uptake compared to hydroxyacid-based systems (ie, PLGA, poly(caprolactone)),31 whereas the PEO block (a long chain form of PEG) creates a corona with the potential to repel opsonin proteins, reducing immunogenicity and increasing circulation times.32 This approach is distinct from methods that encapsulate chemically similar polymers in micelles, such as those incorporating homopolymers or BCPs with common block(s),33,34 as PLGA is chemically distinct from both PS and PEO blocks. This approach is also different from admicellar polymerization processes,35 which involve in situ polymerization of adsolubilized monomers in the hydrophobic region of micelles or bilayers, as already synthesized PLGA chains are employed. Using this approach, we evaluated the effect of PLGA:PS-b-PEO ratio on resultant NP size and polydispersity, resistance to degradation in acidic medium, drug encapsulation and release, cellular trafficking, and ability to preserve encapsulants following lyophilization.
Materials and methods
Materials
Carboxyl-terminated PS-b-PEO (molecular weight 9500-b-18000 Da) (Cat No P5755-SEOCOOH) was purchased from Polymer Source Inc. (Montreal, QC, Canada). Poly(DL-lactide-co-glycolide) (molecular weight 50–70 kDa, Cat No B6010-3) was purchased from Lactel Absorbable Polymers. Poly(vinyl alcohol) (PVA; 13,000–23,000 g/gmol) (Cat No 363170) was purchased from Sigma-Aldrich, and chloroform (Cat No C606SK-4) was purchased from Thermo Fisher Scientific. Dexamethasone (DEX; Cat No BML-EI126-0001) was purchased from Enzo Life Sciences. Coumarin 6 dye was purchased from Sigma-Aldrich (Cat No 442631). All chemicals were used as received without further purification.
PolyDot synthesis
PolyDots were prepared by an emulsion-based procedure described by Geng and Discher36 and Zhu and Hayward33 to yield BCP micelles loaded with hydrophobic polymer and drug. Two model drugs were investigated: DEX, a steroidal anti-inflammatory drug, and coumarin 6, a hydrophobic fluorescent dye. Two approaches were used to generate emulsions: sonication, consistent with that previously employed,33 and electrospray, developed by us, that can be scaled to commercial volumes.37
Sonication
PLGA (20, 50, 100, and 200 μL) at a fixed stock concentration of 50 mg/mL was added to DEX (500 μL, 5 mg/mL) and carboxylated PS-b-PEO (200 μL, 1 mg/mL) in chloroform. Thus, the four PolyDot samples examined had PLGA:PS-b-PEO weight ratios of 5, 12.5, 25, and 50, respectively. Solutions were mixed by vortexing for 30 seconds and added to 8 mL of aqueous PVA solution (5 mg/mL). These biphasic solutions were then sonicated using a probe sonicator (Branson Sonifier 450) at a constant duty cycle for 5 minutes. As controls, PS-b-PEO micelles were prepared via the same procedure without any PLGA in the starting organic phase, and PVA-stabilized PLGA NPs were synthesized by sonicating an organic phase consisting of PLGA (200 μL, 50 mg/mL) in chloroform with PVA (8 mL, 5 mg/mL) under the same conditions. The milky white emulsions obtained after sonication were transferred to aluminum dishes with an internal diameter of 5 cm and placed on a rocker plate for 2.5 hours to allow evaporation of the chloroform solvent. After 2.5 hours, clear and transparent NP solutions were obtained for the PS-b-PEO micelles and PolyDots with low initial PLGA:PS-b-PEO ratios (20 and 50 μL), whereas those with higher PLGA:PS-b-PEO ratios (100 and 200 μL) and the control PLGA NPs appeared opaque, indicating highly concentrated solutions or the presence of some micron-sized particles. All samples were prepared in triplicate.
Electrospray
Coaxial electrospray was performed using a concentric needle connected to two syringes, a grounded copper ring, and an aluminum dish to collect the aerosol droplets, similar to that described previously.37 PolyDot samples with a PLGA:PS-b-PEO weight ratio of 12.5 were synthesized in semicontinuous mode using electrospray. Organic solution, prepared by mixing PS-b-PEO (200 μL, 1 mg/mL) and PLGA (50 μL, 50 mg/mL) in chloroform, was passed through the inner needle at 0.5 mL/h. Aqueous solution containing 50 mg/mL PVA was passed through the outer needle at 2.5 mL/h. The organic and aqueous solutions first emerged as compound droplets at the exit of the concentric needle. An electrostatic potential of ~3.5 kV was applied between the concentric needle and the grounded copper ring. The electrostatic potential was adjusted so that a concave jet was obtained at the exit of the needle. This jet further broke down into aerosol droplets with fine internal mixing, forming an emulsion. The resulting emulsion droplets were collected for 30 minutes in an aluminum dish with an internal diameter of 5 cm containing 10 mL of water. The spray duration was selected to match the batch size produced via sonication. Then, the emulsion was left on a rocker plate to allow chloroform to evaporate. A clear and transparent solution was obtained in ~2 hours.
Nanocarrier size analysis
Nanocarrier sizes were measured using two complementary approaches described as follows.
Dynamic light scattering
Particle hydrodynamic diameters were measured using dynamic light scattering (DLS) (BI 200SM; Brookhaven Instruments Corporation). Particle concentration in aqueous solution was adjusted so as to obtain a scattered light intensity between 10 and 200 kilocounts per second. Laser illumination was applied at 633 nm, and the detector was set to 150°. Each particle sample was measured in triplicate.
Transmission electron microscopy (TEM)
Negatively stained TEM images were collected using an FEI Tecnai G2 BioTWIN transmission electron microscope. Sample droplets (20 μL) were placed on silicone pads. TEM grids (Formvar/carbon-coated nickel) were then placed on these droplets with carbon side down. After 3 minutes, the TEM grids were lifted, and excess solution was wicked away with a filter paper. Uranyl acetate (UA, 1%, 20 μL) was used for negative staining. Excess UA was wicked away as described. Size measurements were recorded for 100–2,000 particles per sample using ImageJ image analysis software with the analyzed particles feature. Size histograms were plotted using Igor Pro software (Wavemetrics, Tigard, OR, USA). The size distributions were approximated by either log normal or Gaussian distributions. The distribution that minimized the chi-squared value was used for fitting the entire data. Means and standard deviations of the data were thus extracted.
Nanocarrier degradation studies
PolyDot stability in acidic environments was tested by acidifying freshly prepared PolyDot 50 samples to pH 2 using concentrated hydrochloric acid. The PS-b-PEO micelles and PLGA NPs served as negative and positive controls, respectively, and were subjected to the same conditions. To increase the rate of degradation, the acidified sample was further incubated at 80°C for 9 hours which, given the small size of PLGA NPs, was sufficient to enable nearly complete degradation. TEM and image analysis were performed to observe the morphology and particle size of samples exposed to acid attack.
Drug encapsulation
Drug encapsulation in PS-b-PEO micelles, PolyDots, and PLGA NPs was evaluated using DEX as a model therapeutic drug. Samples for drug encapsulation measurement were synthesized in triplicate by sonication using 500 μL (5 mg/mL) DEX in the initial chloroform solution. Following synthesis, un-encapsulated DEX was removed by repeated washing with distilled, deionized (DI) water. For PolyDots and PLGA NPs, centrifugation (1 hour, 2330 g) was used to induce pellet formation; then, 90% of the supernatant was discarded and replaced with DI water. This washing cycle was repeated two more times (30 minutes each). Low speeds were used during centrifugation to prevent aggregation during pellet formation. PS-b-PEO micelles not containing PLGA failed to form pellets and were therefore subjected to ultrafiltration (molecular weight cutoff 100 kDa; Amicon Ultra Centrifugal filter) at the same centrifugation settings and washing conditions. The washed pellets or micelle concentrates were dried overnight, and resuspended in 0.9 mL chloroform. This dissolved the pellets, releasing the entrapped DEX into the chloroform solution. DEX content was measured in chloroform solution by UV–Vis spectrophotometry at 278 nm (peak position of DEX in chloroform) and quantified by comparing to a standard curve.
Drug release
Drug release from PS-b-PEO micelles, PolyDots, and PLGA NPs was evaluated over the course of 38 days using DEX as a model therapeutic drug. Samples were synthesized via sonication using 500 μL DEX (5 mg/mL) in the initial chloroform solution. Un-encapsulated DEX was removed by centrifugation/ultrafiltration as described. Then, 1 mL samples were incubated in Milli-Q water at 37°C. To minimize water evaporation, tubes were closed and sealed with Parafilm. At predetermined time points, pellets or concentrates were formed by centrifugation or ultrafiltration (10 minutes, 4,000 g), as described. The supernatant containing the released DEX was collected and analyzed by UV–Vis spectroscopy by comparing absorbance at 242 nm (peak position of DEX in water) to a standard curve. The pellets were then reconstituted in fresh Milli-Q water and incubated until the next time point. All measurements were performed in triplicate.
Lyophilization
PolyDots were synthesized via sonication, as described, at a PLGA:PS-b-PEO ratio of 12.5 with 50 μL of fluorescent coumarin 6 as a model drug. Immediately following synthesis, samples were frozen solid at −80°C and freeze-dried overnight (Labconco FreeZone 4.5, 0°C, 10 Pa). Lyophilized samples were stored at −20°C until testing. Prior to testing, lyophilized samples were resuspended in DI water at a concentration consistent with the original. Negatively stained TEM images were obtained for freshly synthesized and lyophilized samples. In addition, particle integrity for freshly prepared and lyophilized samples was verified by measuring the fluorescence emission spectra over a range of 460–640 nm at an excitation wavelength of 420 nm using a PTI spectrofluorometer.
PolyDot cellular uptake
To visualize PolyDot cellular uptake, lyophilized PolyDots were synthesized as described via sonication using coumarin 6. As a model system, LN229 and U87 glioma cells (ATCC) were cultured to 60%–80% confluency using complete medium (DMEM/F12, 10% fetal bovine serum, 100 IU/mL penicillin, 100 μg/mL streptomycin, 2 mM glutamine). PolyDot medium was prepared by resuspending PolyDots in a volume of complete medium equivalent to the original PolyDot solution volume (estimated PS-b-PEO concentration 23 μg/mL). Cells were incubated with PolyDot-containing medium and evaluated at 30 minutes, 90 minutes, 3 hours, and 24 hours as follows. Medium was aspirated, and cells were briefly washed with phosphate-buffered saline (PBS) at room temperature. Paraformaldehyde (4% in PBS) was used to fix the cells at room temperature for 20 minutes. Cells were rinsed with PBS, stained with DAPI (Thermo Fisher Scientific 62248), and mounted using ProLong Antifade mounting agent (Thermo Fisher Scientific). Images were acquired with a Zeiss Axiovert upright confocal microscope at a magnification of 63×.
Statistics and curve fitting
Statistical analyses were performed using JMP Pro 12 software (SAS Institute Inc., Cary, NC, USA) using the ANOVA test. p-values <0.05 were interpreted as statistically significant. Size distribution histograms of particle sizes measured by ImageJ were constructed using Igor Pro 6.37 software (Wavemetrics). Data were fit using either log normal or Gaussian functions. For log normal fits, the location parameter and scale parameter were converted to mean size and standard deviation. For all samples, data are reported as mean size ± standard deviation.
Results and discussion
PolyDot size distribution
PS-b-PEO micelles without PLGA (control) and loaded with PLGA chains (molecular weight 50,000–70,000 Da, PolyDots) were prepared via the interfacial instability mechanism as described by Zhu and Hayward.33 We have previously shown that this approach can be used to encapsulate large hydrophobic payloads, such as NPs.27,28 To examine the ability of this method to encapsulate high-molecular-weight PLGA chains in the hydrophobic core, PolyDots were first created with a PLGA:PS-b-PEO weight ratio of 5. Resultant PolyDot morphology was evaluated using TEM and DLS (Figure 1). Accounting for hydration differences, both methods yielded a consistent diameter of ~50 nm for PolyDot 5 samples. We noted that two potential PolyDot populations were obtained, with larger and smaller diameters, respectively.
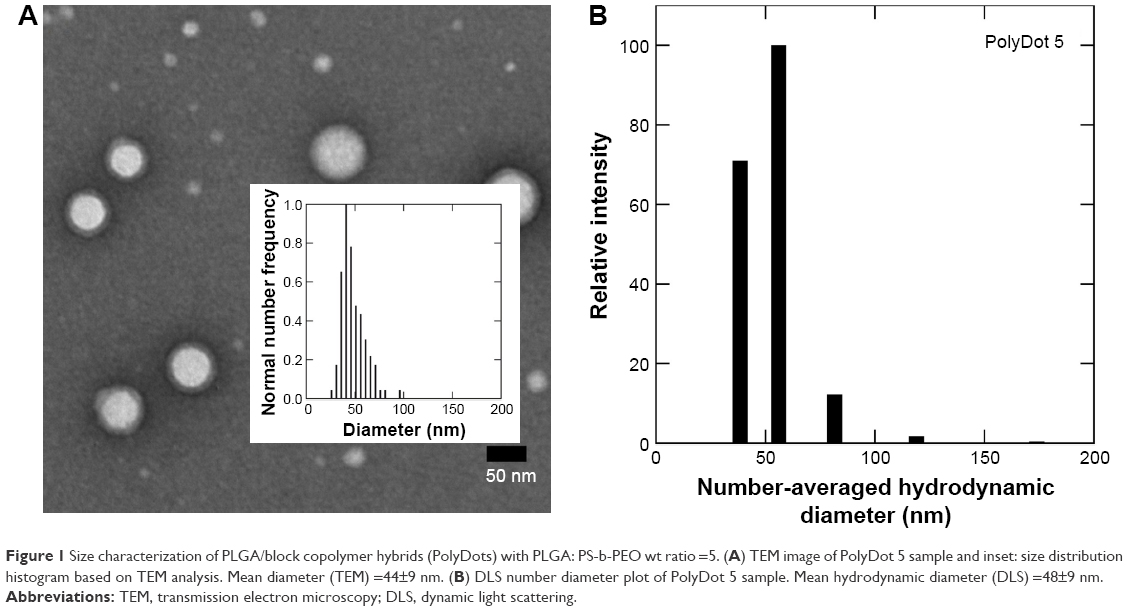 | Figure 1 Size characterization of PLGA/block copolymer hybrids (PolyDots) with PLGA: PS-b-PEO wt ratio =5. (A) TEM image of PolyDot 5 sample and inset: size distribution histogram based on TEM analysis. Mean diameter (TEM) =44±9 nm. (B) DLS number diameter plot of PolyDot 5 sample. Mean hydrodynamic diameter (DLS) =48±9 nm. |
Next, we evaluated the effect of PLGA:PS-b-PEO weight ratio (5, 12.5, 25, 50) on PolyDot morphology (Figure 2) and size distribution (Figure 3). The PS-b-PEO micelles and all PolyDot samples (PLGA:PS-b-PEO=5, 12.5, 25, and 50) exhibited log normal size distributions, whereas the PLGA NPs displayed a Gaussian distribution in size (Figure 3 and Table 1). Mean diameter ± standard deviation and mode diameters were extracted by curve-fitting size distribution histograms (Table 1). Polydispersity values, defined as standard deviation/mean diameter, were also calculated (Table 1). As represented by an increasing tail to larger sizes, PolyDots continued to exhibit two potential populations with different sizes, whereas PLGA NPs generally displayed a single population with large size variation.
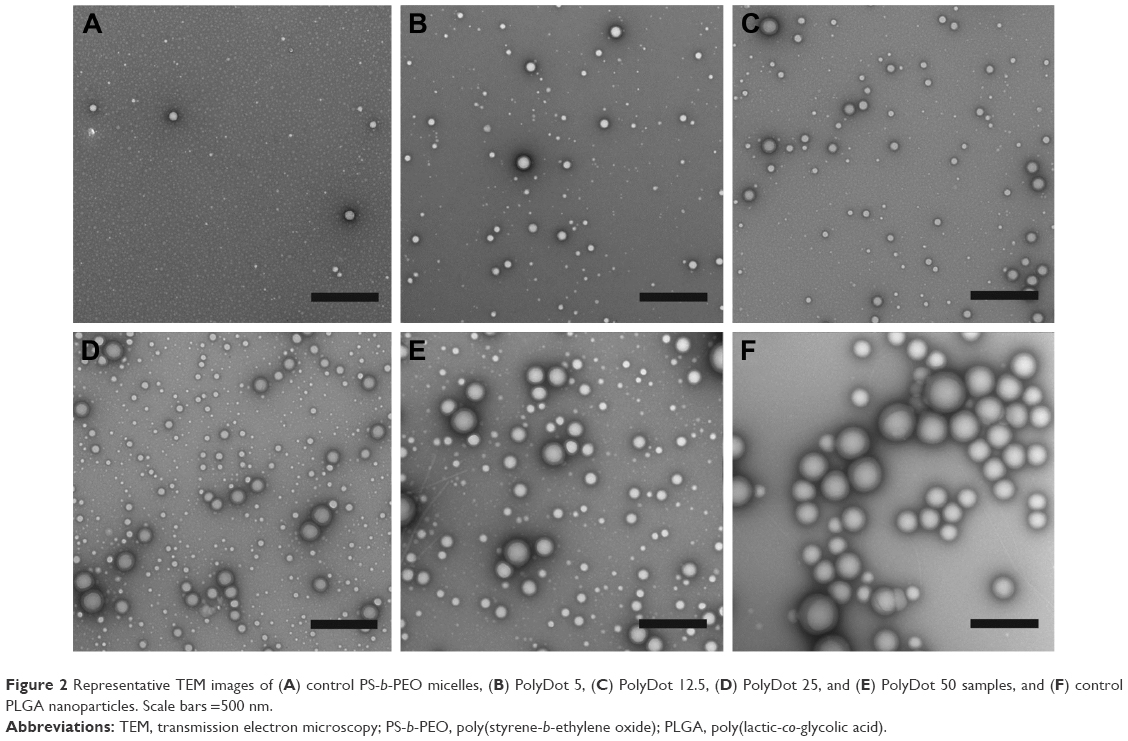 | Figure 2 Representative TEM images of (A) control PS-b-PEO micelles, (B) PolyDot 5, (C) PolyDot 12.5, (D) PolyDot 25, and (E) PolyDot 50 samples, and (F) control PLGA nanoparticles. Scale bars =500 nm. |
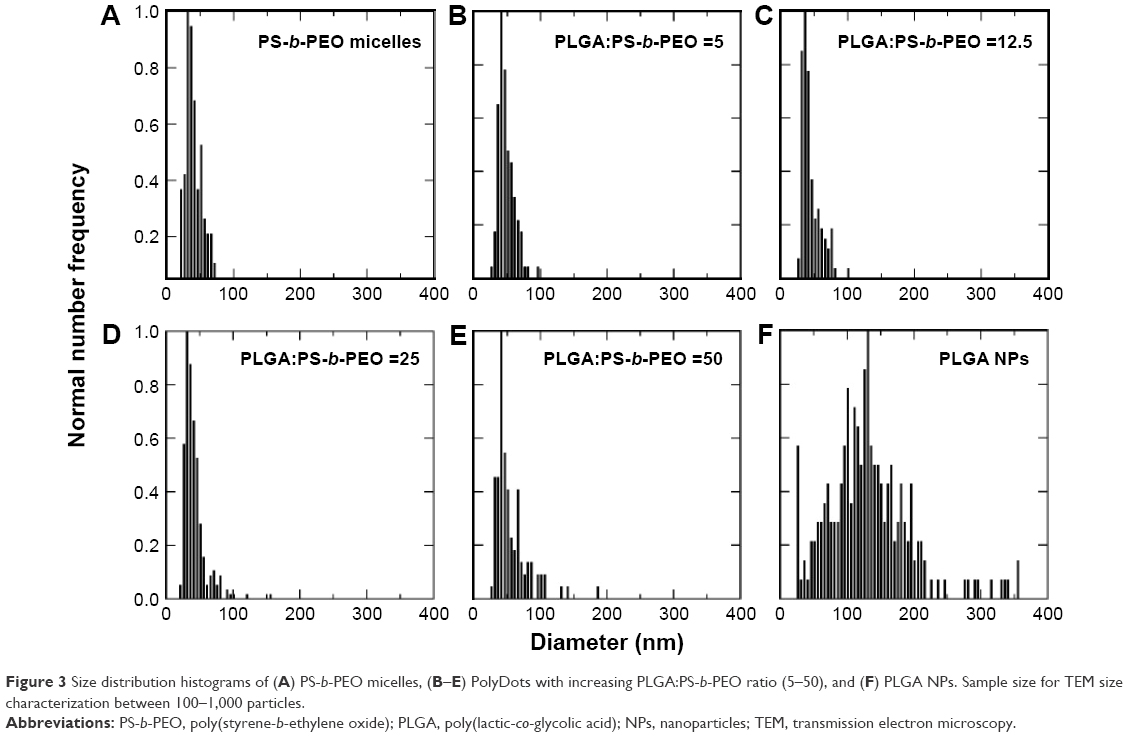 | Figure 3 Size distribution histograms of (A) PS-b-PEO micelles, (B–E) PolyDots with increasing PLGA:PS-b-PEO ratio (5–50), and (F) PLGA NPs. Sample size for TEM size characterization between 100–1,000 particles. |
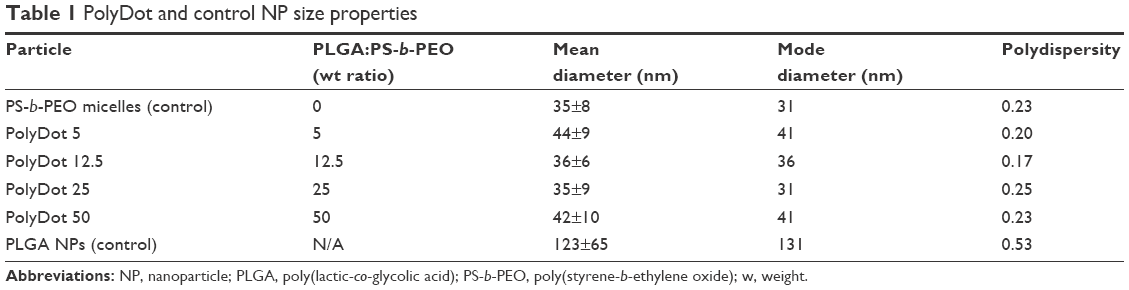 | Table 1 PolyDot and control NP size properties |
PS-b-PEO micelles not containing PLGA exhibited a mean size of 35 nm with a polydispersity of ~20%, consistent with the literature.38 PolyDots, containing both PS-b-PEO and PLGA, exhibited a mean size of 35–45 nm with a polydispersity of ~20%. Increasing PLGA:PS-b-PEO weight ratio from 5 to 50 increased the distribution tail toward larger diameters (Figure 3), and more structures could be seen in the TEM images (Figure 2B–E), but the mode diameters for all PolyDots ranged from 31 to 41 nm. Of particular note were PolyDot 50 samples (Figure 2E), which were synthesized with the same PLGA mass (10 mg) as control PLGA NPs and the same PS-b-PEO mass (0.2 mg) as control PS-b-PEO micelles. The PolyDot 50 size distribution (Figure 3 and Table 1) more closely resembled that of PS-b-PEO micelles than PLGA NPs, with a peak centered at ~40 nm. Over 93% of PolyDot 50 structures had sizes <100 nm. In contrast, PLGA NPs had much higher polydispersity (ie, ~0.5) with an average size of 123 nm, consistent with prior reports.39 The drastic reduction in size and polydispersity of PolyDot 50 structures compared to PLGA NPs suggests that PolyDots are not stabilized by PVA alone and supports the hypothesis that PLGA is incorporated within the micelle structure.
In addition, increasing PLGA:PS-b-PEO ratio did not noticeably impact the shape or smoothness of PolyDots (Figure 2). These observations are in contrast to micelles encapsulating NPs, which undergo changes in size and shape at high NP loadings.40 In NP-loaded micelles, BCPs suffer loss in conformational entropy during NP encapsulation resulting from NP rigidity, giving rise to phase transitions or surface defects.41 PLGA copolymers are softer and more compressible than NPs, which may have enabled PLGA loading in the micelle core or micelle structure without affecting shape.
PolyDot degradation
Low intra-tumoral pH is regarded as a cancer hallmark.42 PLGA degrades rapidly at low pH because of the acid-catalyzed degradation of its ester backbone.43 Thus, stability in acidic environments constitutes an important criterion in evaluating the potential of PLGA-based nanocarriers for chemotherapy. In addition, PLGA NPs degrade via ester hydrolysis, which is accelerated in acidic conditions,14 whereas PEO-based micelles are reportedly stable in acidic environments.44 Thus, degradation behavior can provide insight into the PolyDot structure. In vitro degradation studies of PLGA-based materials can require days to weeks for completion at physiologic temperature. Several groups have examined degradation behavior of PLGA-based substrates at elevated temperatures to increase degradation rates, thereby reducing experiment time.45 Thus, degradation behavior of PS-b-PEO micelles, PLGA NPs, and PolyDot 50 carriers was examined at low pH (pH 2) and high temperature (T =80°C) over 9 hours (Figure 4 and Table 2).
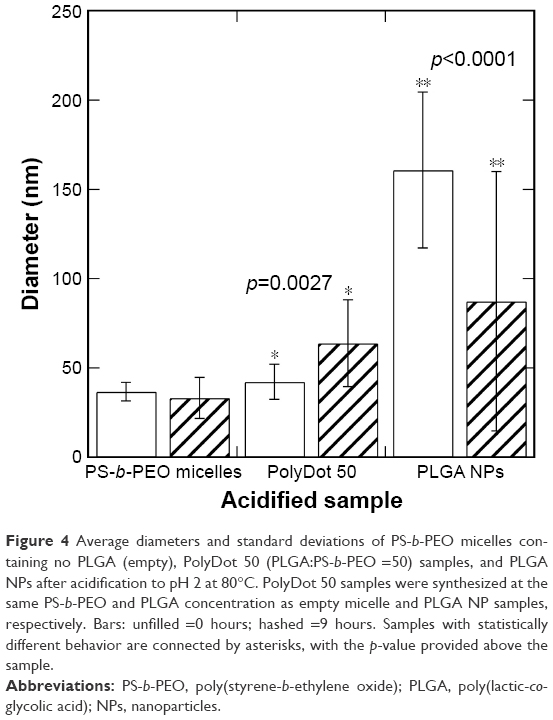 | Figure 4 Average diameters and standard deviations of PS-b-PEO micelles containing no PLGA (empty), PolyDot 50 (PLGA:PS-b-PEO =50) samples, and PLGA NPs after acidification to pH 2 at 80°C. PolyDot 50 samples were synthesized at the same PS-b-PEO and PLGA concentration as empty micelle and PLGA NP samples, respectively. Bars: unfilled =0 hours; hashed =9 hours. Samples with statistically different behavior are connected by asterisks, with the p-value provided above the sample. |
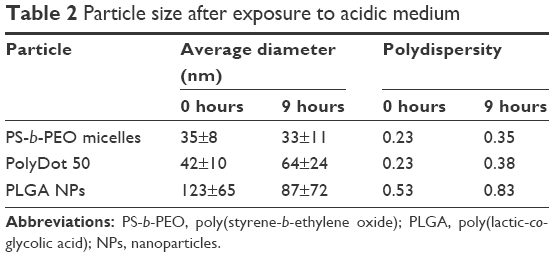 | Table 2 Particle size after exposure to acidic medium |
PLGA NPs displayed a statistically significant decrease in mean particle size following exposure to acid (p<0.0001). Decreases in diameter were more significant for larger PLGA NPs, suggesting higher degradation rates, consistent with the expected bulk erosion mechanism and enhanced autocatalytic degradation of larger structures.46 With prolonged acid exposure (48 hours), nearly all the PLGA NPs dissolved, most likely as a result of their small size and the elevated temperatures employed. The high temperature maintained throughout the experiment increased the degradation rate, but did not increase the aggregation of the PLGA NPs. De and Robinson have noted that high temperature can facilitate the diffusion of entrapped solvent to the surface of PLGA NPs, where it can dissolve the surface and facilitate aggregation.47 The absence of PLGA NP aggregation in our experiments suggests that the amount of solvent (chloroform) in the sample after interfacial instability is too low to enable temperature-induced aggregation.
In contrast, PS-b-PEO micelles not containing PLGA remained stable in acidic medium throughout the observation period with no statistically significant change in diameter. This is consistent with previous reports that similar PCL-b-PEO micelles remain structurally stable at pH 2.5 for up to a week.44 In those materials, a slight increase in size was observed, attributed to PEO coil swelling and increased repulsion between the PEO chains.44 Similar mechanisms are likely involved in PS-b-PEO micelles and may explain the observed increase in polydispersity. Further, the observed stability at high temperature is also not surprising as previous studies indicate that the hydrophobic PS blocks remain immobile even when heated to temperatures close to its Tg in pure water.48 PolyDots displayed a statistically significant increase in average size in acidic environments (p=0.0027), with few PolyDots observed after 9 hours of exposure to acidic medium. These findings suggest that PolyDots present a unique structure that is distinct from pure PS-b-PEO micelles and PLGA NPs.
Proposed PolyDot structure
Three structures are proposed for PolyDot (PS-b-PEO:PLGA) composites (Figure 5): 1) a heterogeneous population of PLGA NPs and PS-b-PEO micelles (Figure 5A); 2) PS-b-PEO micelle with intercalating PLGA chains (Figure 5B); or 3) PS-b-PEO micelles encapsulating PLGA chains within the micelle core (Figure 5C). The first possibility, in which self-assembly of a composite fails and separate populations of PS-b-PEO micelles and PLGA NPs form, is not supported by our observations. The size distribution of such a system would be distinctly bimodal with separate peaks associated with the PLGA NPs and the much smaller PS-b-PEO micelles. Although there is some suggestion of two size populations in PolyDots samples (Figure 3), most samples present a single peak at 31–41 nm, regardless of the PLGA:PS-b-PEO ratio. This finding does not provide strong support for the possibility of a separate population of PLGA NPs, as much larger sizes (>100 nm) indicative of PLGA NP formation are not observed. The observed smaller particles may represent general variation in the micelle size distribution. In Figure 2A, we see similar, but less pronounced, size distribution in control, unloaded PS-b-PEO micelles. The second possibility is that these smaller particles may represent PS-b-PEO micelles not loaded with PLGA. It is known that hydrophobic loading in the core, for example, with drug49 or NPs,27 can increase micelle size. Degradation tests in acidic media further suggest that PLGA NPs are not present in solution. PLGA NPs formed in conjunction with PS-b-PEO micelles would be expected to degrade similarly to PLGA NP controls and result in a size reduction with acid exposure (Figure 4 and Table 2). In stark contrast, PolyDot size increased with acid exposure, suggesting that PolyDots present a mixed micelle morphology, consisting of hydrophobic PLGA copolymers entrapped within or intercalating with the PS-b-PEO micelles.
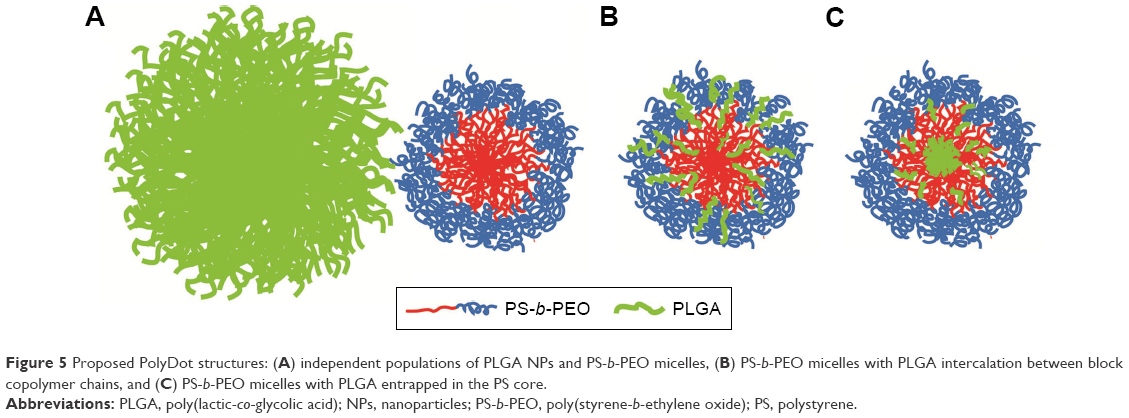 | Figure 5 Proposed PolyDot structures: (A) independent populations of PLGA NPs and PS-b-PEO micelles, (B) PS-b-PEO micelles with PLGA intercalation between block copolymer chains, and (C) PS-b-PEO micelles with PLGA entrapped in the PS core. |
Previous reports of mixed micelles have primarily focused on BCP/polymer systems, in which the polymer is a homopolymer of one of the blocks constituting the BCP or a small-chain surfactant. In these systems, the distribution of hydrophobic polymer encapsulants within micelles is loading dependent.50–52 Uniform distribution of polymer encapsulants throughout the micelle or via “intercalation” stretches the core blocks and is thus permitted only at relatively low encapsulant loadings. At higher loadings, the hydrophobic polymer encapsulants first form aggregates that are then stabilized by the adsorption of the BCP.50 These macroscopically separated structures can reach larger sizes than uniformly mixed micelles. Although PLGA copolymers have higher molecular weights than the surfactants used in previous studies and are chemically distinct from PS-b-PEO, their entrainment within PS-b-PEO micelles likely proceeds through similar mechanisms. At low PLGA:PS-b-PEO loadings, PLGA copolymers may intercalate within the micelle core giving rise to smaller structures with sizes controlled by PS-block stretching. This would explain the tightly controlled average diameters (35–45 nm) seen in PolyDot samples with different PLGA:PS-b-PEO loadings (Table 1). This configuration would allow degradation of some of the PLGA chains that are exposed to the surrounding aqueous environments and cause eventual destabilization of the PolyDots as seen in the acid exposure studies. In addition, some of the larger structures seen at high PLGA:PS-b-PEO loadings (Figure 2D and E) may be macroscopically separated structures in which a PLGA-aggregated core is stabilized by PS-b-PEO adsorption. Scattering data (ie, small-angle X-ray scattering or small-angle neutron scattering) would provide more detail on the final structure.
It should be noted that PLGA encapsulation in a micellar structure is fundamentally different from admicellar polymerization processes. In the admicellar approach, adsolubilized monomers are polymerized in situ, in the hydrophobic regions of surfactant micelles or bilayers.35 In contrast, in our approach, the hydrophobic core does not serve as a reaction center for polymerization, as the encapsulant is an already polymerized PLGA chain. Thus, the encapsulation of PLGA is a physical process as opposed to a chemical reaction.
Drug encapsulation efficiency
To further evaluate PolyDots as potential drug release vehicles, the ability to encapsulate a model drug, DEX, was evaluated as a function of PGLA:PS-b-PEO ratio. As controls, PS-b-PEO micelles containing no PGLA and PLGA NPs were employed. DEX encapsulation efficiency (DEXEE) was calculated as follows:53
|
|
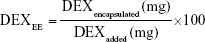
DEXEE in PS-b-PEO micelles was only ~7% (w/w) with significant variability between samples, indicating the poor reproducibility of these systems. DEXEE in PLGA NPs was ~30% (Figure 6). PolyDots prepared with varying PLGA:PS-b-PEO ratios also displayed DEXEE of ~30%, consistent with those of PLGA NPs. There were no statistically significant differences in DEXEE between the four PolyDot samples and PLGA NPs, but all samples were statistically different from PS-b-PEO micelles (p<0.05 for all). Thus, PolyDots display quantitatively different encapsulation behaviors to PS-b-PEO micelles despite having similar size (Table 1).
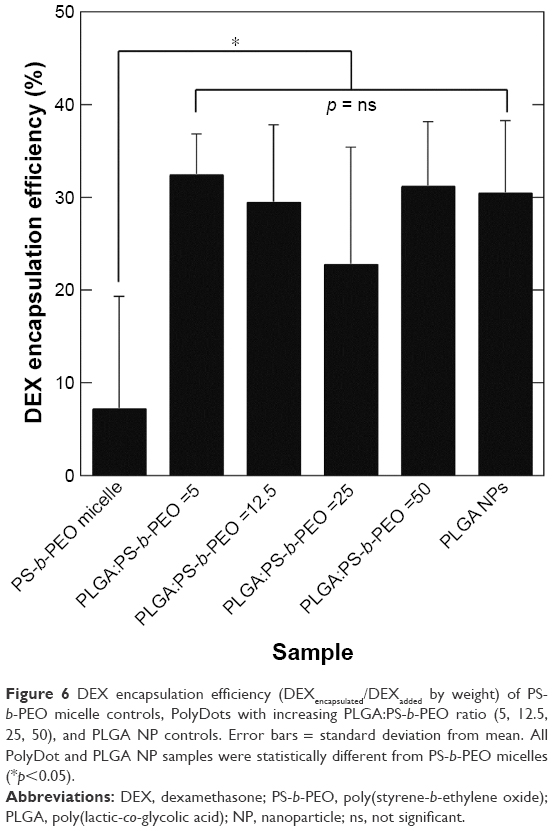 | Figure 6 DEX encapsulation efficiency (DEXencapsulated/DEXadded by weight) of PS-b-PEO micelle controls, PolyDots with increasing PLGA:PS-b-PEO ratio (5, 12.5, 25, 50), and PLGA NP controls. Error bars = standard deviation from mean. All PolyDot and PLGA NP samples were statistically different from PS-b-PEO micelles (*p<0.05). |
Drug loading is influenced by micelle core size, core hydrophobicity, polymer–drug compatibility, and the micelle preparation method.54 Higher DEXEE obtained in the PLGA-based systems (PolyDots and PLGA NPs) most likely arises from higher compatibility between DEX and PLGA, compared to PS blocks alone. Hildebrand solubility parameters, δD, δP, and δH representing contributions from dispersion, polar, and hydrogen bonding forces, respectively, have been widely used to assess compatibility between polymers and drugs.55 Solubility parameters (Table 3) were estimated using the group contribution method of Van Krevelen and Te Nijenhuis,70 and the overall solubility parameter δTOTAL calculated using the equation:
|
|
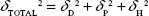
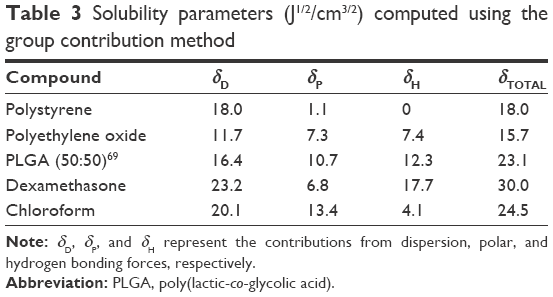 | Table 3 Solubility parameters (J1/2/cm3/2) computed using the group contribution method |
It has been shown that drug–polymer systems with closely matching δTOTAL and δH result in high drug loading.56 In our system, δTOTAL and δH for DEX are closer to those of PLGA than to those of PS (Table 3). Thus, hydrogen bonding forces likely play an important role in enabling higher DEXEE for these PLGA-based systems. Entrapment efficiencies may be further improved by optimizing solvent, emulsification, and solvent evaporation rates.57 Further, it is likely that PolyDots form first by the interaction of DEX with PLGA chains, which are in turn encapsulated into self-assembled PS-b-PEO micelles. DEX and PS-b-PEO have very different δTOTAL and δH compared to chloroform solvent, and it is therefore likely that some phase separation occurs during the evaporation process.54
Drug release
Release of model drug DEX was measured over a period of 38 days (Figure 7). However, incremental release was negligible for all systems after 22 days. Cumulative drug release (Figure 7A) was highest for PolyDot 5 samples (PLGA:PS-b-PEO =5), a factor of 1.2 higher than that of PLGA NPs and 5.9 times higher compared to PS-b-PEO micelles not containing PLGA. Fractional release curves (Figure 7B; Mt/M∞, where Mt is the mass released at time t and M∞ is the mass released at infinite time, that is, after additional release can no longer be detected) exhibit very similar behaviors for all samples.
 | Figure 7 (A) Cumulative drug release and (B) fractional drug release from PS-b-PEO micelles (○), PolyDots (PLGA:PS-b-PEO =5 (Δ), 12.5 (▽), 25 (◊), 50 (*)), and PLGA NPs (□). Inset: Mt/M∞ for days 0–7. |
With the exception of PS-b-PEO micelles, all solutions contain nearly the same total amount of drug encapsulated (ie, DEXEE is ~30% for all PolyDots and PLGA NPs). There was no statistically significant difference in the weight of drug released from all the PolyDots and the PLGA NPs from day 2 to day 38 of release. To investigate drug release mechanisms, including the role of carrier degradation versus diffusion, the data shown in Figure 7B were fit to the Peppas equation for the first 60% of the released drug (Mt/M∞ ≤0.6):58
|
|
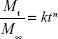
where k is the rate constant, correlated to diffusion rate, and n is the diffusional exponent, representing the mode of release.
The diffusional exponent, n, offers insight into the molecular mechanism governing the release phenomenon (Table 4). Control PS-b-PEO micelles and PLGA NPs are physicochemically distinct, with different mechanisms governing release behavior. PS-b-PEO micelles had an n equal to 1.15 ± 0.43, consistent with previous reports59,60 and indicating Super Case II transport (ie, for spherical geometries, n>0.85).61 Super Case II transport is primarily associated with release of drug from glassy polymers via chain relaxation. As water enters the glassy phase, it plasticizes the core, lowering Tg. When Tg reaches the system temperature, the polymer chains relax, increasing volume and promoting drug mobility.62 The Tg of PS blocks employed is ~80°C–90°C.63 PLGA NPs displayed a diffusional exponent of 0.73 ± 0.10, indicating anomalous diffusion (0.43<n<0.85), consistent with previous reports.64 PLGA NPs release drug by a combination of diffusional release and bulk degradation of the PLGA ester linkages via hydrolysis.14
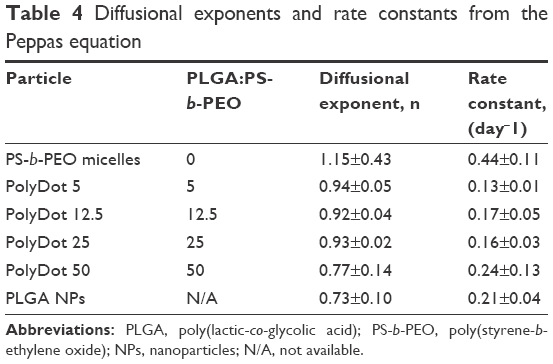 | Table 4 Diffusional exponents and rate constants from the Peppas equation |
PolyDots are composites of PS-b-PEO and PLGA. Hence, their release characteristics are expected to reflect combined attributes of both control systems. PolyDots displayed variation in n values depending on PLGA:PS-b-PEO weight ratio. PolyDots with lower ratios (ie, 5, 12.5, and 25) displayed diffusional exponents ranging from 0.92 to 0.94 – which are classified as Super Case II, similar to PS-b-PEO micelles not containing PLGA – whereas PolyDots at the highest PLGA:PS-b-PEO ratio of 50 (with the weight of PLGA same as PLGA NPs during synthesis) displayed n values of 0.77±0.14, similar to PLGA NPs and consistent with anomalous diffusion. The PLGA employed (50:50, molecular weight 50–70 kDa) is reported by the manufacturer to have a Tg of 40°C–50°C, much lower than that of PS. Thus, PLGA chains likely partially plasticize the PS core, reducing n from values observed for pure PS-b-PEO materials.
Rate constant k values support these observations. PS-b-PEO micelles displayed the highest k values, consistent with previous reports indicating rapid drug leakage from micelles resulting from poor drug–polymer interaction.25 PolyDots displayed lower k values relative to the PS-b-PEO micelles because of the stronger interaction between DEX and PLGA as indicated by their δ values (Table 3). Thus, the presence of PLGA within the PolyDots served to control release and prevent rapid leakage of DEX from the carriers. PolyDot 50 and PLGA NPs exhibited similar rate constants exceeding 0.2 day−n suggesting that the PLGA content in PolyDot 50 samples may be sufficient to fully plasticize PS cores at the temperatures investigated. Nonetheless, these differences do not result in statistically significant differences in release amounts, excepting PS-b-PEO micelles.
Lyophilization
For clinical translation, shelf life and stability are critical to any drug delivery system. To determine the impact of standard storage conditions on PolyDot performance, the ability to lyophilize PolyDots containing the model coumarin 6 drug was investigated (Figure 8). Freshly synthesized and lyophilized, reconstituted PolyDots displayed similar morphology with slightly reduced size post-lyophilization (Figure 8A and B). Fluorescence spectra were similar (Figure 8C), although there was a slight increase in fluorescence (~5%) from lyophilized PolyDots (Figure 8D). This may result from changes in the solvent environment, particularly water content. Thus, changes in morphology and encapsulant properties were modest in response to lyophilization.
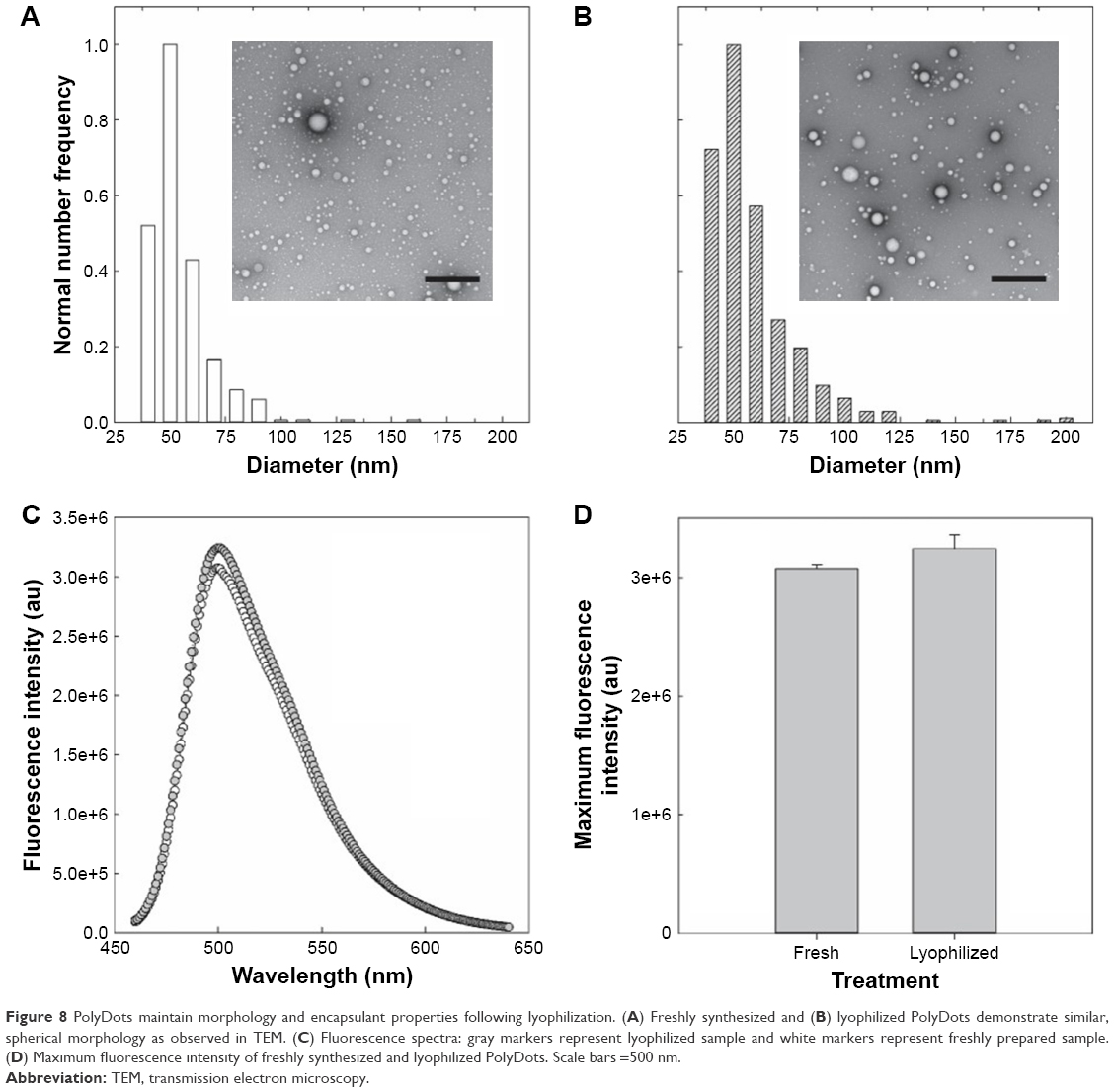 | Figure 8 PolyDots maintain morphology and encapsulant properties following lyophilization. (A) Freshly synthesized and (B) lyophilized PolyDots demonstrate similar, spherical morphology as observed in TEM. (C) Fluorescence spectra: gray markers represent lyophilized sample and white markers represent freshly prepared sample. (D) Maximum fluorescence intensity of freshly synthesized and lyophilized PolyDots. Scale bars =500 nm. |
Cellular uptake
To confirm capability of PolyDots for intracellular drug delivery, cellular uptake of lyophilized and reconstituted, coumarin 6 dye-loaded PolyDots was examined. Two model glioma cell lines were employed, as glioma is a potential target for hydrophobic drug delivery (eg, vorinostat65). Both LN229 and U87 glioma cells demonstrated the uptake of PolyDots via endocytotic mechanisms, as evidenced by punctate staining at short time points (Figure 9). PolyDots initially clustered at the cell surface (Figure 9A and E, arrowheads), and within 90 minutes, these clusters were internalized into large vesicles (Figure 9B and F, arrows). These vesicles remained present within the cells for several hours (Figure 9C and G, arrows), and then dwindled in number by 24 hours (Figure 9D and H, arrows). In addition, vesicle location is variable throughout the cytoplasm. Taken together, these data strongly support the idea that PolyDots enter and move throughout the cell using preexisting endocytic and trafficking pathways. We also observe an increase in diffuse green staining (Figure 9D, F, and G), potentially associated with coumarin 6 release, which could indicate acidic degradation of PolyDots within the lysosome. These data are supported by our observations in acidic degradation studies that most PolyDots are degraded after 9 hours of exposure to acidic media. Additionally, cytotoxicity was not observed over the course of observation, consistent with our observations that PolyDots at concentrations of up to ~150 μg/mL (unpublished) PS-b-PEO do not illicit significant cell death.
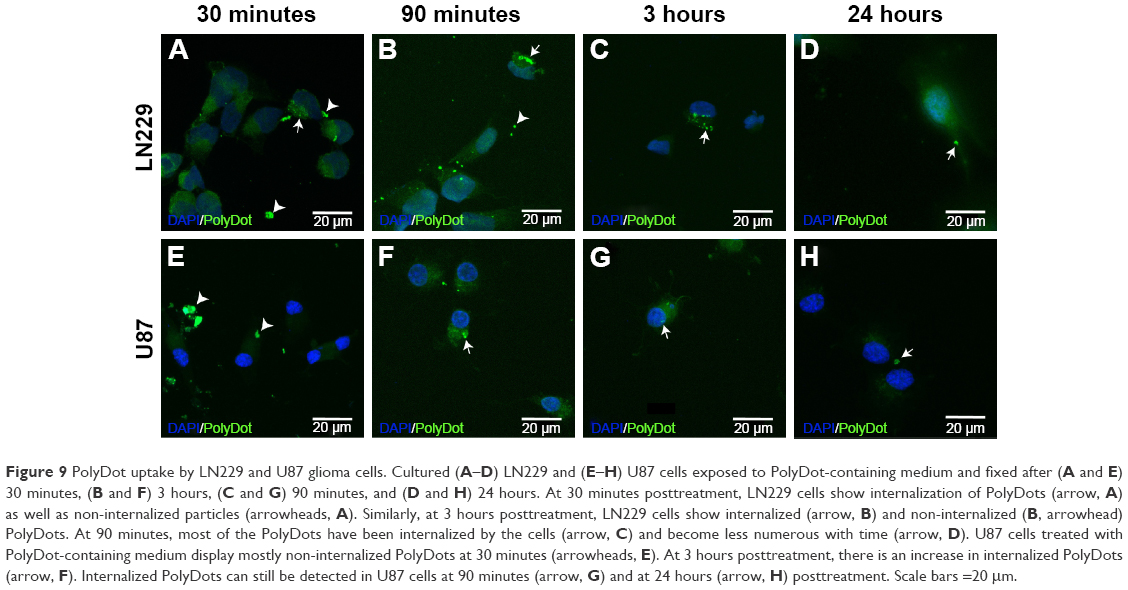 | Figure 9 PolyDot uptake by LN229 and U87 glioma cells. Cultured (A–D) LN229 and (E–H) U87 cells exposed to PolyDot-containing medium and fixed after (A and E) 30 minutes, (B and F) 3 hours, (C and G) 90 minutes, and (D and H) 24 hours. At 30 minutes posttreatment, LN229 cells show internalization of PolyDots (arrow, A) as well as non-internalized particles (arrowheads, A). Similarly, at 3 hours posttreatment, LN229 cells show internalized (arrow, B) and non-internalized (B, arrowhead) PolyDots. At 90 minutes, most of the PolyDots have been internalized by the cells (arrow, C) and become less numerous with time (arrow, D). U87 cells treated with PolyDot-containing medium display mostly non-internalized PolyDots at 30 minutes (arrowheads, E). At 3 hours posttreatment, there is an increase in internalized PolyDots (arrow, F). Internalized PolyDots can still be detected in U87 cells at 90 minutes (arrow, G) and at 24 hours (arrow, H) posttreatment. Scale bars =20 μm. |
Electrospray
Finally, we evaluated the ability to produce PolyDots using a commercial-scale process: micellar electrospray.37 Electrospray is a well-established, energy-efficient technique for the commercial-scale production of micron-sized particles for pharmaceutical applications.66 In electrospray, electro-hydrodynamic forces stretch the chloroform–water interface until capillary instabilities induce stream breakup into monodisperse emulsion droplets.67 Further scaleup can be achieved by multiplexing the coaxial electrospray system.68 Previously, we showed that BCPs and hydrophobic NPs can self-assemble within these emulsion droplets to form micellar nanocomposites.37
Here, electrospray synthesis of PolyDot 12.5 samples was demonstrated (Figure 10), resulting in a solution with high monodispersity (polydispersity =0.14). In addition, the potential bimodal distribution of two particles sizes seen in Figures 1–3 was not observed. Lower polydispersity observed in electrospray-generated samples compared to samples generated via conventional sonication approaches may be attributed to the finer size of emulsion droplets generated by electrospray. Additionally, electrospray may be advantageous to sonication approaches as ultrasonic cavitation produces local high-temperature regions, which may be detrimental for heat-sensitive pharmaceutics. Thus, electrospray offers an attractive method of scalable production of PolyDots, minimizing polydispersity and potentially reducing drug denaturation.
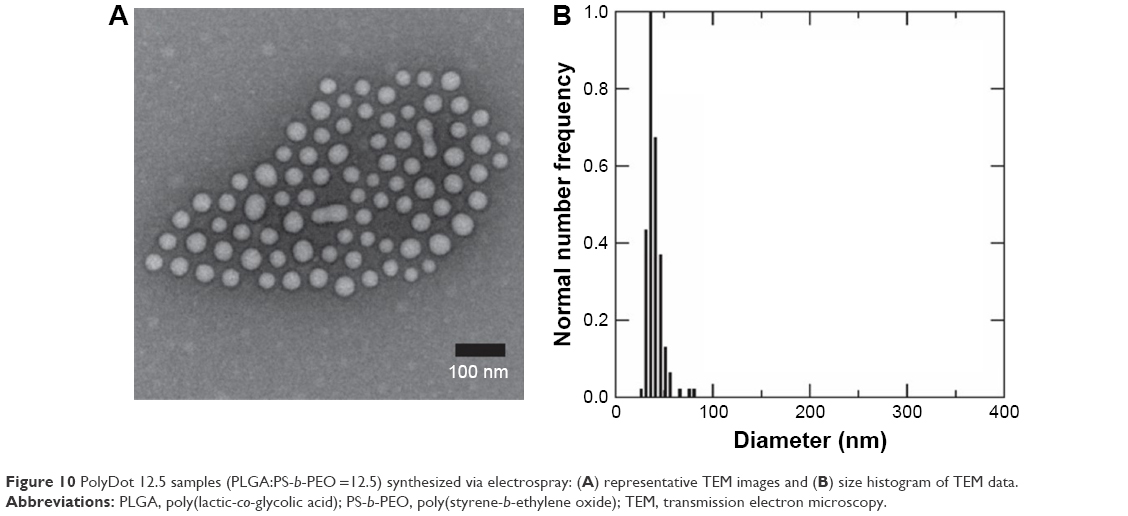 | Figure 10 PolyDot 12.5 samples (PLGA:PS-b-PEO =12.5) synthesized via electrospray: (A) representative TEM images and (B) size histogram of TEM data. |
Conclusion
Here, we report the synthesis of PolyDots, polymer drug release carriers that combine features of PS-b-PEO micelles and PLGA NPs. PolyDots were formed using the interfacial instability approach, with PLGA added during the micelle formation process. This approach permits the formation of PLGA-containing drug carriers that are much smaller and with narrower size distributions than those synthesized via emulsion routes. PolyDots exhibited hydrophobic drug encapsulation efficiencies comparable to much larger PLGA NPs, and much higher than PS-b-PEO micelles not containing PLGA. This, most likely, results from increased hydrogen bonding provided by PLGA chains. PolyDot drug release was dependent on PLGA:PS-b-PEO ratio, with lower loadings exhibiting Super Case II release, consistent with chain relaxation, and higher loadings exhibiting anomalous diffusion, consistent with a combination of PLGA degradation and Fickian diffusion. These data support a possible structure in which PLGA is either encapsulated in the PS core or interspersed with amphiphile chains. PolyDots are taken up by cells, most likely through endocytosis, and release drug cargoes in the cytoplasm. PolyDots can be lyophilized with minimal changes in size or encapsulant properties and can be produced through scalable nanomanufacturing processes, enabling potential clinical translation. Thus, controlled size distributions, high drug loading, and tunable release characteristics of PolyDots make them promising candidates for hydrophobic drug delivery.
Acknowledgments
This work was supported by the National Science Foundation (grant number CMMI-1344567, awarded to GMN, KDM, ADD, MSS, BEW, JOW), the National Institutes of Health (grant number NIH K24 CA160777, awarded to VKP), and the Pelotonia Cancer Research program (awarded to MAC, JX, CC, VKP, JJO, JOW). Any opinions, findings, and conclusions expressed in this material are those of the author(s) and do not necessarily reflect those of the Pelotonia Cancer Research Program.
Author contributions
GMN, KDM, and JOW performed, designed, and interpreted PolyDot synthesis, characterization, encapsulation, and release experiments. GN and JOW performed, designed, and interpreted degradation experiments. MC and JOW performed, designed, and interpreted lyophilization experiments. ADD, MSS, and BEW performed, designed, and interpreted electrospray experiments. JX, CC, VKP, and JJO performed, designed, and interpreted cellular trafficking experiments. All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work. JOW was the primary editor.
Disclosure
The authors report no conflicts of interest in this work.
References
Torchilin VP. Micellar nanocarriers: pharmaceutical perspectives. Pharm Res. 2007;24(1):1–16. | ||
Singla AK, Garg A, Aggarwal D. Paclitaxel and its formulations. Int J Pharm. 2002;235(1–2):179–192. | ||
Kedar U, Phutane P, Shidhaye S, Kadam V. Advances in polymeric micelles for drug delivery and tumor targeting. Nanomedicine. 2010;6(6):714–729. | ||
Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. | ||
Choi HS, Liu W, Misra P, et al. Renal clearance of quantum dots. Nat Biotechnol. 2007;25(10):1165–1170. | ||
Yoo JW, Chambers E, Mitragotri S. Factors that control the circulation time of nanoparticles in blood: challenges, solutions and future prospects. Curr Pharm Des. 2010;16(21):2298–2307. | ||
Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–151. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Sinha VR, Trehan A. Biodegradable microspheres for parenteral delivery. Crit Rev Ther Drug Carrier Syst. 2005;22(6):535–602. | ||
Lu JM, Wang X, Marin-Muller C, et al. Current advances in research and clinical applications of PLGA-based nanotechnology. Expert Rev Mol Diagn. 2009;9(4):325–341. | ||
Govender T, Stolnik S, Garnett MC, Illum L, Davis SS. PLGA nanoparticles prepared by nanoprecipitation: drug loading and release studies of a water soluble drug. J Control Release. 1999;57(2): 171–185. | ||
Mundargi RC, Babu VR, Rangaswamy V, Patel P, Aminabhavi TM. Nano/micro technologies for delivering macromolecular therapeutics using poly(D,L-lactide-co-glycolide) and its derivatives. J Control Release. 2008;125(3):193–209. | ||
Kwon HY, Lee JY, Choi SW, Jang YS, Kim JH. Preparation of PLGA nanoparticles containing estrogen by emulsification-diffusion method. Colloids Surf A Physicochem Eng Asp. 2001;182(1–3):123–130. | ||
Makadia HK, Siegel SJ. Poly Lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers (Basel). 2011;3(3):1377–1397. | ||
Jiang XY, Zhou CS, Tang KW. Preparation of PLA and PLGA nanoparticles by binary organic solvent diffusion method. J Cent South Univ Technol. 2003;10(3):202–206. | ||
Ahlin P, Kristl J, Kristl A, Vrecer F. Investigation of polymeric nanoparticles as carriers of enalaprilat for oral administration. Int J Pharm. 2002;239(1–2):113–120. | ||
Julienne MC, Alonso MJ, Amoza JLG, Benoit JP. Preparation of poly (D,L-lactide/glycolide) nanoparticles of controlled particle size distribution: application of experimental designs. Drug Dev Ind Pharm. 1992;18(10):1063–1077. | ||
Astete CE, Sabliov CM. Synthesis and characterization of PLGA nanoparticles. J Biomater Sci Polym Ed. 2006;17(3):247–289. | ||
Song CX, Labhasetwar V, Murphy H, et al. Formulation and characterization of biodegradable nanoparticles for intravascular local drug delivery. J Control Release. 1997;43(2–3):197–212. | ||
Nishiyama N, Kataoka K. Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmacol Ther. 2006;112(3):630–648. | ||
Oerlemans C, Bult W, Bos M, Storm G, Nijsen JFW, Hennink WE. Polymeric micelles in anticancer therapy: targeting, imaging and triggered release. Pharm Res. 2010;27(12):2569–2589. | ||
Zhang K, Tang X, Zhang J, et al. PEG-PLGA copolymers: their structure and structure-influenced drug delivery applications. J Control Release. 2014;183:77–86. | ||
Kim S, Shi Y, Kim JY, Park K, Cheng JX. Overcoming the barriers in micellar drug delivery: loading efficiency, in vivo stability, and micelle-cell interaction. Expert Opin Drug Deliv. 2010;7(1):49–62. | ||
Johnston AP, Such GK, Ng SL, Caruso F. Challenges facing colloidal delivery systems: from synthesis to the clinic. Curr Opin Colloid Interface Sci. 2011;16(3):171–181. | ||
Yoo HS, Park TG. Biodegradable polymeric micelles composed of doxorubicin conjugated PLGA-PEG block copolymer. J Control Release. 2001;70(1–2):63–70. | ||
Mahajan KD, Vieira GB, Ruan G, et al. A Magdot nanoconveyer assay detects and isolates molecular biomarkers. Chem Eng Prog. 2012;108:41–51. | ||
Ruan G, Vieira G, Henighan T, et al. Simultaneous magnetic manipulation and fluorescent tracking of multiple individual hybrid nanostructures. Nano Lett. 2010;10(6):2220–2224. | ||
Ruan G, Winter JO. Alternating-color quantum dot nanocomposites for particle tracking. Nano Lett. 2011;11(3):941–945. | ||
Mahajan KD, Ruan G, Dorcéna J, et al. Steering microtubule shuttle transport with dynamically controlled magnetic fields. Nanoscale. 2016;8:8641–8649. | ||
Nance E, Zhang C, Shih TY, Xu QG, Schuster BS, Hanes J. Brain-penetrating nanoparticles improve paclitaxel efficacy in malignant glioma following local administration. ACS Nano. 2014;8(10):10655–10664. | ||
Sun X, Rossin R, Turner JL, et al. An assessment of the effects of shell cross-linked nanoparticle size, core composition, and surface PEGylation on in vivo biodistribution. Biomacromolecules. 2005;6(5):2541–2554. | ||
Eugene M. Polyethyleneglycols and immunocamouflage of the cells tissues and organs for transplantation. Cell Mol Biol (Noisy-le-grand). 2004;50(3):209–215. | ||
Zhu J, Hayward RC. Spontaneous generation of amphiphilic block copolymer micelles with multiple morphologies through interfacial instabilities. J Am Chem Soc. 2008;130(23):7496–7502. | ||
Sens P, Marques CM, Joanny JF. Mixed micelles in a bidisperse solution of diblock copolymers. Macromolecules. 1996;29(14):4880–4890. | ||
Magaraphan R, Srinarang V. Admicellar polymerization of polystyrene on natural rubber particles. J Elastom Plast. 2009;41(5):457–477. | ||
Geng Y, Discher DE. Hydrolytic degradation of poly(ethylene oxide)-block-polycaprolactone worm micelles. J Am Chem Soc. 2005;127(37):12780–12781. | ||
Duong AD, Ruan G, Mahajan K, Winter JO, Wyslouzil BE. Scalable, semicontinuous production of micelles encapsulating nanoparticles via electrospray. Langmuir. 2014;30(14):3939–3948. | ||
Hurtrez G, Dumas P, Riess G. Polystyrene-poly(ethylene oxide) diblock copolymers micelles in water. Polym Bull (Berlin). 1998;40(2–3):203–210. | ||
Murakami H, Kobayashi M, Takeuchi H, Kawashima Y. Preparation of poly(DL-lactide-co-glycolide) nanoparticles by modified spontaneous emulsification solvent diffusion method. Int J Pharm. 1999;187(2):143–152. | ||
Wang J, Li W, Zhu J. Encapsulation of inorganic nanoparticles into block copolymer micellar aggregates: strategies and precise localization of nanoparticles. Polymer. 2014;55(5):1079–1096. | ||
Sarkar B, Alexandridis P. Block copolymer-nanoparticle composites: structure, functional properties, and processing. Prog Polym Sci. 2015;40:33–62. | ||
Swietach P, Vaughan-Jones RD, Harris AL, Hulikova A. The chemistry, physiology and pathology of pH in cancer. Philos Trans R Soc Lond B Biol Sci. 2014;369(1638):20130099. | ||
Gasmi H, Danede F, Siepmann J, Siepmann F. Does PLGA microparticle swelling control drug release? New insight based on single particle swelling studies. J Control Release. 2015;213:120–127. | ||
He WN, Xu JT, Du BY, Fan ZQ, Sun FL. Effect of pH on the micellar morphology of semicrystalline PCL-b-PEO block copolymers in aqueous solution. Macromol Chem Phys. 2012;213(9):952–964. | ||
Agrawal CM, Huang D, Schmitz JP, Athanasiou KA. Elevated temperature degradation of a 50:50 copolymer of PLA-PGA. Tissue Eng. 1997;3(4):345–352. | ||
Klose D, Siepmann F, Elkharraz K, Krenzlin S, Siepmann J. How porosity and size affect the drug release mechanisms from PLIGA-based microparticles. Int J Pharm. 2006;314(2):198–206. | ||
De S, Robinson DH. Particle size and temperature effect on the physical stability of PLGA nanospheres and microspheres containing Bodipy. AAPS PharmSciTech. 2004;5(4):18–24. | ||
Bhargava P, Tu Y, Zheng JX, Xiong H, Quirk RP, Cheng SZ. Temperature-induced reversible morphological changes of polystyrene-block-poly(ethylene oxide) micelles in solution. J Am Chem Soc. 2007;129(5):1113–1121. | ||
Sharma PK, Reilly MJ, Jones DN, Robinson PM, Bhatia SR. The effect of pharmaceuticals on the nanoscale structure of PEO-PPO-PEO micelles. Colloids Surf B Biointerfaces. 2008;61(1):53–60. | ||
Zhang L, Eisenberg A. Multiple morphologies and characteristics of “crew-cut” micelle-like aggregates of polystyrene-b-poly(acrylic acid) diblock copolymers in aqueous solutions. J Am Chem Soc. 1996;118(13):3168–3181. | ||
Hashimoto T, Tanaka H, Hasegawa H. Ordered structure in mixtures of a block copolymer and homopolymers. 2. Effects of molecular weights of homopolymers. Macromolecules. 1990;23(20):4378–4386. | ||
Winey KI, Thomas EL, Fetters LJ. Swelling a lamellar diblock copolymer with homopolymer: influences of homopolymer concentration and molecular weight. Macromolecules. 1991;24(23):6182–6188. | ||
Judefeind A, de Villiers MM. Drug loading into and in vitro release from nanosized drug delivery systems. In: de Villiers MM, Aramwit P, Kwon GS, editors. Nanotechnology in Drug Delivery, Vol. 10. Biotechnology: Pharmaceutical Aspects. New York: Springer; 2009:129–162. | ||
Liu J, Lee H, Allen C. Formulation of drugs in block copolymer micelles: drug loading and release. Curr Pharm Des. 2006;12(36):4685–4701. | ||
Liu J, Xiao Y, Allen C. Polymer-drug compatibility: a guide to the development of delivery systems for the anticancer agent, ellipticine. J Pharm Sci. 2004;93(1):132–143. | ||
Zhang J, Li S, Li X, Li X, Zhu K. Morphology modulation of polymeric assemblies by guest drug molecules: TEM study and compatibility evaluation. Polymer. 2009;50(7):1778–1789. | ||
Sharma H, Sharma SN, Kumar U, et al. Formation of water-soluble and biocompatible TOPO-capped CdSe quantum dots with efficient photoluminescence. J Mater Sci Mater Med. 2009;20(Suppl 1):S123–S130. | ||
Ritger PL, Peppas NA. A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J Control Release. 1987;5(1):23–36. | ||
Vaishya RD, Gokulgandhi M, Patel S, Minocha M, Mitra AK. Novel dexamethasone-loaded nanomicelles for the intermediate and posterior segment uveitis. AAPS PharmSciTech. 2014;15(5):1238–1251. | ||
Morán MC, Pais AA, Ramalho A, Miguel MG, Lindman B. Mixed protein carriers for modulating DNA release. Langmuir. 2009;25(17):10263–10270. | ||
Peppas NA, Brannon-Peppas L. Water diffusion and sorption in amorphous macromolecular systems and foods. J Food Eng. 1994;22(1–4):189–210. | ||
Siepmann J, Peppas NA. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv Drug Deliv Rev. 2001;48(2–3):139–157. | ||
Rieger J. The glass transition temperature of polystyrene. J Therm Anal Calorim. 1996;46(3–4):965–972. | ||
Schliecker G, Schmidt C, Fuchs S, Ehinger A, Sandow E, Kissel T. In vitro and in vivo correlation of buserelin release from biodegradable implants using statistical moment analysis. J Control Release. 2004;94(1):25–37. | ||
Xu J, Sampath D, Lang FF, et al. Vorinostat modulates cell cycle regulatory proteins in glioma cells and human glioma slice cultures. J Neurooncol. 2011;105(2):241–251. | ||
Wu Y, Chalmers JJ, Wyslouzil BE. The use of electrohydrodynamic spraying to disperse hydrophobic compounds in aqueous media. Aerosol Sci Technol. 2009;43(9):902–910. | ||
Loscertales IG, Barrero A, Guerrero I, Cortijo R, Marquez M, Ganan-Calvo AM. Micro/nano encapsutation via electrified coaxial liquid jets. Science. 2002;295(5560):1695–1698. | ||
Deng WW, Waits CM, Morgan B, Gomez A. Compact multiplexing of monodisperse electrosprays. J Aerosol Sci. 2009;40(10):907–918. | ||
Schenderlein S, Lück M, Müller BW. Partial solubility parameters of poly(D,L-lactide-co-glycolide). Int J Pharm. 2004;286(1–2):19–26. | ||
Van Krevelen DW, Te Nijenhuis K. Cohesive properties and solubility. In: Properties of Polymers: Their Correlation with Chemical Structure; Their Numerical Estimation and Prediction from Additive Group Contributions. Elsevier. 2009;189–227. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.