")
Back to Journals » Cancer Management and Research » Volume 10
Methyl jasmonate enhances the radiation sensitivity of esophageal carcinoma cells by inhibiting the 11-ketoprostaglandin reductase activity of AKR1C3
Authors Li XY, Hong X, Gao XS , Gu XB, Xiong W, Zhao J, Yu HL, Cui M, Xie M, Bai Y, Sun SQ
Received 28 February 2018
Accepted for publication 14 May 2018
Published 31 August 2018 Volume 2018:10 Pages 3149—3158
DOI https://doi.org/10.2147/CMAR.S166942
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kenan Onel
Xiaoying Li,1,* Xin Hong,2,* Xianshu Gao,1 Xiaobin Gu,1 Wei Xiong,3 Jing Zhao,4 Hongliang Yu,5 Ming Cui,1 Mu Xie,1 Yun Bai,1 Shaoqian Sun6
1Department of Radiation Oncology, Peking University First Hospital, Peking University, Beijing, China; 2Department of Urology, Peking University International Hospital, Peking University, Beijing, China; 3Department of Oncology, Tangshan People’s Hospital, Hebei, China; 4Department of Oncology, Beijing Shijitan Hospital, Capital Medical University, Beijing, China; 5Department of Radiation Oncology, Jiangsu Cancer Hospital and Jiangsu Institute of Cancer Research, The Affiliated Cancer Hospital of Nanjing Medical University, Nanjing, China; 6College of Biochemical Engineering, Beijing Union University, Beijing, China
*These authors contributed equally to this work
Purpose: In our previous study, we found that AKR1C3 was a radioresistance gene in KY170R cells. Downregulating the expression of AKR1C3 could enhance the radiosensitivity of esophageal carcinoma cells. In this study, we investigated whether methyl jasmonate (MeJ), an inhibitor of Aldo-keto reductase family1 member C3 (AKR1C3), could overcome radiation resistance in AKR1C3 highly expressed cells.
Patients and methods: We used clone formation assays to detect radiosensitivity effects. Flow cytometry assays were used to detect reactive oxygen species (ROS) accumulation and apoptosis. Enzyme linked immunosorbent assays (ELISAs) were used to detect the concentrations of prostaglandin F2 (PGF2) and prostaglandin D2 (PGD2) in the cells after incubation with MeJ. Western blotting was used to detect AKR1C3 and peroxisome proliferator-activated receptor gamma (PPARγ) expression.
Results: We found that AKR1C3 was highly expressed in radioresistant esophageal carcinoma cells. MeJ inhibited the expression of AKR1C3 and enhanced the radiation sensitivity of esophageal carcinoma cells expressing high levels of AKR1C3 (P<0.05). MeJ could inhibit the 11-ketoprostaglandin reductase activity of AKR1C3 in a dose-dependent manner in KY170R cells. Incubation of KY170R cells with 200 µmol/L of MeJ for 24 h reduced the expression of PGF2 by roughly 30% (P<0.05). The PPAR pathway inhibitor GW9662 prevented the radiation sensitivity enhancement imparted by MeJ. After adding GW9662, there were no significant differences between the radiation sensitivities of MeJ-treated and -untreated KY170R cells (P>0.05).The radiation sensitivity effect of MeJ also depended upon the generation of ROS in KY170R cells; 48 h after irradiation, ROS levels in the MeJ group was twofold higher than in the untreated KY170R cells (P<0.05). The ROS scavenger, N-acetyl cysteine, could reverse the radiosensitivity effects of MeJ (P>0.05).
Conclusion: Our results indicate that MeJ can increase the radiation sensitivity of AKR1C3-overexpressing KY170R cells by inhibiting the 11-ketoprostaglandin reductase activity of AKR1C3 and increasing cellular ROS levels.
Keywords: radiosensitivity, esophageal carcinoma, methyl jasmonate, AKR1C3
Introduction
Esophageal cancer is one of the most common cancers in China. The 5-year survival rate of esophageal cancer patients after radical surgery is about 20–30%.1,2 Radical radiochemotherapy is a critical therapy for patients with unresectable cancer. After radiation, the 5-year survival rate can reach up to 27%,3 which is comparable to surgery. Disease persistence or recurrent cancer has typically been the most common reason for radiation treatment failure. Cancer recurs in about 37% of patients with unresectable esophageal cancer after radiochemotherapy.3 This suggests that radioresistant cells may exist within the tumor tissue. Radioresistance can limit radiotherapy efficacy in esophageal carcinoma patients. The addition of a radiation sensitizer can increase the radiation sensitivity of resistant cells and may increase its clinical potency and efficacy in esophageal cancer patients.4
The aldo-keto reductase (AKR) superfamily is comprised of NADPH-linked oxidoreductases.5 Of these, AKR1C3 catalyzes androgen, estrogen, prostaglandin (PG), and xenobiotic metabolism.6,7 In our previous study, we found that AKR1C3 was a radioresistance gene. AKR1C3 was highly expressed in KY170R cells (an esophageal cancer cell line), which were more radioresistant than cells that only express AKR1C3 at low levels. Downregulating the expression of AKR1C3 could enhance the radiosensitivity of esophageal carcinoma cells.8 The mechanism by which this occurs involved downregulation of the expression of AKR1C3, which can increase reactive oxygen species (ROS) levels in radioresistant cells. Another study produced similar results, showing that overexpression of AKR1C3 significantly enhanced the radioresistance of human prostate cancer cells.9 The mechanism was related to the accumulation of PGF2 and involved inhibition of the expression of PPARγ.
There are some commercially available inhibitors of AKR1C3. Whether these inhibitors can induce radiotherapy sensitization remains to be investigated. Based on our previous studies,8,9 we wanted to find an inhibitor target related to the prostaglandin metabolism of AKR1C3, which would also be capable of increasing cellular ROS levels. Methyl jasmonate (MeJ) was an effective small molecule inhibitor of the PGD2 11-keto reductase activity of the AKR1C3 enzyme.10 MeJ also increased ROS levels in many types of human cancer cells.10–12 Therefore, in this study we wanted to investigate whether MeJ could increase the radiation sensitivity of KY170R cells.
Patients and methods
Cell culture and irradiation
KY170R is an esophageal squamous cancer cell line. It was provided as a gift by Professor Joe Y Chang (Department of Radiology, MD Anderson Cancer Center, USA). The use of the gifted cells was approved by Department of Radiation Oncology of the First Affliated Hospital School of Medicine, Beijing University. Cells were passaged for less than 1 month before experimentation. Cells were cultured in high glucose RPMI 1640 (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (HyClone) with 50 units/mL penicillin and 50 mg/mL streptomycin. Cells were maintained in a humidified incubator with 5% CO2 at 37°C and split every 3–4 days. Tumor cell irradiation was carried out with a 6 MV X-ray linear accelerator.
Construction of AKR1C3-silencing stable cells
Construction of AKR1C3-low sh-KY170R cells was performed following the method described in our previous study.8 We used polymerase chain reaction (PCR) to construct the upstream primer (hU6 promoter) of the AKR1C3-targeting shRNA: 5′-TGGATCCAAGGTCGGGCAGGAAGAG. The downstream primer of hU6-scramble was 5′-AGGATCCAAAAACAACAAGAT GAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTT GTTGGGTGTTTCGTCCTTTCCAC-3′. The downstream primer of shRNA was: 5′-AGGATCCAAAAACCTAGACAGAAATCTCCACTACTCG AGTAGTGGAGATTTCTGTCTGGCCGGTGTTTCGTCCTTTCCAC-3′. The cassettes obtained were cloned into pSD31 for constitutive expression of shRNA. Lentiviral vector production was performed according to standard protocol (Thermo Fisher Scientific). Experiments for lentivector stable transduction were carried out as follows: 1×106 of replicon cells in a T25 flask were seeded and transduced on Day 2 in the presence of 8 mg/mL of polybrene. Selection was performed after Day 3 by using 800 ng/mL of puromycin until the parental cells in parallel experiments completely died.
Proliferation assay
KY170R cells and sh-KY170R cells were plated in 96-well plates at a density of 3×104 cells/well. Cells were treated with different concentrations of MeJ (1, 10, 100, 1000, and 2500 µmol/L) in 100 μL of medium. After incubating the cells for different lengths of times (20, 44, and 68 h), we added 10 μL Alamar Blue to each well. We then incubated the cells with Alamar Blue at 37°C for 4 h and used fluorescence at an excitation/emission of 535/595 nm to test cell viability.
KY170R cells and sh-KY170R cells were plated in six-well plates for 24 h. Then the cells were incubated with different concentrations of MeJ (0, 50, 100, 200, and 400 μmol/L) for 7 days. The surviving cells were stained with crystal violet and then counted. Cell clusters with more than 50 cells were counted as one clone. Each group had five copies, and the experiment was repeated three times.
Western blots
Cultured cells were washed once with PBS buffer and then lysed in lysis buffer for 5 min. The sample was placed on ice for 30 min, and then centrifuged at 12,000 ×g for 30 min. We used the Bio-Rad DC protein assay to determine the protein concentration of each sample. Equal amounts of protein were separated by sodium salt-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes. We washed the PVDF membranes in Tween and Tris-Buffered Saline (TTBS) (20 mM Tris, 0.5 M NaCl, 0.05% Tween-20) for 10 min, and then blocked the membranes in 10% skim milk for 1 h. The primary antibodies were diluted with TTBS as follows: anti-AKR1C3 (1:4000, A6229–200U; Sigma-Aldrich Co.), anti-PPARγ (1:1000; Sigma-Aldrich Co.), β-actin (1:10,000), and anti-human GAPDH (1:3000). We used the Gel Doc analysis system (Bio-Rad) to scan the gray level of each band. Each Western blot was repeated at least twice.
Colony formation assays
KY170R cells and sh-KY170R cells were plated in six-well plates for 24 h. Cells were treated in culture media with or without MeJ. After incubation for 24 h, the cells were exposed to different dosages of 6 MV X-rays (0, 2, 4, 6, and 8 Gy). One hour after irradiation, cells were seeded into six-well plates at different cell densities. In the 0 Gy group, we seeded 500 cells, in the 2 Gy group 800 cells, in the 4 Gy group 1000 cells, in the 6 Gy group 2500 cells; and in the 8 Gy group 4000 cells. The cells were then incubated in culture media with or without MeJ (200 μmol/L for 7–10 days, and the surviving cells were stained with crystal violet and then counted. Cell clusters with more than 50 cells were counted as one clone. Plating efficiency (PE) = numbers of colonies formed (control group)/numbers of cells plated × 100%. Surviving fractions (SF) = number of colonies formed/ [number of cells plated (irradiated group) × plating efficiency (control group)]. Each group had triplicate samples, and the experiment was repeated three times.
Apoptotic assays
KY170R cells were plated in six-well plates at a density of 2 × 105 cells/mL. Cells were treated in culture media with or without 200 μmol/L of MeJ. After incubation for 24 h, the cells were exposed to 4 Gy X-ray radiation. Then we collected the cells before and 48 h after radiation. Cells were collected, washed twice with cold PBS, resuspended with 100 mL of binding buffer at a density of 2×105 cells/mL density, and incubated with annexin V-FITC for 10 min. Then cells were washed with binding buffer and resuspended with 400 mL of binding buffer containing 10 mL of propidium iodide (PI; 20 mg/mL) and incubated on ice for 15 min. Apoptosis was analyzed by a flow cytometer (BD-LSR flow cytometer; BD Biosciences, Franklin Lakes, NJ, USA) at an excitation wavelength of 488 nm and an emission wavelength of 585 nm. Each group had triplicate samples, and the experiment was repeated three times.
ROS production assays
Flow cytometry measurements of dichloro-dihydro-fluorescein diacetate (DFCH-DA) were used to measure the cellular ROS levels of cells. KY170R cells were plated in six-well plates at a density of 2×105 cells/mL. Cells were treated in culture media with or without 200 μmol/L of MeJ. After incubation for 24 h, the cells were exposed to 4 Gy X-ray radiation. Cells were exposed to radiation for different lengths of times (24, 48, and 72 h). After radiation, cells were treated with 3 μmol DCFH-DA (D6883/50 mg; Sigma-Aldrich Co.) for 15 min and then washed with ice-cold PBS three times before testing for ROS by flow cytometry (excitation wavelength 488 nm, emission 585 nm). Each group had triplicate samples, and the experiment was repeated three times.
Prostaglandin assessment
Prostaglandin concentrations were assessed by an ELISA kit (Cayman Chemical, Ann Arbor, MI, USA). Cells were seeded in 96-well plates at a density of 3×104 cells/mL. When cells grew to 60% density, we changed the medium to fresh media containing MeJ (0, 200, 500, 1000, and 1500 µmol/L) and incubated for 48 h. We then collected the supernatants from each well and determined prostaglandin concentrations by ELISA, following manufacturer directions. Each group had triplicate samples, and the experiment was repeated three times.
Statistical analysis
We used the Statistical Package for Social Sciences (SPSS, version 13.0; IBM Corp., Armonk, NY, USA) to analyze all data. Student’s t-tests were used to determine the statistical difference between means of two groups and data. P-values less than 0.05 were considered significant.
Results
Methyl jasmonate suppressed the proliferation of KY170R, sh-KY170R, and scr-KY170R cells independent of the expression of AKR1C3
In our previous study, we found that the expression of AKR1C3 was high in KY170R esophageal cancer cells.8 shRNAs delivered by lentivector pSD31 were used to downregulate AKR1C3 expression. We confirmed that AKR1C3 expression is lower in sh-KY170R cells (AKR1C3 stable transfectants) (Figure 1A). Alamar Blue assay results demonstrated that MeJ can inhibit the proliferation of sh-KY170R, KY170R, and scr-KY170R cells in a dose-dependent manner. As the dose of MeJ was increased from 1 to 2500 µmol/L, cell viability decreased gradually, and there no were no differences in the inhibitory rates of MeJ between the three cells lines (Figure 1B, P>0.05). We incubated the three cell lines with various concentrations of MeJ for 7 days. Both the cell index and colony formation assays showed that there were no significant differences in the inhibitory rates of MeJ on KY170R, sh-KY170R, and scr-KY170R cells (Figure 1C and D, P>0.05). The cytotoxic effects of MeJ on cells did not depend upon the expression of AKR1C3. Incubation with 200 µmol/L MeJ for 7 days had little effect on cell growth (Figure 1C and D).
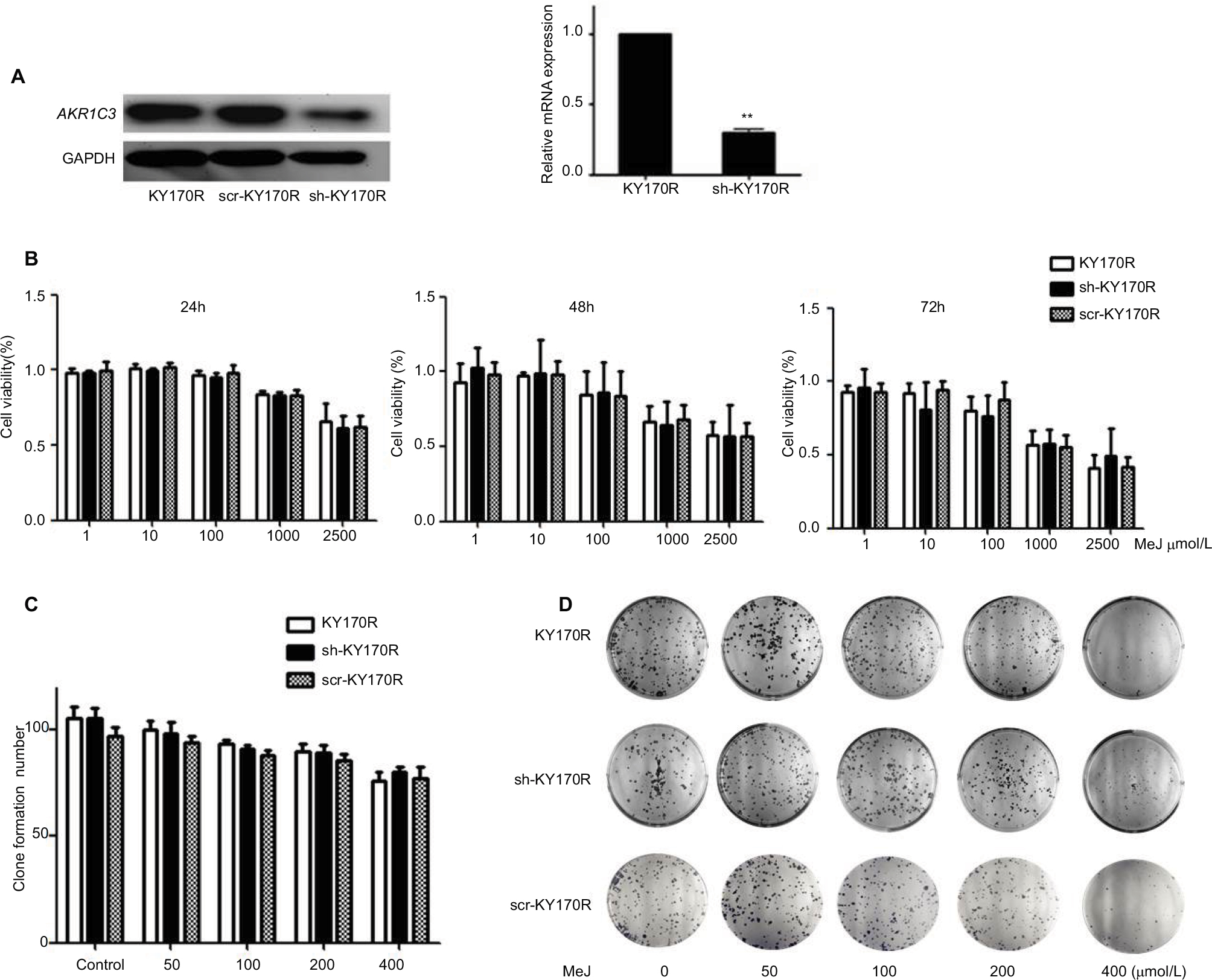 | Figure 1 Methyl jasmonate (MeJ) suppressed the proliferation of KY170R, sh-KY170R, and scr-KY170R cells independent of the expression of AKR1C3. Notes: (A) The construction of stable low expression of AKR1C3 in KY170R cells (sh-KY170R). Western blot (left) and real-time RT-PCR (right) showed AKR1C3 expression was lower in sh-KY170R cells. (B) The proliferation of KY170R, sh-KY170R, and scr-KY170R cells was analyzed by Alamar Blue assay after incubation with different concentrations of MeJ for 24, 48, or 72 h. There were no significant differences between the three cells (P>0.05). (C) Incubation of KY170R, sh-KY170R, and scr-KY170R cells with indicated concentrations of MeJ for 24 h; the inhibition rates of the three cell lines showed no differences (P>0.05). (D) The colony formation of KY170R, sh-KY170R, and scr-KY170R cells were counted after cells were treated with the indicated concentrations of MeJ for 7 days. A concentration of 200 μmol/L of MeJ had little effect on the growths of the three cells. Abbreviations: AKR1C3, aldo-keto reductase family 1 member 3; MeJ, methyl jasmonate; RT-PCR, reverse-transcription PCR. |
Effects of methyl jasmonate on the radiation sensitivity of KY170R and sh-KY170R cells
Clone formation assays showed that 200 µmol/L of MeJ had little effect on the growth of cells. Therefore, we incubated KY170R and sh-KY170R cells with 200 µmol/L of MeJ for 24 h, and then irradiated the cells with increasing doses of radiation (0, 2, 4, 6, and 8 Gy). In KY170R cells, expressing high levels of AKR1C3, combining MeJ with radiation suppressed colony formation (P<0.05, Figure 2A and C). However, in sh-KY170R cells, where AKR1C3 expression was low, 200 µmol/L of MeJ treatment did not alter radiation sensitivity (P>0.05, Figure 2B and C). These results indicated that the radiosensitivity effects of MeJ were dependent upon the expression of AKR1C3.
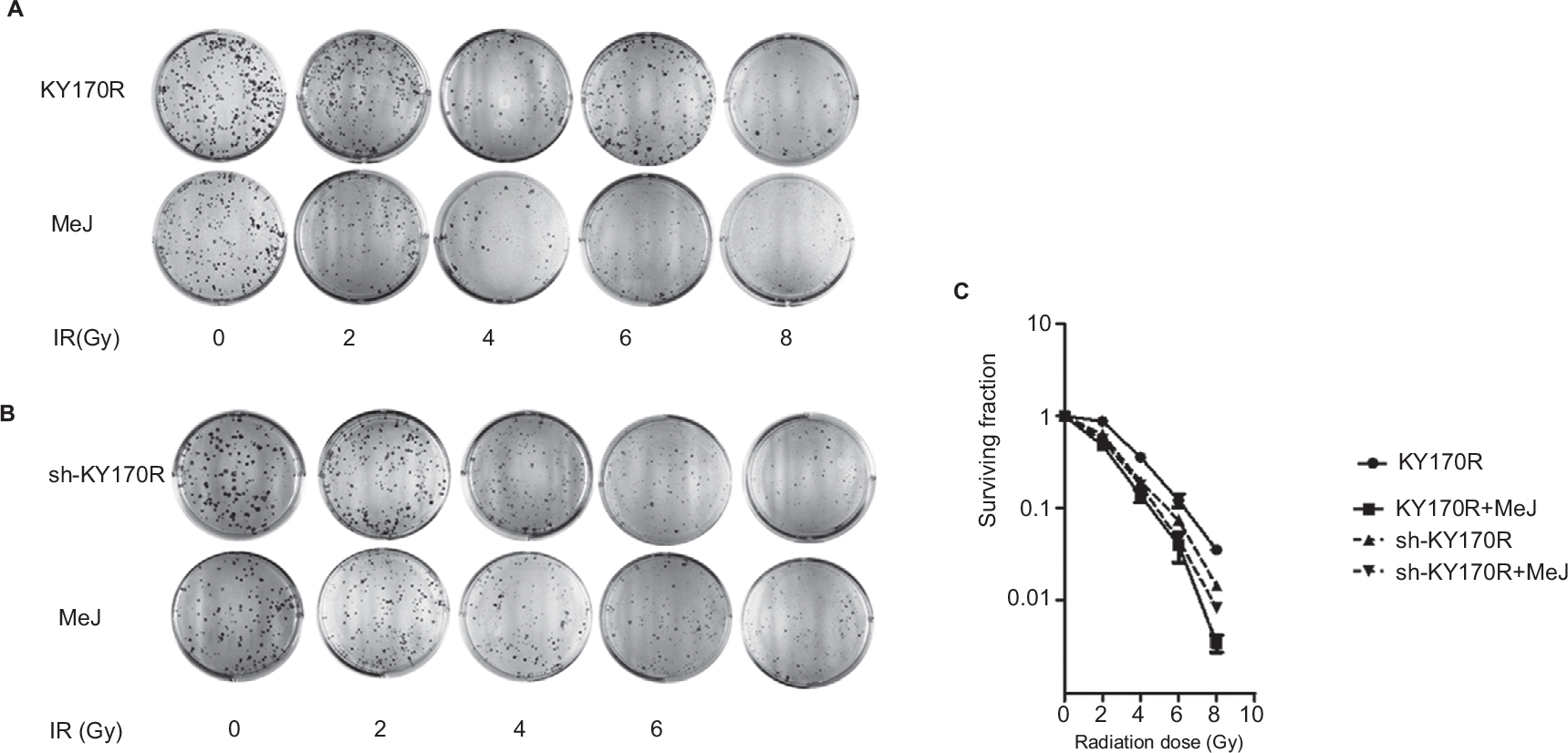 | Figure 2 Radiosensitivity effect of MeJ depended on the expression of AKR1C3. Notes: (A) KY170R cells (highly expressing AKR1C3) incubated with 200 µmol/L MeJ for 24 h before radiation were more sensitive to radiation than their untreated counterparts. MeJ could enhance the radiation sensitivity of KY170R cells that highly expressed AKR1C3. (B) sh-KY170R cells (not or lowly expressing AKR1C3) incubated with 200 µmol/L MeJ for 24 h were equally resistant to radiation as their untreated counterparts. (C) The survival curves of sh-KY170R and KY170R cells incubated with or without MeJ. Abbreviations: AKR1C3, aldo-keto reductase family 1 member 3; IR, irradiation; MeJ, methyl jasmonate. |
Methyl jasmonate can inhibit the AKR1C3 enzyme expression and the 11-ketoprostaglandin reductase activity of AKR1C3
We incubated KY170R cells with different concentrations (0, 100, and 200 µmol/L) of MeJ for 24 h. Western blot assays showed MeJ could reduce AKR1C3 protein expression in KY170R cells in a dose-dependent manner. A concentration of 200 µmol/L of MeJ could reduce the protein expression of AKR1C3 (Figure 3A). AKR1C3 is a multifunctional enzyme, which possesses 11-ketoprostaglandin reductase activity. It can convert PGD2 to PGF2. In our previous study,9 we found overexpression of AKR1C3 in DU145 cells increased the amount of PGF2. In the current study, we incubated different concentrations of MeJ with KY170R cells for 24 h. Then, we used an ELISA kit to measure the PGF2 and PGD2 content of the cells. With increasing MeJ concentrations, PGD2 levels increased, while PGF2 levels decreased (Figure 3B). MeJ could inhibit the 11-ketoprostaglandin reductase activity of AKR1C3 in a dose-dependent manner in KY170R cells. Incubation of KY170R cells with 200 µmol/L MeJ for 24 h had a significant effect (P<0.05), reducing the expression of PGF2 by roughly 30%.
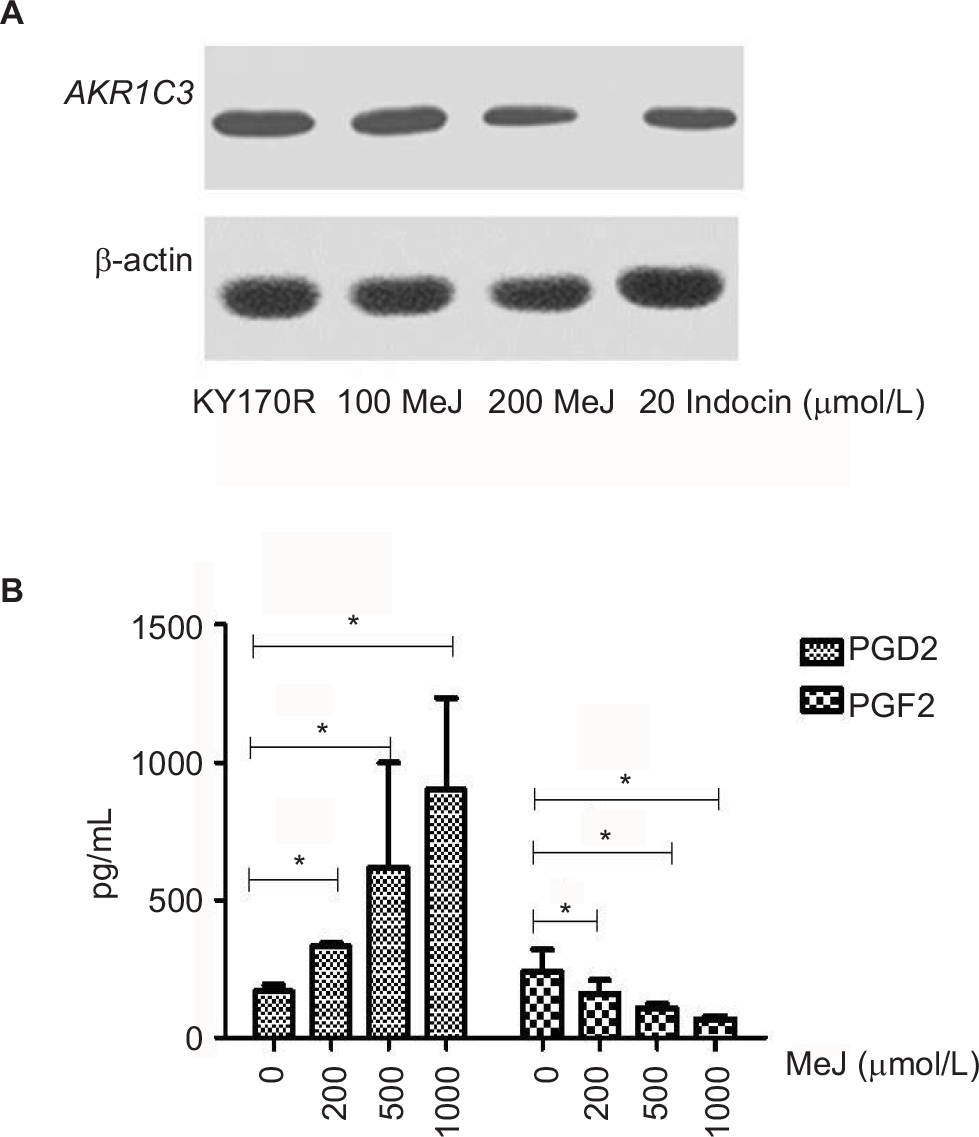 | Figure 3 Methyl jasmonate (MeJ) could inhibit AKR1C3 enzyme expression and the 11-ketoprostaglandin reductase activity of AKR1C3. Notes: (A) Western blots showed MeJ could reduce AKR1C3 protein expression in KY170R cells in a dose-dependent manner (indocin was a positive drug). (B) ELISA results showed that MeJ could increase the levels of PGD2, while decreasing the levels of PGF2. MeJ could inhibit the 11-ketoprostaglandin reductase activity of AKR1C3 in a dose-dependent manner; *P<0.05. Abbreviations: AKR1C3, aldo-keto reductase family 1 member 3; PG, prostaglandin; ELISA, enzyme-linked immunosorbent assay; MeJ, methyl jasmonate. |
Radiation sensitivity effects of methyl jasmonate depend upon activation of the PPARγ pathway
MeJ could elicit the accumulation of PGD2 in KY170R cells by inhibiting the 11-ketoprostaglandin reductase activity of AKR1C3. However, PGD2 is unstable and can divert to PGJ2, and then activate the PPARγ pathway.13 Therefore, we investigated whether the radiation sensitivity of MeJ depended upon PPAR pathway activation. We incubated KY170R cells with different densities (0,100, and 200 µmol/L) of MeJ for 24 h. We found that MeJ treatment increased PPARγ protein expression in KY170R cells. A dose of 200 µmol/L MeJ could double PPARγ protein levels (Figure 4A). We incubated KY170R cells with both 200 µmol/L MeJ and 1 μM GW9662 (a PPARγ antagonist) for 24 h. Then, we irradiated the cells with 4 Gy X-ray radiation. Immediately after radiation, we seeded the cells at 600 cells/well and incubated for 7–10 days. Clone formation results showed that GW9662 decreased the radiation sensitivity effects of MeJ. After adding GW9662, there were no significant differences between the radiation sensitivities of MeJ and KY170R cells (P>0.05, Figure 4B and C). This indicated that the radiation sensitivity effects of MeJ depended upon the activation of the PPARγ pathway.
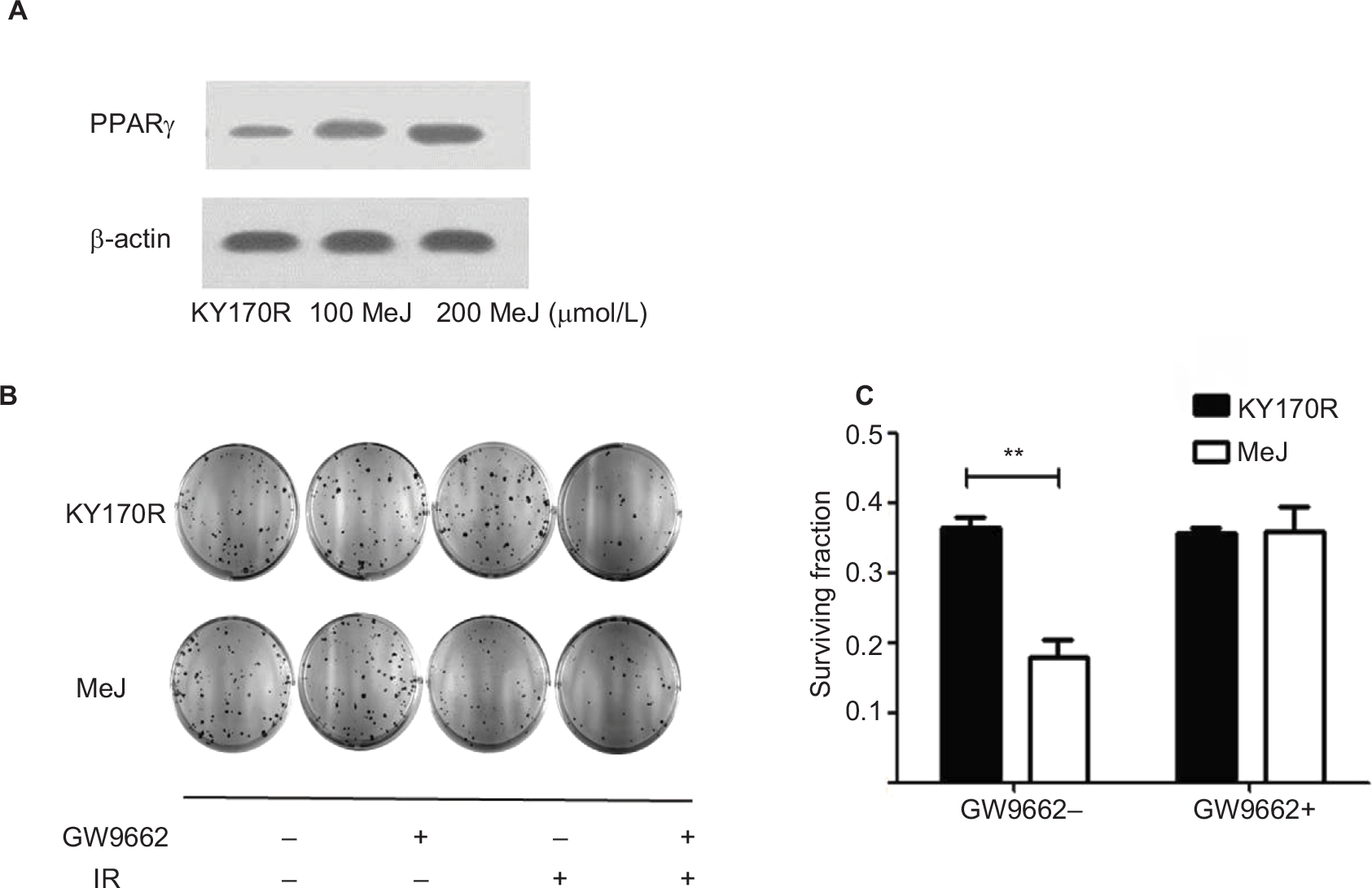 | Figure 4 Radiation sensitivity effects of MeJ depended on activation of the PPARγ pathway. Notes: (A) KY170R cells incubated with different densities (0,100, and 200 µmol/L) of MeJ for 24 h. Expression levels of PPARγ in KY170R cells were detected by Western blot analysis. MeJ could activate the PPARγ pathyway. (B) Clone formation assays showed GW9662 can weaken the radiation sensitivity effects of MeJ on KY170R cells. The cloning numbers were significantly higher after cells were incubated with both MeJ and GW9662. (C) Surviving fraction results showed that cells incubated with GW9662 were no longer sensitive to the effects of MeJ (P>0.05); **P<0.01. Abbreviations: PPARγ, peroxisome proliferator-activated receptor gamma; IR, irradiation; MeJ, methyl jasmonate. |
Methyl jasmonate cannot increase the apoptosis rate of KY170R cells
In bladder cancer cells, MeJ could sensitize gambogic acid-induced apoptosis.14 MeJ could also induce apoptosis and pro-apoptotic autophagy in non-small cell lung cancer cells.15 Another study found the apoptotic effects of MeJ depended on P53 expression in cells.16 Here, we used flow cytometry to assess the apoptotic rates of cells. Apoptotic ratio of cells increased when MeJ density increased (from 0 to 5000 µmol/L; Figure 5A). Radiation slightly increased the apoptotic rate of KY170R cells (from 4.37% to 15.16%; Figure 5A and B). A concentration of 200 µmol/L of MeJ only slightly increased the apoptotic ratio of KY170R cells (5.63% vs 4.37%, P>0.05; Figure 5A). However, there were no significant differences between the control group and 200 µmol/L MeJ group 48 h after 4 Gy radiation (15.89% vs 15.16%, P>0.05; Figure 5B). A combination of 200 µmol/L of MeJ and radiation did not increase the apoptotic cell percentage compared to radiation alone (P>0.05; Figure 5C).
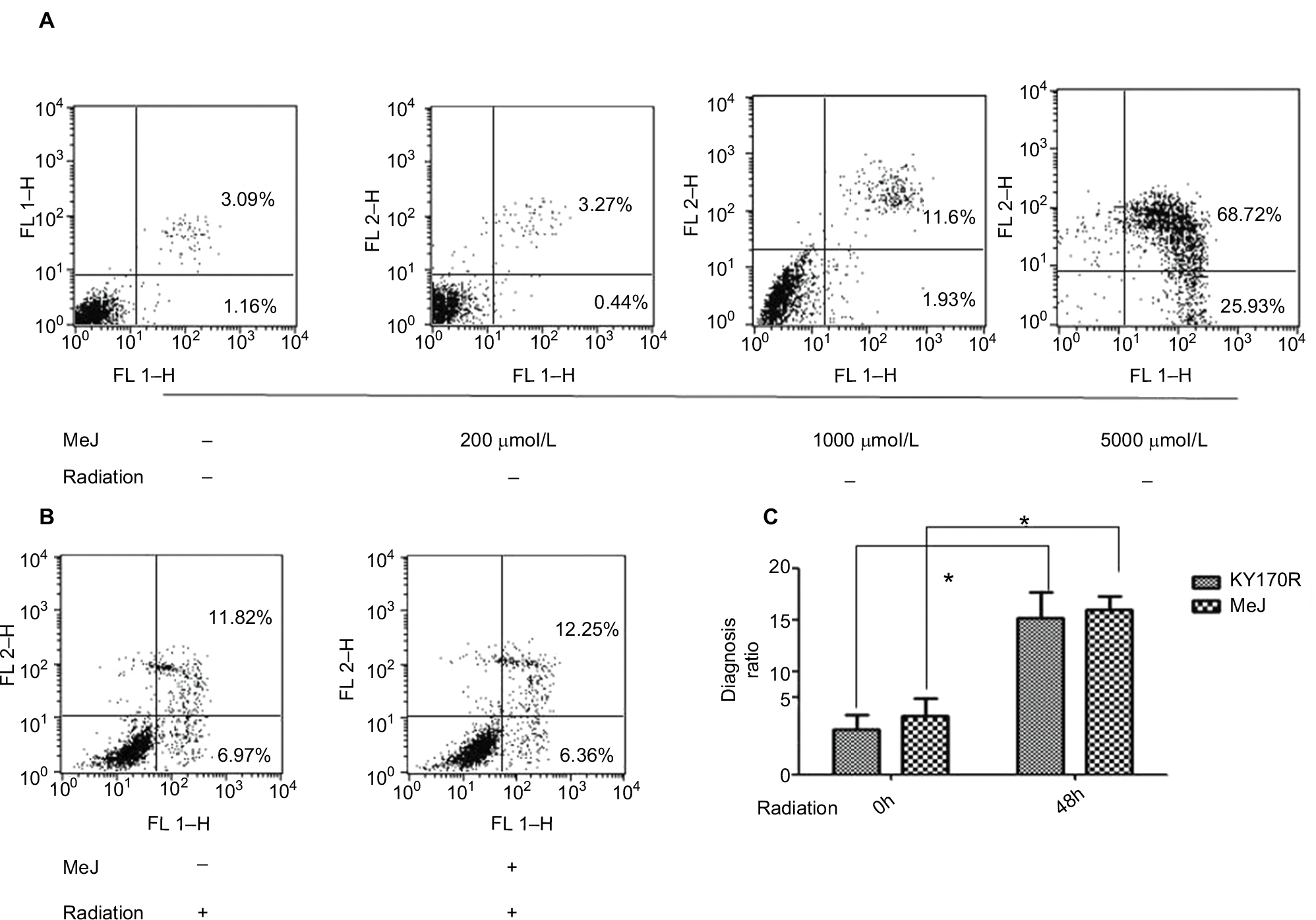 | Figure 5 The distribution of apoptotic KY170R cells before and after radiation. Notes: (A) The cell apoptosis analysis of KY170R cells treated before radiation (n=3). Apoptotic ratio of cells increased when MeJ density increased (from 0 to 5000 µmol/L). Apoptotic rates between 200 µmol/L and control group were not significantly different (P>0.05). (B) The cell apoptosis analysis of KY170R cells treated 48 h after radiation (n=3). Apoptotic rates between drug group and control group were not significantly different (P>0.05). (C) Radiation of 4 Gy could increase the apoptotic rate of cells, while cells pretreated with 200 µmol/L MeJ did not exhibit an increase in the apoptotic rate (P>0.05); *P<0.05. Abbreviations: MeJ, methyl jasmonate; IR, irradiation. |
The radiation sensitivity effects of methyl jasmonate depend upon the generation of ROS in KY170R cells
ROS can increase the radiation sensitivity of cells. Therefore, we set out to explore ROS changes in KY170R cells after incubation with MeJ. After 24 h of incubation with MeJ, we irradiated the cells with 4 Gy X-ray. We measured the ROS levels of KY170R cells before and at 24, 48, and 72 h after radiation. Cells treated with 200 µmol/L of MeJ had increased ROS levels before radiation (P<0.05, Figure 6A).
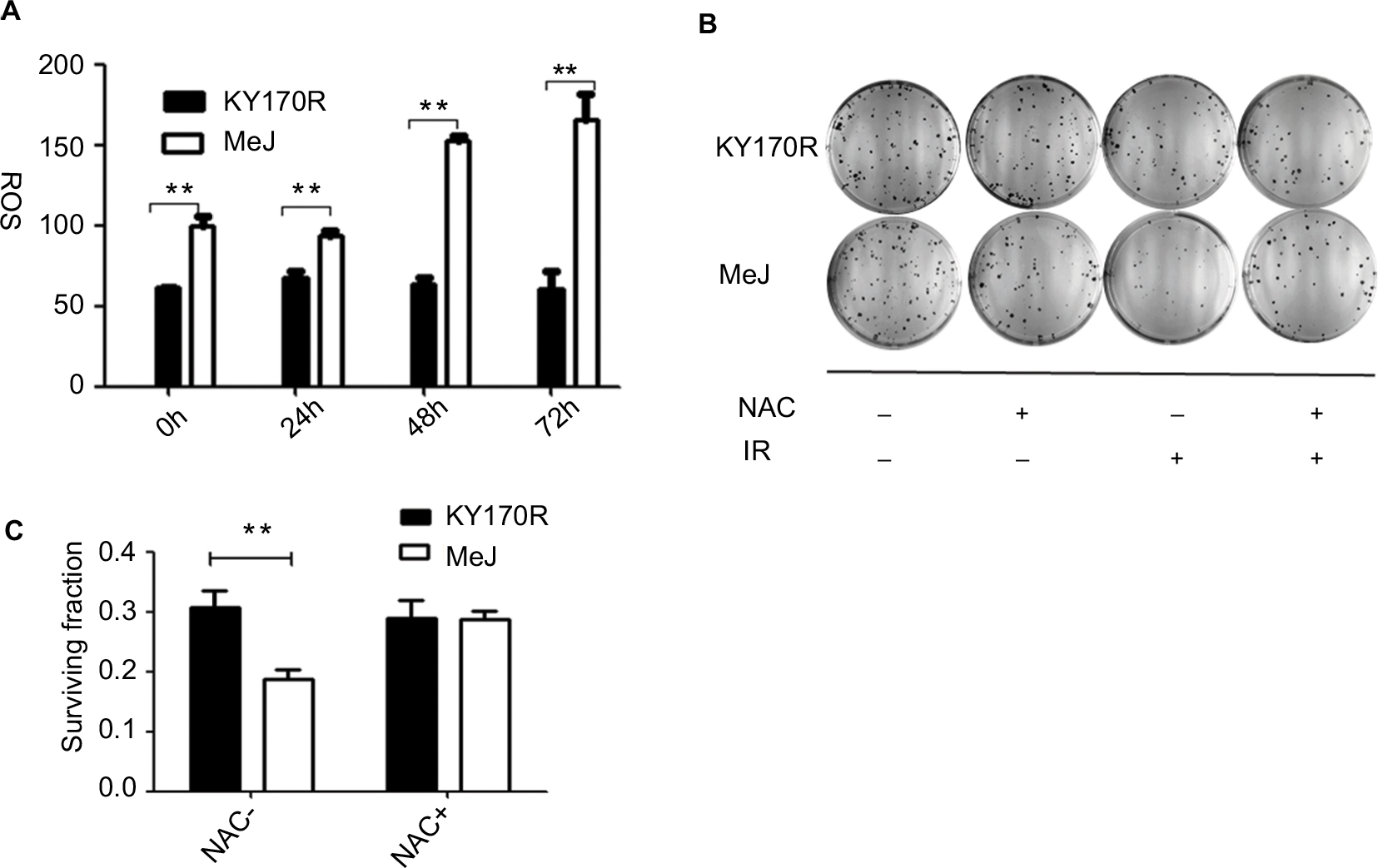 | Figure 6 Radiation sensitivity effects of methyl jasmonate depended on the generation of ROS in KY170R cells. Notes: (A) MeJ could increase the ROS levels in KY170R cells both before and after radiation (P>0.05). (B, C) Clone formation assays of KY170R cells. The ROS scavenger NAC could reverse the radiosensitivity effects of MeJ (P>0.05); **P<0.001. Abbreviations: ROS, reactive oxygen species; NAC, N-acetyl cysteine; IR; MeJ, methyl jasmonate. |
Pre-incubation with MeJ (prior to radiation) resulted in a significant increase in the ROS levels of KY170R cells after 4 Gy radiation (P<0.05, Figure 6A). Forty-eight hours after irradiation, ROS levels in the MeJ group were twofold higher than in untreated KY170R cells (Figure 6A). N-Acetyl cysteine (NAC) is an ROS scavenger. To verify that MeJ increased radiation sensitivity in KY170R cells through ROS pathways, we also investigated whether NAC could reverse the radiosensitivity effects of MeJ. Clone formation assays indicated that the radiosensitivity effects of MeJ were negated with the addition of NAC (5 mmol/L NAC, P>0.05; Figure 6B and C).
Discussion
AKRs are a superfamily of NAD(P)H-linked oxidoreductases that are generally cytosolic monomeric (37 kDa) proteins.17 The AKR superfamily is overexpressed in various cancers occurring in the central nervous system,18 kidney,19 pancreas, lung, and gastrointestinal tract.20,21 AKR1C3 is a multifunctional enzyme that can catalyze androgen, estrogen, prostaglandin, and xenobiotic metabolism.7,22,23 A key activity of AKR1C3 is the metabolism of prostaglandin D2 (PGD2) to prostaglandin F2 (PGF2).23 PGF2 is a radioresistant substrate and can increase the radioresistance of cells.9 The relationship of AKR1C3 and tumor radiotherapy had been studied in recent years. Overexpression of the AKR1C3 gene has been implicated in the radioresistance of several tumor cell types. Downregulated expression of AKR1C3 (accomplished with RNAi) could increase the radiosensitivity of cancer cells,8,9 the mechanism involved in decreasing the intracellular ROS in radioresistant cells.8 AKR1C3 overexpression could cause the accumulation of PGF2 (radioresistant substrate) and inhibition of the expression of PPARγ compared with AKR1C3 low expression cells.9
Overexpression of AKR1C3 is also related to drug resistance in tumor cells. In cisplatin, cis-diamminedichloroplatinum (II) (CDDP)-resistant human cancer cell lines, AKR1C3 expression is high. Inhibitors of AKR1C3 can enhance sensitivity to CDDP and 5-fluorouracil.24 In prostate cancer cells, overexpression of AKR1C3 confers resistance to abiraterone treatment. An AKR1C3 inhibitor, indomethacin, can overcome resistance.25
MeJ belongs to the jasmonate family.26 It is produced by plants as a defensive mechanism. It is a small molecule inhibitor of AKR1C3. In recent years, MeJ has been shown to induce apoptosis in cancer cells.27–29 It can inhibit the migration, invasion, and angiogenesis of cancer cells.30 It exerts anti-cancer effects and is a promising and novel anti-cancer drug. However, the therapeutic dose of MeJ is relatively high, which is one potential drawback for its clinical use. Using it in combination with other therapies may be beneficial, allowing for the effective dose to be substantially reduced. Some studies have demonstrated that MeJ has synergistic effects when combined with other anti-tumor drugs. MeJ demonstrated cooperative activity with several other anti-cancer drugs, such as adriamycin, taxol, carmustine, and cisplatin.31 MeJ could increase the cytotoxic effects of X-ray radiation and cisplatin therapies in cervical cancer cell lines.32 MeJ could increase the radiosensitivity of a human prostate adenocarcinoma cell line (PC-3) by inhibiting the expression of the anti-apoptotic protein Bcl-2.33 In our study, we found that sub-cytotoxic MeJ could enhance the radiosensitivity of KY170R cells, and the effects were dependent upon the expression of AKR1C3.
In this study, we found that MeJ could increase the radiosensitivity of KY170R cells, which highly express AKR1C3, while it did not affect the sensitivity of sh-KY170R cells which expressed AKR1C3 at low levels. These results showed that the radiation sensitivity effects of MeJ depended upon the expression of AKR1C3. MeJ can inhibit the 11-ketoprostaglandin reductase activity of AKR1C3 and decrease PGF2.10 In our study, we showed that MeJ decreased the PGF2 content of KY170R cells while increasing PGD2. PGD2 is a relatively unstable lipid, and it can be non-enzymatically converted to PGJ2.13 Inhibition of AKR1C3 activity can skew PGD2 metabolism toward 15d-PGJ2, which can activate the PPARγ pathway.23,34,35 In our study, MeJ increased PPARγ protein expression in KY170R cells.
PPARγ is a ligand-activated transcription factor, which is involved in cell differentiation, proliferation, survival, apoptosis, and motility.36,37 PPARγ pathway activation was found to have a synergistic effect with chemotherapy and radiation therapy. In breast cancer cells, increasing the expression of PPARγ could enhance the cytotoxic effect of cisplatin.38 PPARγ activation could also increase the effects of radiation-induced heart injury.39 The role of PPAR agonists as anti-cancer agents had been well characterized in the treatment of colon, gastric, breast, and lung cancers.40–44 In human squamous cell carcinoma (SCC) cells with high AKR1C3 gene expression, activation of PPARγ could decrease SCC proliferation. The presence of 1 μM GW9662 partially restored the proliferation rate of SCC cells.45 In our study, the PPARγ pathway inhibitor, GW9662, decreased the radiosensitivity effects of MeJ. MeJ exerted its radiation sensitivity effect via activation of the PPARγ pathway.
ROS is critical in irradiation-induced cell death, and it can cause cell death via apoptosis, autophagy, and various other pathways.46 In addition to direct ROS, indirect ROS can also cause damage to the mitochondria, leading to prolonged oxidative stress that can further damage cellular DNA.47 Therefore, an increase in ROS can lead to an enhancement in radiation sensitivity. Low ROS levels in tumor cells following radiation are indicative of radioresistance.48 MeJ not only inhibited the 11-ketoprostaglandin reductase activity of AKR1C3 but also increased cellular ROS levels.10 In our study, MeJ significantly increased ROS generation in KY170R cells, an effect which lasted for up to 72 h after radiation. NAC, an ROS scavenger, reversed the radiosensitivity effects of MeJ.
AKR1C3 was found to be a radioresistant gene in our previous studies.8,9 There are no published studies that demonstrate MeJ can increase cell radiation sensitivity by inhibiting the AKR1C3 enzyme. Our study, for the first time, showed that MeJ can increase the radiosensitivity of esophageal carcinoma cells highly expressing AKR1C3 by inhibiting 11-ketoprostaglandin reductase activity. As the current study was conducted in vitro, future studies should be performed to determine the radiosensitization effect of MeJ in vivo to further evaluate the efficiency of this type of combined therapy.
Conclusion
We have identified and characterized a new small molecule, MeJ, which targets AKR1C3 and inhibits11-ketoprostaglandin reductase activity. MeJ affected two important pathways: ROS generation and PPARγ activation. Treatment of KY170R cells with a minimally cytotoxic concentration of MeJ dramatically enhanced clonogenic cell death in response to radiation. ROS generation and PPARγ pathway activation were parts of the mechanism underlying MeJ radiation enhancement. MeJ represents a promising new radiation enhancer.
Disclosure
The authors report no conflicts of interest in this work.
References
Kelsen DP, Winter KA, Gunderson LL, et al. Long-term results of RTOG trial 8911 (USA Intergroup 113): a random assignment trial comparison of chemotherapy followed by surgery compared with surgery alone for esophageal cancer. J Clin Oncol. 2007;25(24):3719–3725. | ||
Pottgen C, Stuschke M. Radiotherapy versus surgery within multimodality protocols for esophageal cancer--a meta-analysis of the randomized trials. Cancer Treat Rev. 2012;38(6):599–604. | ||
Cooper JS, Guo MD, Herskovic A, et al. Chemoradiotherapy of locally advanced esophageal cancer: long-term follow-up of a prospective randomized trial (RTOG 85-01). Radiation Therapy Oncology Group. JAMA. 1999;281(17):1623–1627. | ||
Good JS, Harrington KJ. The hallmarks of cancer and the radiation oncologist: updating the 5Rs of radiobiology. Clin Oncol (R Coll Radiol). 2013;25(10):569–577. | ||
Jez JM, Bennett MJ, Schlegel BP, Lewis M, Penning TM. Comparative anatomy of the aldo-keto reductase superfamily. Biochem J. 1997;326 (Pt 3):625–636. | ||
Labrie F, Luu-The V, Lin SX, et al. Intracrinology: role of the family of 17 beta-hydroxysteroid dehydrogenases in human physiology and disease. J Mol Endocrinol. 2000;25(1):1–16. | ||
Lin HK, Jez JM, Schlegel BP, Peehl DM, Pachter JA, Penning TM. Expression and characterization of recombinant type 2 3 alpha-hydroxysteroid dehydrogenase (HSD) from human prostate: demonstration of bifunctional 3 alpha/17 beta-HSD activity and cellular distribution. Mol Endocrinol. 1997;11(13):1971–1984. | ||
Xiong W, Zhao J, Yu H, et al. Elevated expression of AKR1C3 increases resistance of cancer cells to ionizing radiation via modulation of oxidative stress. PLoS One. 2014;9(11):e111911. | ||
Sun SQ, Gu X, Gao XS, et al. Overexpression of AKR1C3 significantly enhances human prostate cancer cells resistance to radiation. Oncotarget. 2016;7(30):48050–48058. | ||
Davies NJ, Hayden RE, Simpson PJ, et al. AKR1C isoforms represent a novel cellular target for jasmonates alongside their mitochondrial-mediated effects. Cancer Res. 2009;69(11):4769–4775. | ||
Kim JH, Lee SY, Oh SY, et al. Methyl jasmonate induces apoptosis through induction of Bax/Bcl-XS and activation of caspase-3 via ROS production in A549 cells. Oncol Rep. 2004;12(6):1233–1238. | ||
Oh SY, Kim JH, Park MJ, et al. Induction of heat shock protein 72 in C6 glioma cells by methyl jasmonate through ROS-dependent heat shock factor 1 activation. Int J Mol Med. 2005;16(5):833–839. | ||
Desmond JC, Mountford JC, Drayson MT, et al. The aldo-keto reductase AKR1C3 is a novel suppressor of cell differentiation that provides a plausible target for the non-cyclooxygenase-dependent antineoplastic actions of nonsteroidal anti-inflammatory drugs. Cancer Res. 2003;63(2):505–512. | ||
Peng Z, Zhang Y. Methyl jasmonate induces the apoptosis of human colorectal cancer cells via downregulation of EZH2 expression by microRNA101. Mol Med Rep. 2017;15(2):957–962. | ||
Zhang M, Su L, Xiao Z, Liu X, Liu X. Methyl jasmonate induces apoptosis and pro-apoptotic autophagy via the ROS pathway in human non-small cell lung cancer. Am J Cancer Res. 2016;6(2):187–199. | ||
Fingrut O, Reischer D, Rotem R, et al. Jasmonates induce nonapoptotic death in high-resistance mutant p53-expressing B-lymphoma cells. Br J Pharmacol. 2005;146(6):800–808. | ||
Jez JM, Flynn TG, Penning TM. A new nomenclature for the aldo-keto reductase superfamily. Biochem Pharmacol. 1997;54(6):639–647. | ||
Park AL, Lin HK, Yang Q, et al. Differential expression of type 2 3alpha/type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3) in tumors of the central nervous system. Int J Clin Exp Pathol. 2010;3(8):743–754. | ||
Azzarello JT, Lin HK, Gherezghiher A, et al. Expression of AKR1C3 in renal cell carcinoma, papillary urothelial carcinoma, and Wilms’ tumor. Int J Clin Exp Pathol. 2009;3(2):147–155. | ||
Chang TS, Lin HK, Rogers KA, et al. Expression of aldo-keto reductase family 1 member C3 (AKR1C3) in neuroendocrine tumors & adenocarcinomas of pancreas, gastrointestinal tract, and lung. Int J Clin Exp Pathol. 2013;6(11):2419–2429. | ||
Miller VL, Lin HK, Murugan P, et al. Aldo-keto reductase family 1 member C3 (AKR1C3) is expressed in adenocarcinoma and squamous cell carcinoma but not small cell carcinoma. Int J Clin Exp Pathol. 2012;5(4):278–289. | ||
Penning TM, Burczynski ME, Jez JM, et al. Human 3alpha-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J. 2000;351(Pt 1):67–77. | ||
Matsuura K, Shiraishi H, Hara A, et al. Identification of a principal mRNA species for human 3alpha-hydroxysteroid dehydrogenase isoform (AKR1C3) that exhibits high prostaglandin D2 11-ketoreductase activity. J Biochem. 1998;124(5):940–946. | ||
Shiiba M, Yamagami H, Yamamoto A, et al. Mefenamic acid enhances anticancer drug sensitivity via inhibition of aldo-keto reductase 1C enzyme activity. Oncol Rep. 2017;37(4):2025–2032. | ||
Liu C, Armstrong CM, Lou W, Lombard A, Evans CP, Gao AC. Inhibition of AKR1C3 activation overcomes resistance to abiraterone in advanced prostate cancer. Mol Cancer Ther. 2017;16(1):35–44. | ||
Ahmad P, Rasool S, Gul A, et al. Jasmonates: multifunctional roles in stress tolerance. Front Plant Science. 2016;7:813. | ||
Wang JJ, Mak OT. Induction of apoptosis by 15d-PGJ2 via ROS formation: an alternative pathway without PPARgamma activation in non-small cell lung carcinoma A549 cells. Prostaglandins Other Lipid Mediat. 2011;94(3-4):104–111. | ||
Kniazhanski T, Jackman A, Heyfets A, Gonen P, Flescher E, Sherman L. Methyl jasmonate induces cell death with mixed characteristics of apoptosis and necrosis in cervical cancer cells. Cancer Lett. 2008;271(1):34–46. | ||
Wang Y, Xiang W, Wang M, et al. Methyl jasmonate sensitizes human bladder cancer cells to gambogic acid-induced apoptosis through down-regulation of EZH2 expression by miR-101. Br J Pharmacol. 2014;171(3):618–635. | ||
Zheng L, Li D, Xiang X, et al. Methyl jasmonate abolishes the migration, invasion and angiogenesis of gastric cancer cells through down-regulation of matrix metalloproteinase 14. BMC Cancer. 2013;13:74. | ||
Heyfets A, Flescher E. Cooperative cytotoxicity of methyl jasmonate with anti-cancer drugs and 2-deoxy-D-glucose. Cancer Lett. 2007;250(2):300–310. | ||
Milrot E, Jackman A, Flescher E, et al. Enhanced killing of cervical cancer cells by combinations of methyl jasmonate with cisplatin, X or alpha radiation. Invest New Drugs. 2013;31(2):333–344. | ||
Ezekwudo D, Shashidharamurthy R, Devineni D, Bozeman E, Palaniappan R, Selvaraj P. Inhibition of expression of anti-apoptotic protein Bcl-2 and induction of cell death in radioresistant human prostate adenocarcinoma cell line (PC-3) by methyl jasmonate. Cancer Lett. 2008;270(2):277–285. | ||
Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83(5):803–812. | ||
Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83(5):813–819. | ||
Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer. 2004;4(1):61–70. | ||
Michalik L, Wahli W. PPARs Mediate lipid signaling in inflammation and cancer. PPAR Res. 2008;2008:134059. | ||
Kumar P, Barua CC, Sulakhiya K, Sharma RK. Curcumin ameliorates cisplatin-induced nephrotoxicity and potentiates its anticancer activity in SD rats: potential role of curcumin in breast cancer chemotherapy. Front Pharmacol. 2017;8:132. | ||
Gao S, Wu R, Zeng Y. Up-regulation of peroxisome proliferator-activated receptor gamma in radiation-induced heart injury in rats. Radiat Environ Biophys. 2012;51(1):53-59. | ||
Tachibana K, Yamasaki D, Ishimoto K, Doi T. The role of PPARs in cancer. PPAR Res. 2008;2008:102737. | ||
Lea MA, Sura M, Desbordes C. Inhibition of cell proliferation by potential peroxisome proliferator-activated receptor (PPAR) gamma agonists and antagonists. Anticancer Res. 2004;24(5a):2765–2771. | ||
Keshamouni VG, Reddy RC, Arenberg DA, et al. Peroxisome proliferator-activated receptor-gamma activation inhibits tumor progression in non-small-cell lung cancer. Oncogene. 2004;23(1):100–108. | ||
Wick M, Hurteau G, Dessev C, et al. Peroxisome proliferator-activated receptor-gamma is a target of nonsteroidal anti-inflammatory drugs mediating cyclooxygenase-independent inhibition of lung cancer cell growth. Mol Pharmacol. 2002;62(5):1207–1214. | ||
Kim KY, Kim SS, Cheon HG. Differential anti-proliferative actions of peroxisome proliferator-activated receptor-gamma agonists in MCF-7 breast cancer cells. Biochem Pharmacol. 2006;72(5):530–540. | ||
Mantel A, Carpenter-Mendini A, VanBuskirk J, Pentland AP. Aldo-keto reductase 1C3 is overexpressed in skin squamous cell carcinoma (SCC) and affects SCC growth via prostaglandin metabolism. Exp Dermatol. 2014;23(8):573–578. | ||
Kim R, Emi M, Tanabe K, Murakami S, Uchida Y, Arihiro K. Regulation and interplay of apoptotic and non-apoptotic cell death. J Pathol. 2006;208(3):319–326. | ||
Spitz DR, Azzam EI, Li JJ, Gius D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metastasis Rev. 2004;23(3–4):311–322. | ||
Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780–783. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.