")
Back to Journals » OncoTargets and Therapy » Volume 9
Methotrexate induces DNA damage and inhibits homologous recombination repair in choriocarcinoma cells
Authors Xie L, Zhao T, Cai J , Su Y, Wang Z , Dong W
Received 4 July 2016
Accepted for publication 20 September 2016
Published 17 November 2016 Volume 2016:9 Pages 7115—7122
DOI https://doi.org/10.2147/OTT.S116387
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Min Li
Lisha Xie,1,* Tiancen Zhao,1,2,* Jing Cai,1 You Su,1 Zehua Wang,1 Weihong Dong1
1Department of Obstetrics and Gynecology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, 2Department of Obstetrics and Gynecology, Central Hospital of Wuhan, Wuhan, China
*These authors contributed equally to this work
Objective: The objective of this study was to investigate the mechanism of sensitivity to methotrexate (MTX) in human choriocarcinoma cells regarding DNA damage response.
Methods: Two choriocarcinoma cancer cell lines, JAR and JEG-3, were utilized in this study. An MTX-sensitive osteosarcoma cell line MG63, an MTX-resistant epithelial ovarian cancer cell line A2780 and an MTX-resistant cervical adenocarcinoma cell line Hela served as controls. Cell viability assay was carried out to assess MTX sensitivity of cell lines. MTX-induced DNA damage was evaluated by comet assay. Quantitative reverse transcription polymerase chain reaction was used to detect the mRNA levels of BRCA1, BRCA2, RAD51 and RAD52. The protein levels of γH2AX, RAD 51 and p53 were analyzed by Western blot.
Results: Remarkable DNA strand breaks were observed in MTX-sensitive cell lines (JAR, JEG-3 and MG63) but not in MTX-resistant cancer cells (A2780 and Hela) after 48 h of MTX treatment. Only in the choriocarcinoma cells, the expression of homologous recombination (HR) repair gene RAD51 was dramatically suppressed by MTX in a dose- and time-dependent manner, accompanied with the increase in p53.
Conclusion: The MTX-induced DNA strand breaks accompanied by deficiencies in HR repair may contribute to the hypersensitivity to chemotherapy in choriocarcinoma.
Keywords: choriocarcinoma, chemotherapy hypersensitivity, DNA double-strand break, RAD51, p53
Introduction
Gestational trophoblastic neoplasia (GTN) is a spectrum of pregnancy-related malignant disorders, including invasive mole, gestational choriocarcinoma, placental-site trophoblastic tumor and epithelioid trophoblastic tumor. Despite the extremely low incidence of choriocarcinoma, it grows rapidly and tends to widely metastasize at an early stage.1,2 Unlike other gynecological malignancies, such as ovarian cancer and cervix cancer, that tend to develop resistance to chemotherapy, choriocarcinoma responds extremely well to chemotherapy and can be cured in most women. A better understanding of the mechanism of the chemosensitivity in choriocarcinoma may help improve the treatments for chemo-resistant tumors.
Methotrexate (MTX) is a common chemotherapeutic drug used in treating multiple cancers, such as human leukemia, non-small-cell lung cancer and choriocarcinoma.3 Single-agent MTX chemotherapy and MTX combined with other cytotoxic drugs (eg, EMA/CO [etoposide, MTX, actinomycin D, cyclophosphamide and vincristine]) were recommended as the first-line treatments for low- and high-risk choriocarcinoma, respectively, which achieve a high survival rate (>90%).4,5
As a structural analog of folic acid, MTX is widely thought to act through competitively inhibiting dihydrofolate reductase (DHFR) to affect cell proliferation. DHFR catalyzes the reduction of dihydrofolate to tetrahydrofolate, which is indispensable for DNA synthesis.6 Previous studies found that DHFR gene variants’ amplification and expression levels were associated with the response to MTX.7–9 However, Han et al10 observed that the DHFR expression in choriocarcinoma was not always correlated with MTX sensitivity, suggesting that additional mechanisms are involved in the hypersensitivity in choriocarcinoma to MTX. In some drug toxicity tests, MTX was shown to induce genetic injury, such as chromosomal aberration, gene mutation and DNA damage.11,12 There is also evidence that MTX causes DNA damage in cancer cells, such as oxidative damage in colon cancer and double-strand breaks (DSBs) in non-small-cell lung cancer.13,14 When DNA damage occurs, the intrinsic repair mechanisms are initiated to maintain genomic integrity and stability, which are important determinants of cellular response to chemotherapy.15 For example, MTX selectively led to cell death depending on the presence of defects in DNA mismatch repair.13 The defects in DNA base excision repair played a positive role in the MTX treatment of acute lymphoblastic leukemia.16 However, the role of unbalanced DNA damage and repair in the MTX sensitivity of choriocarcinoma remains to be elucidated. In addition, there are concerns that the increased rate of second primary tumors (SPTs) after choriocarcinoma may be associated with MTX chemotherapy.17 Thus, study on the mechanisms underlying MTX effects is of great clinical relevance.
DSB is a kind of DNA damage that leads to chromosomal rearrangements and is highly hazardous to cells. It arises from single-strand breaks, the most frequent DNA lesion, or directly from various exogenous or endogenous exposures.18,19 Homologous recombination (HR) is a major repair pathway for DSB and proceeds accurately with sequential involvements of multiple proteins. BRCA1 functions in the initial stage of HR, where a DSB undergoes 5′–3′ end resection to generate 3′ single-stranded DNA (ssDNA) tails. Then, the important recombinase RAD51 polymerizes forming nucleoprotein filaments on ssDNA and catalyzes DNA strand exchange between a damaged sequence and its intact homologue. Some ancillary factors are indispensable for RAD51 activity, such as BRCA2 serving as a main mediator for facilitating RAD51 assembly on ssDNA. In addition to promoting resection, BRCA1 is required for the interaction between BRCA2 and RAD51. RAD52 is another recombination mediator that possesses non-conserved functions among different species. In mammal cells, RAD52 is responsible for ssDNA annealing to synthesize new double-strand DNA, whereas in yeast, RAD52 has the similar function to that of mammal BRCA2.20,21
In order to investigate the implication of DNA damage and repair in choriocarcinoma chemosensitivity, in the present study, we analyzed the MTX-induced DNA strand breaks and alterations in the expression of HR-related genes in two choriocarcinoma cell lines JAR and JEG-3 and three non-GTN cancer cell lines MG63, A2780 and Hela. We found that defects in DNA damage repair associated with suppressed RAD51 contributed to the MTX sensitivity in choriocarcinoma.
Materials and methods
Cell lines
The present study was approved by the Ethical Committee of Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, China. The human placental choriocarcinoma cell lines JAR and JEG-3 were, respectively, purchased from the Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China) and the Institute of Basic Medical Sciences of the Chinese Academy of Medical Sciences (Beijing, China). The human osteosarcoma cell line MG63, the human epithelial ovarian cancer cell line A2780 and the human cervical adenocarcinoma cancer cell line Hela were purchased from the China Center for Type Culture Collection (Wuhan, China).
Drug sensitivity assay
The sensitivity to MTX was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells were seeded in a 96-well plate at a density of 3,000 cells/well and incubated for 24 h. Then, they were treated with different concentrations of MTX (Sigma-Aldrich Co., St Louis, MO, USA) in sextuplicate. After 48 h, cells were incubated with 20 μL MTT (AMRESCO, Solon, OH, USA) per well for additional 4 h, followed by 100 μL triple solution overnight. The cell viability and half maximal inhibitory concentration (IC50) were calculated based on the mean value of optical density at 470 nm measured by iMark Microplate Reader (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Comet assay
Cells were seeded in a six-well plate at a density of 2,000 cells/well and incubated for 24 h, followed by 48 h of MTX treatment. The MTX concentrations were IC50 for JAR, JEG-3 and MG63 and 200 μmol/L for A2780 and Hela. Comet assay was performed based on the protocol of Singh et al22 with slight modifications. The slides were sequentially coated with 100 μL 1% normal melting agarose (Biosharp, Hefei, China) as the first layer and 100 μL 0.5% low melting agarose (Biosharp) as the second layer. The next step was to add 20 μL cell suspension to 160 μL 0.7% low melting agarose, which was distributed on the slides as the third layer. Then the slides were incubated in cell lysis buffer (2.5 M NaCl, 0.2 M NaOH, 100 mM Na2EDTA, 10 mM Tris–HCl, 1% Triton X-100 and 10% dimethyl sulfoxide, pH =10.0) for 90 min. After that, we immersed the slides in ultrapure water for three times followed by 20 min incubation of unwinding solution (3 M NaOH). Subsequently, the slides were placed in a horizontal gel electrophoresis tank containing electrophoresis solution (1 mM Na2EDTA and 300 mM NaOH, pH =13). The electrophoresis was conducted at 25 V (1 V/cm, 300 mA) for 25 min. Then the slides were incubated in neutralization buffer (0.4 M Tris–HCl, pH =7.5) for 10 min followed by immersion in ultrapure water for three times and air-dry. Before being observed under fluorescence microscope, the cells were stained using 50 μL ethidium bromide (2 mg/L; AMRESCO). Comet Assay Software (CaspLab—Comet Assay Software Project Lab, Warsaw, Poland) was used to analyze tail length and tail moment. One hundred cells were evaluated for each sample. All steps were carried out under dim light to minimize extra DNA damage.
RNA extraction and quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted using TRIzol Reagent (Takara, Dalian, China), and the RNA quality and concentration were determined by NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA). The reverse transcription was conducted using Kit (Takara) according to the manufacturer’s protocol. The complementary DNA was quantified by StepOne Detection System (Thermo Fisher Scientific) using the SYBR Green PCR Master Mix (Takara). The quantitative amplification conditions were 95°C for 60 sec, 40 cycles at 95°C for 30 sec, 57°C for 60 sec and 72°C for 30 sec. β-actin was used as the reference gene for normalization. The primer sequences are presented in Table 1.
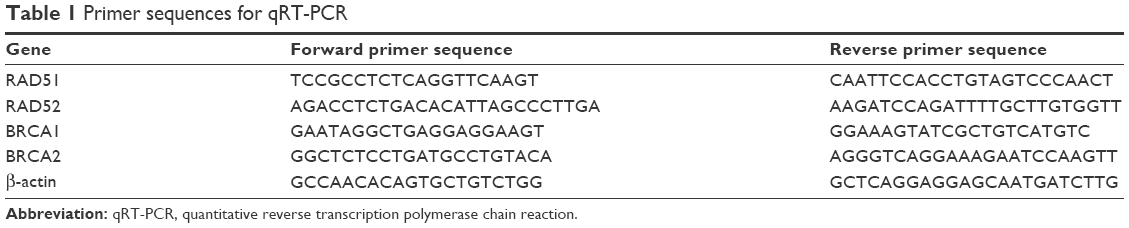 | Table 1 Primer sequences for qRT-PCR |
Protein extraction and Western blot
The cells were dissociated in cold radioimmune precipitation assay buffer for 30 min and centrifuged at 12,000 rpm for 10 min. The supernatants were protein extracts. Protein was quantified by Lowry assay. Subsequently, the samples were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes in the semi-dry system for 20 min. Then, the membranes were blocked using 5% defatted milk for 1 h and incubated with primary antibodies overnight, including rabbit anti-γH2AX (1:1,000 dilution; Cell Signaling Technology, Beverly, MA, USA), rabbit anti-RAD51 (1:1,000 dilution; Santa Cruz Biotechnology Inc., Dallas, TX, USA) and rabbit anti-p53 (1:800 dilution; Cell Signaling Technology), followed by 1.5 h incubation with horseradish peroxidase-conjugated anti-rabbit secondary antibody (1:5,000 dilution; Santa Cruz Biotechnology Inc.). The samples were visualized by ECL substrate kit (Amersham Biosciences, Marlborough, MA, USA), and gray values were calculated by Quantity One software (Bio-Rad Laboratories Inc.). α-Tubulin was used as an internal control.
Statistical analysis
The statistical analysis was performed using SPSS 13.0 software (SPSS Inc., Chicago, IL, USA). Numerical data were expressed as mean ± standard deviation from three independent experiments. The difference among groups was evaluated by Student’s t-test and considered statistically significant with a P-value <0.05.
Results
Sensitivity to MTX of different cancer cell lines
MTT assay was performed to determine the sensitivity to MTX in two choriocarcinoma cancer cell lines, JAR and JEG-3, an osteosarcoma cell line MG63, an epithelial ovarian cancer cell line A2780 and a cervical adenocarcinoma cell line Hela. MTX IC50s in JAR, JEG-3 and MG63 were 28.39±3.02 μM, 23.70±2.58 μM and 30.92±3.51 μM, respectively, whereas the MTX IC50s in A2780 and Hela exceeded 400 μM (Figure 1). These findings verified the sensitivity to MTX in JAR, JEG-3 and MG63 compared to A2780 and Hela.
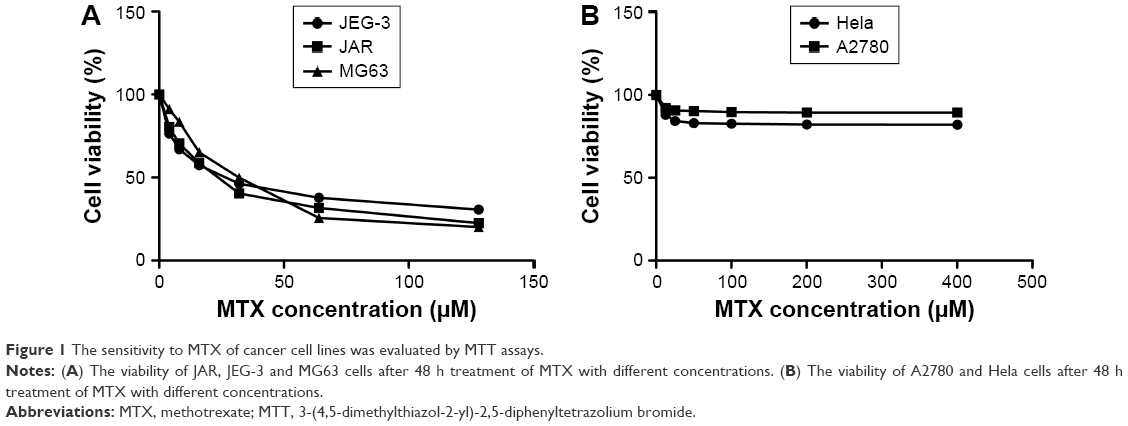 | Figure 1 The sensitivity to MTX of cancer cell lines was evaluated by MTT assays. |
MTX-induced DNA damage in MTX-sensitive cell lines
We evaluated DNA damage using the comet assay. Compared to the control groups, the tail length and tail moment of comet cells were markedly increased after 48 h of MTX treatment in JAR, JEG-3 and MG63 cell lines (all P<0.01). However, we did not observe significant tailing phenomenon in MTX-resistant A2780 and Hela (Figure 2A and B). The phosphorylated histone H2A (γH2AX), which could recruit multiple DNA repair proteins to initiate DNA damage response (DDR) signaling cascade, has been widely acknowledged as a biomarker for evaluating DSB.23 With the MTX treatment, γH2AX was concentration dependently and time dependently upregulated in JAR and JEG-3 cell lines, but not in the MG63 cell line (Figure 2C), indicating MTX-induced DNA damage in choriocarcinoma cells.
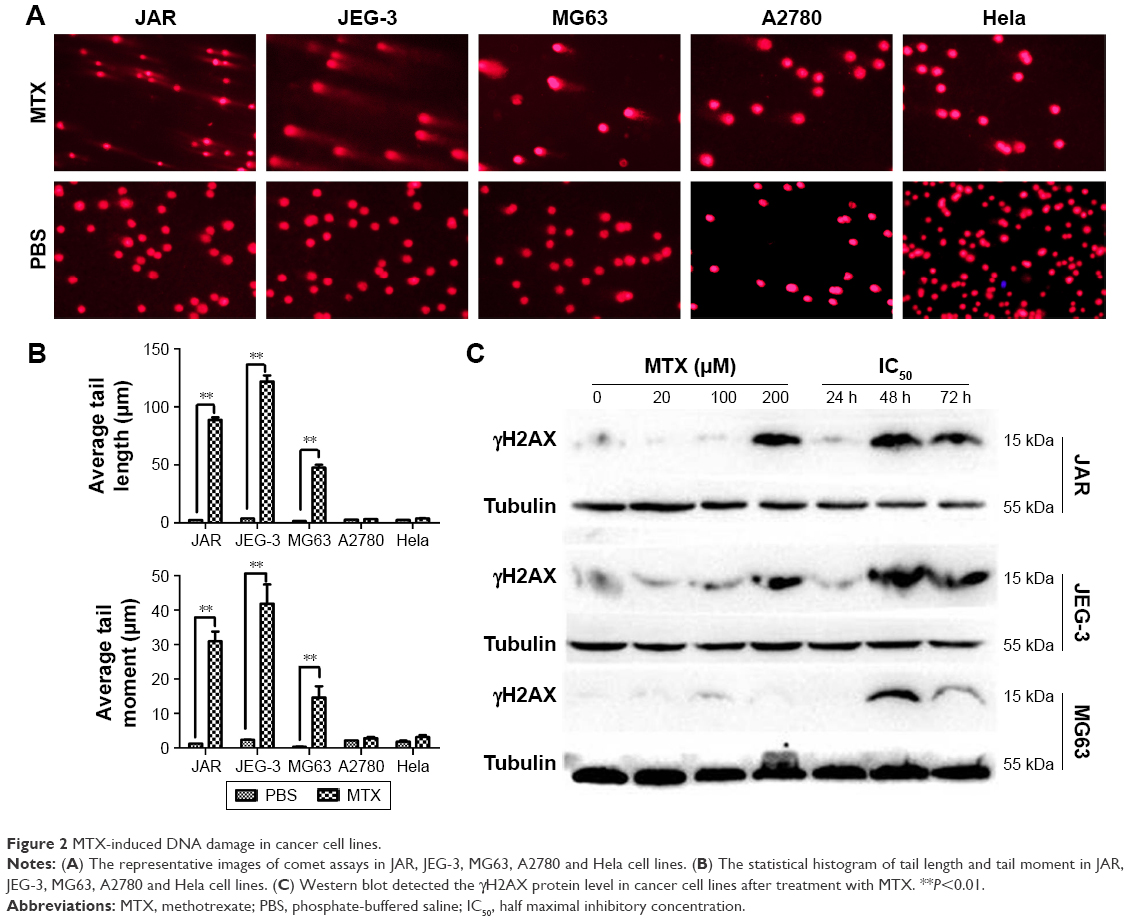 | Figure 2 MTX-induced DNA damage in cancer cell lines. |
MTX suppresses DSB repair mediator RAD51 in choriocarcinoma cells
As HR is a primary pathway for DSB repair, we investigated the effect of MTX on the expression of four HR-related genes, RAD51, RAD52, BRCA1 and BRCA2, in JAR, JEG-3 and MG63 after MTX treatment. The mRNA level of RAD51 was significantly decreased by MTX in a dose-dependent manner in JAR and JEG-3 (P<0.01). In MG63 cell line, MTX merely had a slight inhibitory effect on RAD51. The expression of RAD52, BRCA1 and BRCA2 was not significantly affected by MTX in all the three examined cancer cell lines (Figure 3). Western blot assays demonstrated that MTX suppressed RAD51 in JAR, JEG-3 and MG63 cell lines, but not in MG63, in a dose- and time-dependent manner. On the contrary, p53 expression was significantly increased after MTX treatment, indicating that MTX may inhibit HR through p53-dependent mechanism (Figure 4).
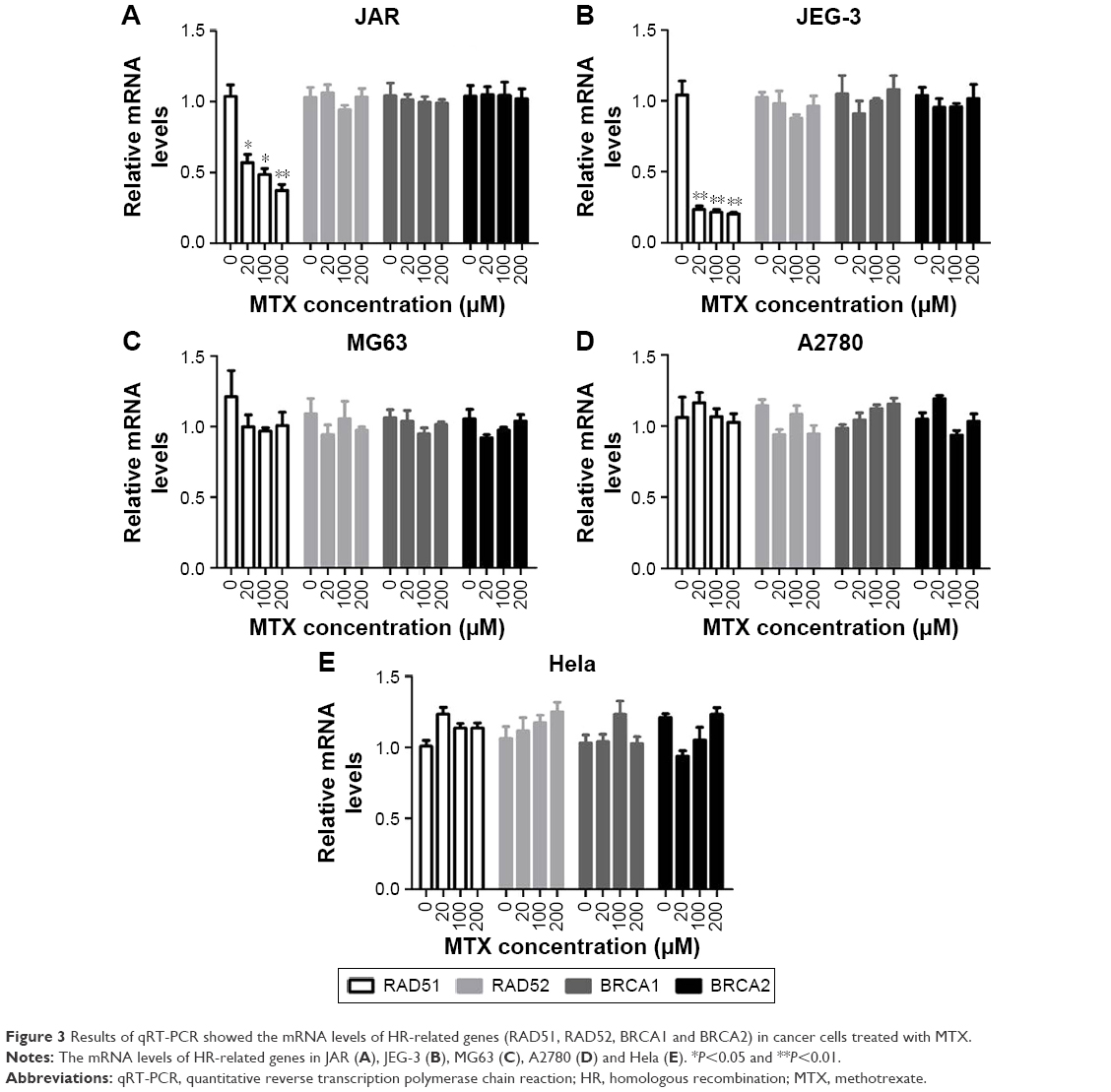 | Figure 3 Results of qRT-PCR showed the mRNA levels of HR-related genes (RAD51, RAD52, BRCA1 and BRCA2) in cancer cells treated with MTX. |
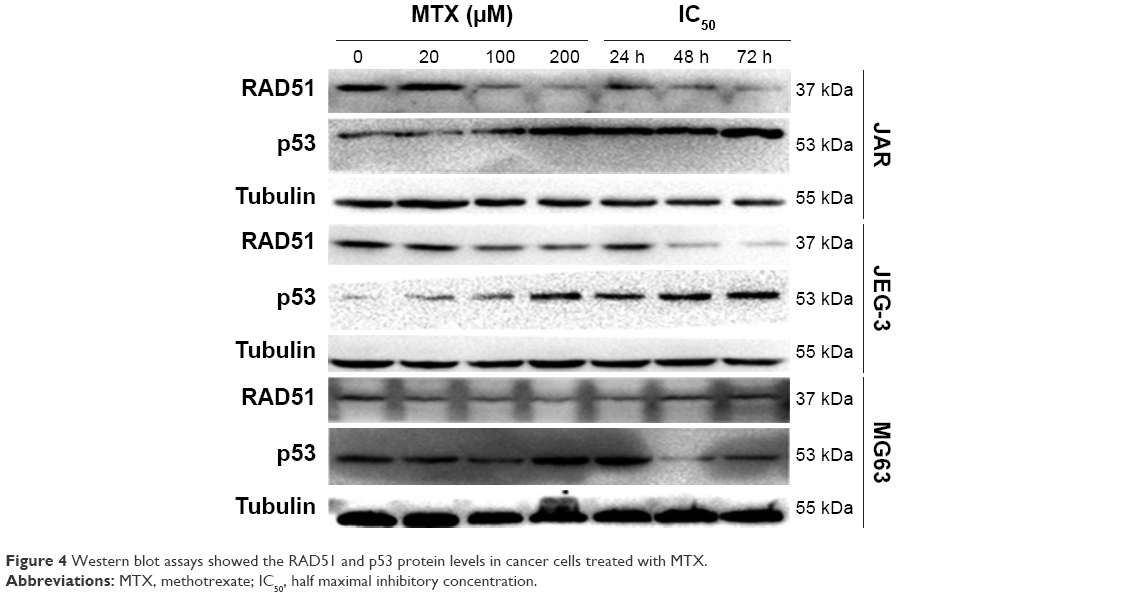 | Figure 4 Western blot assays showed the RAD51 and p53 protein levels in cancer cells treated with MTX. |
Discussion
Choriocarcinoma is prone to early metastasis but can be cured by classical cytotoxic drugs, which makes it an attractive research model. MTX is the first-line chemotherapeutic agent, alone or in combination, for treatment of choriocarcinoma with a response rate of ~90%.24 The mechanism of the hypersensitivity to MTX in choriocarcinoma was partially demonstrated in this study. We showed that MTX-induced DNA damage, particularly DSBs, and suppression of HR in the MTX-sensitive choriocarcinoma and osteosarcoma cell lines but not in the MTX-resistant A2780 and Hela cell lines. These data implicate defects in DNA damage repair in MTX sensitivity.
Our findings suggest that MTX is more lethal to the cancer cells with HR deficiency, which provides some new ideas to improve tumor treatment. For example, concomitant administration of drugs known to inhibit HR may sensitize these MTX-resistant tumors. Also, it is possible to account for interindividual variations in MTX response, but further studies are needed. Besides HR, non-homologous end joining (NHEJ) is another repair pathway for DSB. There is evidence that NHEJ mutant cells are more sensitive to MTX compared to the parental ones, which is consistent with our results that MTX acts through inducing DSBs.25 However, there is evidence that the increased incidence of SPTs (hematologic malignancies, colorectal cancer, thyroid cancer, etc) after choriocarcinoma may be associated with MTX treatment.17 A retrospective cohort study reported that the risk of SPTs in childhood cancer survivors was correlated with the higher baseline and post-irradiation levels of DNA DSBs.26 Further studies are required to answer the question whether the MTX-induced DSB contributes to the SPTs after choriocarcinoma.
RAD51 is a central player of DNA HR for DSB. A number of studies have revealed the notable role of RAD51 in mediating cellular chemoresponsiveness. In soft tissue sarcoma, RAD51 depletion sensitized cells to doxorubicin, and similar results were obtained in a variety of other tumors, such as in colon cancer cells treated with inhibitors of thymidylate synthase and in imatinib-resistant chronic myeloid leukemia cells.27–29 RAD51 has become a promising anticancer target for chemotherapy. We found that MTX treatment gave rise to significantly decreased RAD51 in choriocarcinoma cells, whereas the inhibitory effect was inconspicuous in MG63 cells. However, inconsistent with our results, RAD51 was reported to be dramatically suppressed by MTX in another osteosarcoma cell line HOS, perhaps due to different cell types and MTX treatment conditions.25 Our findings suggest that MTX-induced inhibition on RAD51 led to persistently unrepaired DSBs in choriocarcinoma cell, which consequently result in chemotherapeutic hypersensitivity.
Accumulated studies demonstrated that p53 is a prominent regulator in multiple DDRs, including DNA repair, cell cycle arrest, senescence and apoptosis.30 We found that MTX treatment resulted in a dose- and time-dependent increase in the p53 level, which was reversely correlated with RAD51. In soft tissue sarcoma, p53 inhibits RAD51 expression through suppressing its promoter activities, which presents a possible explanation for their negative relationship in choriocarcinoma. Except for transcriptional regulation, p53 can repress HR repair through physically interacting with RAD51 to abrogate its polymerization.31 The anticancer mechanism of MTX may act through p53-mediated suppression on HR.
Similar to choriocarcinoma, testicular cancer, Hodgkin disease and high-grade lymphomas are also sensitive to chemotherapy, which may be attributed to the nature of tumors themselves rather than the drug properties. Such chemotherapy-curable cancers share a common intrinsic characteristic that they arise from cells, in which most genetic recombination during malignant transformation is taken as part of their normal physiology. Consequently, they may be free from further genetic or epigenetic disorders in apoptosis, DNA repair and senescence, making them retain the natural state of sensitivity to chemotherapy.24
Conclusion
Taken together, the sensitivity of choriocarcinoma to MTX may be due to the MTX-induced DNA damage accompanied with suppressed HR in tumor cells, which is associated with the inhibition of RAD51 expression. These findings highlight the key role of RAD51 in governing the fate of tumor cells suffered from DNA damage induced by chemotherapeutic agents.
Acknowledgment
This study was supported by grant 81272860 from the National Natural Science Foundation of China.
Disclosure
The authors report no conflicts of interest in this work.
References
Froeling FE, Seckl MJ. Gestational trophoblastic tumours: an update for 2014. Curr Oncol Rep. 2014;16(11):408. | ||
Seckl MJ, Sebire NJ, Berkowitz RS. Gestational trophoblastic disease. Lancet. 2010;376(9742):717–729. | ||
Lagarce L, Zenut M, Laine-Cessac P. Pharmacologie du méthotrexate Geschwulste [Methotrexate pharmacology]. J Gynecol Obstet Biol Reprod (Paris): 2015;44(3):203–211. French. | ||
Aghajanian C. Treatment of low-risk gestational trophoblastic neoplasia. J Clin Oncol. 2011;29(7):786–788. | ||
Escobar PF, Lurain JR, Singh DK, Bozorgi K, Fishman DA. Treatment of high-risk gestational trophoblastic neoplasia with etoposide, methotrexate, actinomycin D, cyclophosphamide, and vincristine chemotherapy. Gynecol Oncol. 2003;91(3):552–557. | ||
Goodsell DS. The molecular perspective: methotrexate. Oncologist. 1999;4(4):340–341. | ||
Kodidela S, Pradhan SC, Dubashi B, et al. Influence of dihydrofolate reductase gene polymorphisms rs408626 (-317A>G) and rs442767 (-680C>A) on the outcome of methotrexate-based maintenance therapy in South Indian patients with acute lymphoblastic leukemia. Eur J Clin Pharmacol. 2015;71(11):1349–1358. | ||
Serra M, Reverter-Branchat G, Maurici D, et al. Analysis of dihydrofolate reductase and reduced folate carrier gene status in relation to methotrexate resistance in osteosarcoma cells. Ann Oncol. 2004;15(1):151–160. | ||
Singer MJ, Mesner LD, Friedman CL, Trask BJ, Hamlin JL. Amplification of the human dihydrofolate reductase gene via double minutes is initiated by chromosome breaks. Proc Natl Acad Sci U S A. 2000;97(14):7921–7926. | ||
Han B, Xiang Y, Wang Y, et al. Dihydrofolate reductase transcript level is not suitable for methotrexate-resistance prediction in choriocarcinoma cell line. Int J Gynecol Cancer. 2010;20(7):1259–1263. | ||
Deng H, Zhang M, He J, et al. Investigating genetic damage in workers occupationally exposed to methotrexate using three genetic end-points. Mutagenesis. 2005;20(5):351–357. | ||
Shahin AA, Ismail MM, Saleh AM, Moustafa HA, Aboul-Ella AA, Gabr HM. Protective effect of folinic acid on low-dose methotrexate genotoxicity. Z Rheumatol. 2001;60(2):63–68. | ||
Martin SA, McCarthy A, Barber LJ, et al. Methotrexate induces oxidative DNA damage and is selectively lethal to tumour cells with defects in the DNA mismatch repair gene MSH2. EMBO Mol Med. 2009;1(6–7):323–337. | ||
Huang WY, Yang PM, Chang YF, Marquez VE, Chen CC. Methotrexate induces apoptosis through p53/p21-dependent pathway and increases E-cadherin expression through downregulation of HDAC/EZH2. Biochem Pharmacol. 2011;81(4):510–517. | ||
Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med. 2015;66:129–143. | ||
Stanczyk M, Sliwinski T, Trelinska J, et al. Role of base-excision repair in the treatment of childhood acute lymphoblastic leukaemia with 6-mercaptopurine and high doses of methotrexate. Mutat Res. 2012;741(1–2):13–21. | ||
Sisti G, Kanninen TT, Asciutti S, Sorbi F, Fambrini M. Rate of second primary tumors following diagnosed choriocarcinoma: a SEER analysis (1973–2010). Gynecol Oncol. 2014;134(1):90–95. | ||
Kuzminov A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc Natl Acad Sci U S A. 2001;98(15):8241–8246. | ||
Khanna A. DNA damage in cancer therapeutics: a boon or a curse? Cancer Res. 2015;75(11):2133–2138. | ||
Krejci L, Altmannova V, Spirek M, et al. Homologous recombination and its regulation. Nucleic Acids Res. 2012;40(13):5795–5818. | ||
Prakash R, Zhang Y, Feng W, et al. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 2015;7(4):a016600. | ||
Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175(1):184–191. | ||
Valdiglesias V, Giunta S, Fenech M, et al. GammaH2AX as a marker of DNA double strand breaks and genomic instability in human population studies. Mutat Res. 2013;753(1):24–40. | ||
Savage P, Stebbing J, Bower M, Crook T. Why does cytotoxic chemotherapy cure only some cancers? Nat Clin Pract Oncol. 2009;6(1):43–52. | ||
Du LQ, Du XQ, Bai JQ, et al. Methotrexate-mediated inhibition of RAD51 expression and homologous recombination in cancer cells. J Cancer Res Clin Oncol. 2012;138(5):811–818. | ||
Haddy N, Tartier L, Koscielny S, et al. Repair of ionizing radiation-induced DNA damage and risk of second cancer in childhood cancer survivors. Carcinogenesis. 2014;35(8):1745–1749. | ||
Hannay JA, Liu J, Zhu QS, et al. Rad51 overexpression contributes to chemoresistance in human soft tissue sarcoma cells: a role for p53/activator protein 2 transcriptional regulation. Mol Cancer Ther. 2007;6(5):1650–1660. | ||
Yang Z, Waldman AS, Wyatt MD. Expression and regulation of RAD51 mediate cellular responses to chemotherapeutics. Biochem Pharmacol. 2012;83(6):741–746. | ||
Zhu J, Zhou L, Wu G, et al. A novel small molecule RAD51 inactivator overcomes imatinib-resistance in chronic myeloid leukaemia. EMBO Mol Med. 2013;5(3):353–365. | ||
Speidel D. The role of DNA damage responses in p53 biology. Arch Toxicol. 2015;89(4):501–517. | ||
Bertrand P, Saintigny Y, Lopez BS. p53’s double life: transactivation-independent repression of homologous recombination. Trends Genet. 2004;20(6):235–243. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.