")
Back to Journals » Cancer Management and Research » Volume 10
Metformin sensitizes hypoxia-induced gefitinib treatment resistance of HNSCC via cell cycle regulation and EMT reversal
Authors Yin XT , Wei Z, Song CH , Tang CC, Xu WG, Wang YF, Xie JQ , Lin ZT, Han W
Received 17 June 2018
Accepted for publication 20 August 2018
Published 19 November 2018 Volume 2018:10 Pages 5785—5798
DOI https://doi.org/10.2147/CMAR.S177473
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Beicheng Sun
Xiteng Yin,1,2,* Zheng Wei,1,2,* Chuanhui Song,1,2 Chuanchao Tang,1,2 Wenguang Xu,1,2 Yufeng Wang,1,2 Junqi Xie,1,2 Zitong Lin,2,3 Wei Han1,2
1Department of Oral and Maxillofacial Surgery, Nanjing Stomatological Hospital, Medical School of Nanjing University, Nanjing, China; 2Central Laboratory of Stomatology, Nanjing Stomatological Hospital, Medical School of Nanjing University, Nanjing, China; 3Department of Dentomaxillofacial Radiology, Nanjing Stomatological Hospital, Medical School of Nanjing University, Nanjing, China
*These authors contributed equally to this work
Objectives: The objectives of this study were to explore the mechanisms of metformin sensitization to hypoxia-induced gefitinib treatment in resistant head and neck squamous cell carcinoma (HNSCC) and evaluate the effects of this combined treatment strategy.
Methods: The effects of gefitinib treatment on HNSCC were measured under normoxic and hypoxic conditions. The relationship between hypoxia and cell cycle and epithelial–mesenchymal transition (EMT) in tumor cells were analyzed. Palbociclib and LY294002 were used in combination with gefitinib to evaluate the effects on HNSCC cell cytotoxicity during hypoxia. Finally, metformin was used to evaluate the sensitizing effects of gefitinib treatment on HNSCC in vivo and in vitro.
Results: Cell viability and apoptosis assays demonstrated a significant difference in HNSCC cells treated with gefitinib between the normoxia and hypoxia groups. Hypoxia induced the expression of cyclin D1, decreased the percentage of cells in G1, and promoted the EMT of tumor cells. Both palbociclib and LY294002 enhanced gefitinib-induced cytotoxicity of HNSCC cells under hypoxic conditions. Encouragingly, metformin sensitized HNSCC to gefitinib treatment in vivo and in vitro.
Conclusion: Hypoxia promotes G1–S cell cycle progression and EMT in HNSCC, resulting in gefitinib treatment resistance. Metformin sensitizes HNSCC to gefitinib treatment, which might serve as a novel combined treatment strategy.
Keywords: tumor hypoxia, gefitinib resistance, metformin
Introduction
Head and neck squamous cell carcinoma (HNSCC) ranks as the sixth most common cancer worldwide.1 The main treatment strategy for this cancer includes surgery, chemotherapy, and radiotherapy; however, the 5-year survival rate still remains low, ~50%.2,3 Recently, new therapeutic approaches, such as molecular-targeted therapy, have raised more clinical concern. Until now, molecular targeting strategies included treatments targeting EGFR, vascular endothelial growth factor, and vascular endothelial growth factor receptor, as well as intracellular signaling pathway components associated with the phosphatidylinositol 3-kinase (PI3K)/serine/threonine-specific protein kinase (AKT).4–6 Among the abovementioned treatments, EGFR-targeted therapy has been proven to have great clinical value among various cancers, especially lung cancer.7 Nearly 90% of the HNSCC cases are confirmed to have a high expression of EGFR.8 Theoretically, targeting this molecule should result in significant clinical outcomes and improve the prognosis of HNSCC. Unfortunately, this type of therapy has been shown to have limited benefits.9,10 EGFR tyrosine kinase inhibitors (TKIs), including the first-generation TKI – gefitinib, have not been approved by the Food and Drug Administration (FDA) for use in HNSCC. Based on the results of reported clinical trials, HNSCC cells might apply various mechanisms to attain resistance to gefitinib treatment, which greatly reduces treatment efficacy.11
Currently, the molecular mechanisms underlying gefitinib treatment resistance in HNSCC remain unclear. It was reported that acquired resistance is mainly due to a secondary mutation (threonine-to-methionine substitution at amino acid position 790 [T790M]) and amplification of other oncogenes.12–14 Recently, more and more studies have revealed the importance of the tumor microenvironment in promoting tumor progression and treatment resistance.15,16 Hypoxia is a typical feature in the microenvironment and has been demonstrated by several studies to play a pivotal role in acquired gefitinib resistance.17,18 Our previous work showed that chronic inflammation and hypoxic signaling are involved in crosstalk in the HNSCC tumor microenvironment, consequently decreasing the effect of cisplatin treatment.19 Thus, we hypothesize that hypoxia, a dominant tumor microenvironment feature in HNSCC, might have the same effect on gefitinib resistance. It is therefore imperative to explore the mechanisms and overcome hypoxia-induced treatment resistance. There is an urgent need for clinicians to develop an easy and practical way to re-sensitize HNSCC to the effects of gefitinib.
Metformin is a widely used drug for diabetes, and recent reports have confirmed its effects in combating cancer and improving patient survival rates.20 Tseng reported that metformin use is associated with a significantly lower risk of nasopharyngeal cancer21 and Woo et al showed that metformin inhibits the expression of malic enzyme and sensitizes HNSCC cells to therapy-induced senescence.22 Our group also proved that metformin may synergize the effects of cisplatin and hypoxia via the inhibition of hypoxic inducible factor-1α (HIF-1α) expression.23 Hence, in this study, we explored the mechanisms of hypoxia-induced gefitinib resistance and found that this might be explained by the promotion of G1–S cell cycle progression and epithelial–mesenchymal transition (EMT) in tumor cells. We also provide strong evidence for the combined therapy of metformin and gefitinib in overcoming hypoxia-induced gefitinib resistance in HNSCC.
Materials and methods
Cell culture
The human HNSCC cell lines CAL27, HSC3, and SCC4 were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China); JHU011 was from Johns Hopkins University (Baltimore, MD, USA); Detroit562 and HN4 were purchased from American Type Culture Collection (Manassas, VA, USA). All the cell lines were cultured in DMEM (with 100 µg/mL streptomycin and 100 U/mL penicillin) (KeyGEN Biotech, Nanjing, China) supplemented with 10% FBS (BI, Migdal Haemek, Israel), in a humidified incubator 5% CO2/20% O2 or 5% CO2/1% O2 at 37°C, represented as normoxic and hypoxic tumor environments, respectively. Confluent cells were trypsinized with 0.05% trypsin containing 0.02% EDTA (Thermo Fisher Scientific, Waltham, MA, USA).
Flow cytometry
HNSCC cell lines were harvested and washed with PBS containing 3% BSA. After washing, the monoclonal anti-human PE-conjugated EGFR antibody (BioLegend, San Diego, CA, USA) was added to the cells and incubated for 30 minutes at 4°C in the dark. The cells were washed three times with PBS and analyzed using a FACSVerse flow cytometer (BD Biosciences, San Jose, CA, USA) to measure the expression of EGFR. The data obtained were analyzed by FlowJo (Tree Star, Inc., Ashland, OR, USA). Apoptosis assays and cell cycle analysis were detected by flow cytometry as described in detail below.
Quantitative real-time PCR assay
Total RNA was isolated using TRIzol reagent. Reverse transcription was performed using a PrimeScript™ RT Reagent Kit with gDNA Eraser (Takara Biotechnology, Dalian, China) according to the manufacturer’s instructions. RT-PCR was performed on ViiA™ 7 (Thermo Fisher Scientific) using the ChamQ Universal SYBR qPCR Master Mix (Vazyme Biotech, Nanjing, China) according to the manufacturer’s protocol. β-Actin was used as the reference gene, and results were expressed as the relative expression ratio of target gene to reference gene. Data were analyzed with the 2−ΔΔCt method. The sequences for the primers used in this study are listed in Table 1.
 | Table 1 Sequences of primers used in this study Abbreviation: α-SMA, α-smooth muscle actin. |
Western blot assay
Following the appropriate treatments, the cells were lysed on ice using the mammalian protein extraction reagent RIPA (Beyotime, Shanghai, China) supplemented with a protease inhibitor cocktail (Hoffman-La Roche Ltd, Basel, Switzerland) and phenylmethylsulfonyl fluoride (Beyotime). The supernatants were collected by centrifugation at 12,000× g at 4°C for 25 minutes. Total protein concentrations were determined with a bicinchoninic acid protein assay kit (KeyGEN Biotech). Protein samples were mixed with 5× loading buffer (GenScript, Nanjing, China) and heated at 95°C for 10 minutes. Equal amounts of protein were separated by SDS-PAGE, transferred to a 0.22 mm itrocellulose membrane (EMD Millipore, Billerica, MA, USA) and blocked by incubation with 5% fat-free milk in TBST buffer (150 mM NaCl, 50 mM Tris-HCl, 0.5% Tween 20, pH 7.6) at room temperature for 2 hours. The membranes were incubated with primary antibodies at 4°C overnight and then with horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 2 hours, prior to being exposed with ECL reagent (EMD Millipore). The pictures were captured by a Tanon 6200 Luminescent Imaging Workstation (Tanon, Shanghai, China). The following primary antibodies were used to detect proteins: rabbit anti-cyclin D1 (1:10,000; Abcam, Cambridge, UK), E-cadherin (1:500; Abcam), vimentin (1:2,000; Abcam), slug (1:1,000; Abcam), α-smooth muscle actin (α-SMA; 1:2,000; Abcam), phospho-AKT (1:1,000; Cell Signaling Technology, Danvers, MA, USA), AKT (1:1,000; Cell Signaling Technology), phospho-ERK (1:1,000; Cell Signaling Technology), ERK (1:1,000; Cell Signaling Technology), mouse anti-twist (1:500; Abcam), and anti-β-actin (1:2,000; Proteintech, Rosemont, IL, USA).
Patient cohort
A total of 30 patients diagnosed with HNSCC at the Department of Oral and Maxillofacial Surgery, Nanjing Stomatological Hospital, Medical School of Nanjing University between 2007 and 2011 were included in this study. All patients provided their written informed consent. The mean and median age at diagnosis was 61.17 and 61 years old, respectively (range, 46–81 years). The detailed clinicopathological parameters are provided in Table 2.
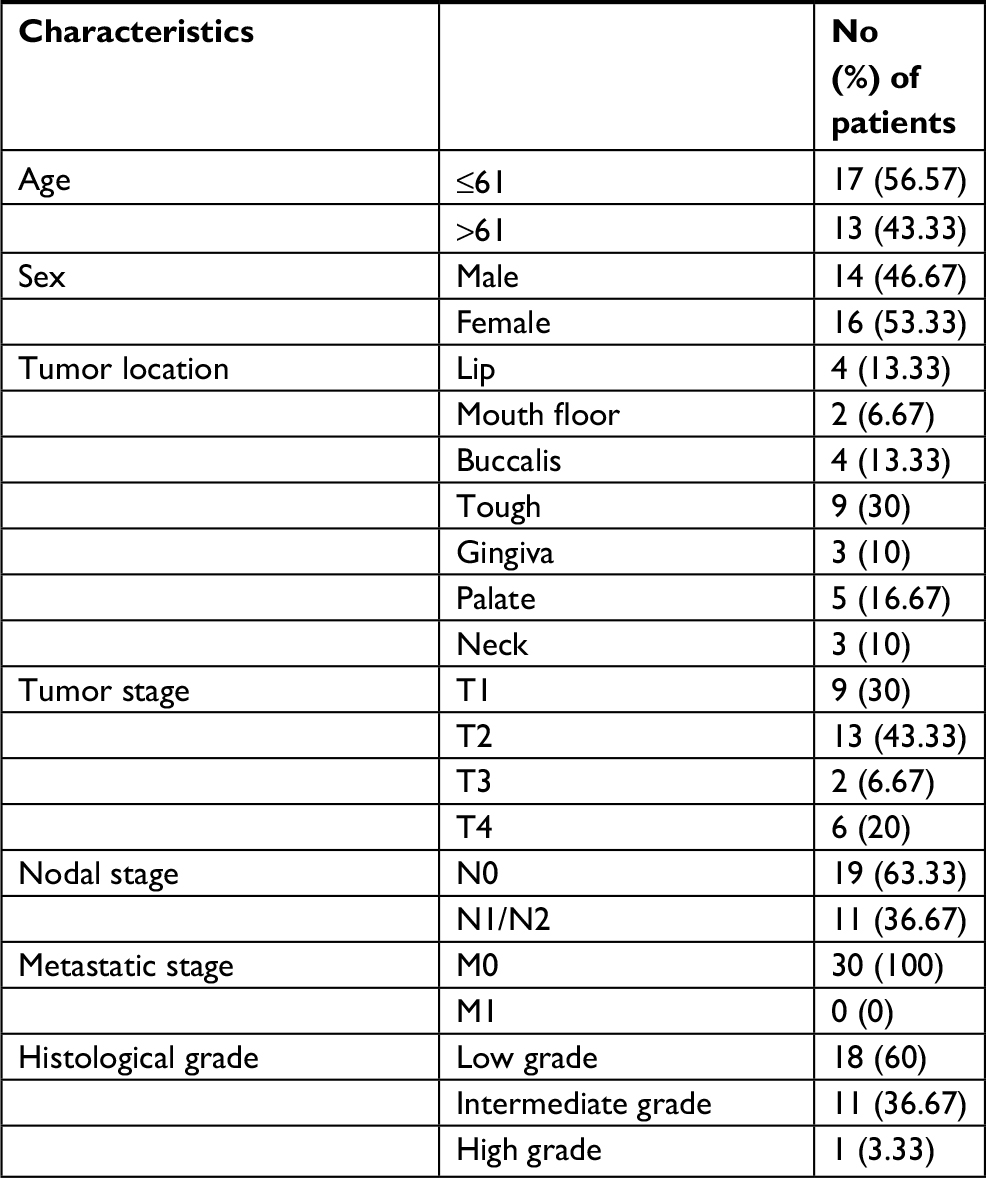 | Table 2 Clinical and pathological characteristics of HNSCC Abbreviation: HNSCC, head and neck squamous cell carcinoma. |
Histopathological analysis and immunohistochemistry
Samples from clinical patients and animal models were collected. Tissue sections (4 μm thick) were obtained, deparaffinized, and subjected to antigen recovery treatment with 100 mM citrate buffer target retrieval solution, pH 6.0 at 95°C, in a water bath for 20 minutes. Endogenous peroxidase activity was blocked by incubating with PBS and 3% hydrogen peroxidase for 30 minutes. After washing with PBS, the sections were incubated with rabbit anti-cyclin D1 (1:500; Abcam), E-cadherin (1:1,600; Abcam), HIF-1α (1:400; Abcam), and Ki67 (Typing, Nanjing, China) overnight at 4°C, followed by the Envision Dual Link System HRP method (Dako Denmark A/S, Glostrup, Denmark). All the antibodies were diluted in Dako antibody diluent. Reactions were revealed by incubating the sections with 3,3´-diaminobenzidine tetrahydrochloride (Dako Denmark A/S). Three pathologists independently scored the immunohistochemically stained slides. The scoring was based on the extent (E) of staining (percentage of positive tumor cells graded on a scale from 0 to 3: 0, none; 1, 1%–25%; 2, 26%–50%; 3, 51%–75%; 4, 75%–100%) and the intensity (I) of staining (graded on a scale of 0–3: 0, none; 1, weak staining; 2, moderate staining; 3, strong staining). Finally, the scores were calculated using the formula: scores = ∑(E×I). In vivo, hypoxia was detected by Hypoxyprobe™-1 Plus Kits (EMD Millipore) according to the manufacturer’s instructions. In brief, 15 minutes before being sacrificed, mice were intraperitoneally injected with a Hypoxyprobe™-1 (pimonidazole HCl) solution at a dosage of 100 mg/kg body weight. The xenograft tumors were removed for formalin fixation, paraffin-embedded, followed by immunostaining analysis with FITC-MAb1 (primary antibody, 1:100) and peroxidase-conjugated anti-FITC secondary reagent (1:100). The remaining steps were carried out as mentioned earlier.
Cell viability assay
The cell viability of HNSCC cells following treatment of gefitinib (Selleck Chemicals, Houston, TX, USA), palbociclib (Selleck Chemicals), LY294002 (Selleck Chemicals), and metformin (Sigma Aldrich Co, St Louis, MO, USA) was tested by using cell counting kit-8 (CCK-8) (Bimake, Houston, TX, USA) following the manufacturer’s instructions. Cells were plated in 96-well plates at a density of 3,000 cells in 100 µL complete culture medium. Blank control wells containing the same volume of complete culture medium were included in each assay. The microplate was incubated overnight at 37°C until cells adhered to the plates. The cells were then washed with PBS and incubated with gefitinib or metformin, which were serially diluted in complete culture medium before use. The plate was further incubated for 48 hours. CCK-8 (10 µL) was added to each well. The OD of formazan at 450 nm was recorded every 0.5 hour until the OD reached 1.0–2.0. Six wells corresponded to each concentration of the abovementioned drugs. Cell viability was calculated as follows: cell viability = ([OD] test − [OD] control)/([OD] control − [OD] blank) ×100%. The IC50 value was calculated using GraphPad Prism 7 (GraphPad Software Inc, La Jolla, CA, USA).
Apoptosis detection
HNSCC cells were seeded into 6-well plates. The cells were incubated overnight and then treated with different agents under normoxic or hypoxic conditions. After 48 hours, the apoptotic cells were quantified by Annexin V-FITC/Propidium Iodide (PI) Cell Apoptosis Detection Kits (KeyGEN Biotech) according to the manufacturer’s protocol. Samples were detected with a FACSVerse flow cytometer (BD Biosciences), and the data obtained were analyzed by FlowJo. The apoptosis rate in every group was described by Annexin V+ PI+ (late apoptosis) + Annexin V+ PI− (early apoptosis).
Cell cycle analysis
HNSCC cells were seeded into 6-well plates. The cells were incubated overnight and then treated with different agents under normoxic or hypoxic conditions. After 48 hours, flow cytometry was used to detect the cell cycle using Cell Cycle Detection Kits (KeyGEN Biotech). Samples were detected with a FACSVerse flow cytometer (BD Biosciences), and the data obtained were analyzed by FlowJo.
In vivo assay
All the animals used in this study were from the Nanjing Biomedical Research Institute of Nanjing University (Nanjing, China). Mice were maintained under specific pathogen-free conditions (N=24, six mice/group). The Immunocompetent nude mice (BALB/cJNju-Foxn1nu/Nju) were treated with 30 μL HSC3 (2×107/mL; six mice/group) injected into the tongue. Tumor volume was measured every 3 days with calipers and calculated using the formula (volume = long diameter × short diameter2/2). After 10 days, the mice were orally gavaged every day with PBS, gefitinib (60 mg/kg), metformin (200 mg/kg), or gefitinib plus metformin. After 3 weeks of treatment, the mice were killed and the xenograft tumors were removed and fixed in formalin, paraffin-embedded, and H&E staining was performed. Immunostaining analysis for HIF-1α, cyclin D1, E-cadherin, and Ki67 protein expression was also carried out, and apoptosis was observed by TUNEL Apoptosis Detection Kits (Sigma-Aldrich Co) according to the manufacturer’s instructions. Hypoxic areas in the tumors were detected by Hypoxyprobe™-1 Plus Kits (EMD Millipore) as previously described.
Statistical analyses
All data are expressed as mean ± SD. Statistical differences were measured using an unpaired two-sided Student’s t-test or one-way ANOVA for multiple comparisons when appropriate. All statistical analyses were performed using SPSS 17.0 statistical software package (SPSS Inc, Chicago, IL, USA). Statistical significance was set at an alpha value of P<0.05. All the graphs were drawn using GraphPad Prism 7 (GraphPad Software Inc).
Results
Hypoxia induced HNSCC gefitinib treatment resistance in vitro
We first evaluated the expression level of EGFR in HNSCC. A total of six HNSCC cell lines (CAL27, Detroit562, JHU011, HN4, HSC3, and SCC4) were included in this experiment. Flow cytometry results showed that all six of the HNSCC cell lines exhibited high expression levels of EGFR (>90%) (Figure 1A), which is in accordance with previous reports.8 Then, we examined the ability of gefitinib to inhibit the proliferation of HNSCC cell lines. Cell viability assay results showed that gefitinib alone could inhibit the proliferation of HNSCC cell lines to a certain degree under normoxic conditions, illustrating the drug’s efficacy and effectiveness in tumor control (Figure 1B). The IC50 values of gefitinib in the HNSCC cell lines, as calculated by GraphPad Prism 7, are listed in Table 3. To further explore the effect of tumor hypoxia on the regulation of gefitinib sensitivity, HNSCC cell lines were cultured with gefitinib under normoxic and hypoxic conditions. The results of the cell viability assay demonstrated a significant increase in cell survival during hypoxia (P<0.05), suggesting the role of hypoxia in desensitizing cells to treatment with gefitinib (Figure 1C). The results of the cell apoptosis assay also showed a significant difference between the normoxic and hypoxic groups (Figure 1D, P<0.05), confirming that hypoxia can induce gefitinib treatment resistance in vitro.
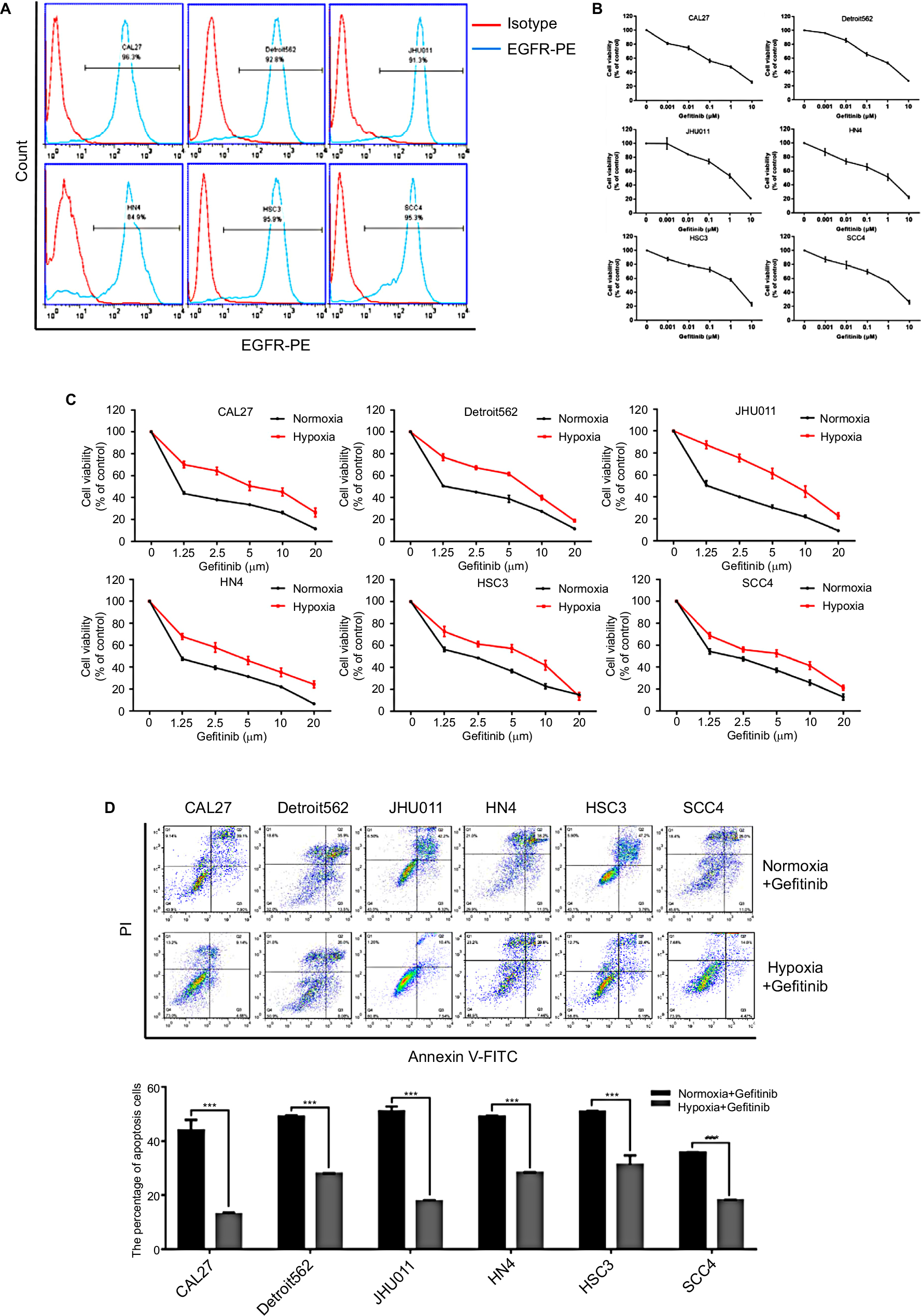 | Figure 1 Hypoxia induced HNSCC gefitinib treatment resistance. Notes: (A) The expression levels of EGFR in HNSCC cell lines were detected by flow cytometry analysis. (B) Cell viability of HNSCC cell lines treated with the indicated doses of gefitinib in normoxia for 48 hours was assessed with the CCK-8 assay. (C) After being treated with the indicated doses of gefitinib in normoxia or hypoxia for 48 hours, cell viability was assessed by the CCK-8 assay. (D) Cell apoptosis of HNSCC cell lines was assessed by flow cytometry following treatment with their own normoxia IC50 value in normoxia or hypoxia for 48 hours. Images are representative of three independent experiments (***P<0.001 compared with normoxia). Abbreviations: HNSCC, head and neck squamous cell carcinoma; CCK-8, cell counting kit-8; HNSCC, head and neck squamous cell carcinoma; PI, propidium iodide; PE, Phycoerythrin.. |
 | Table 3 IC50 values for HNSCC cell lines treated with gefitinib under normoxic conditions Abbreviation: HNSCC, head and neck squamous cell carcinoma. |
Hypoxia regulated HNSCC cell cycle progression and EMT in vitro and in vivo
Since hypoxia improved cell viability in cells co-cultured with gefitinib, we hypothesized that it might function via regulation of the cell cycle. Our in vitro experiment showed that the percentage of HNSCC cells in the G1 phase decreased to varying degrees in response to hypoxic stimulation and 1 µM gefitinib treatment (Figure 2A). It is well known that cyclin D1 is a key factor in cell cycle regulation; therefore, we chose two cell lines among the six available HNSCC cell lines, HN4 and HSC3, to detect the expression of cyclin D1 at the mRNA and protein level when the cells were cultured with gefitinib under hypoxic conditions. The results showed that hypoxia significantly promoted the expression of cyclin D1, which may lead to regulation of the cell cycle (Figure 2B). To further explore the relationship between hypoxia and cyclin D1 expression, 30 HNSCC clinical samples were included in this study. Immunohistochemical staining analysis demonstrated that the expression of HIF-1α was positively correlated with cyclin D1 (R=0.44, P=0.015) (Figure 2C). The representative pictures are shown in Figure 2D. Shien K et al reported that EMT is strongly related to gefitinib resistance;24 Kohnoh et al claimed that hypoxia can induce lung cancer cell EMT.25 Therefore, it was reasonable for us to further analyze the status of EMT treated with gefitinib in cells treated with gefitinib under hypoxic conditions. Encouragingly, compared with normoxic cells, hypoxic HNSCC cells demonstrated significantly higher levels of EMT markers (E-cadherin, vimentin, slug, twist, and α-SMA) at the mRNA and protein level in HN4 and HSC3 cell lines (Figure 2E and F). Further study showed that in clinical samples, the expression of E-cadherin was negatively correlated with the expression of HIF-1α (R=−0.587, P=0.001) (Figure 2G). These data indicated that hypoxia regulates the cell cycle and EMT in vitro and in vivo, which might partially explain the mechanisms behind hypoxia-induced gefitinib resistance.
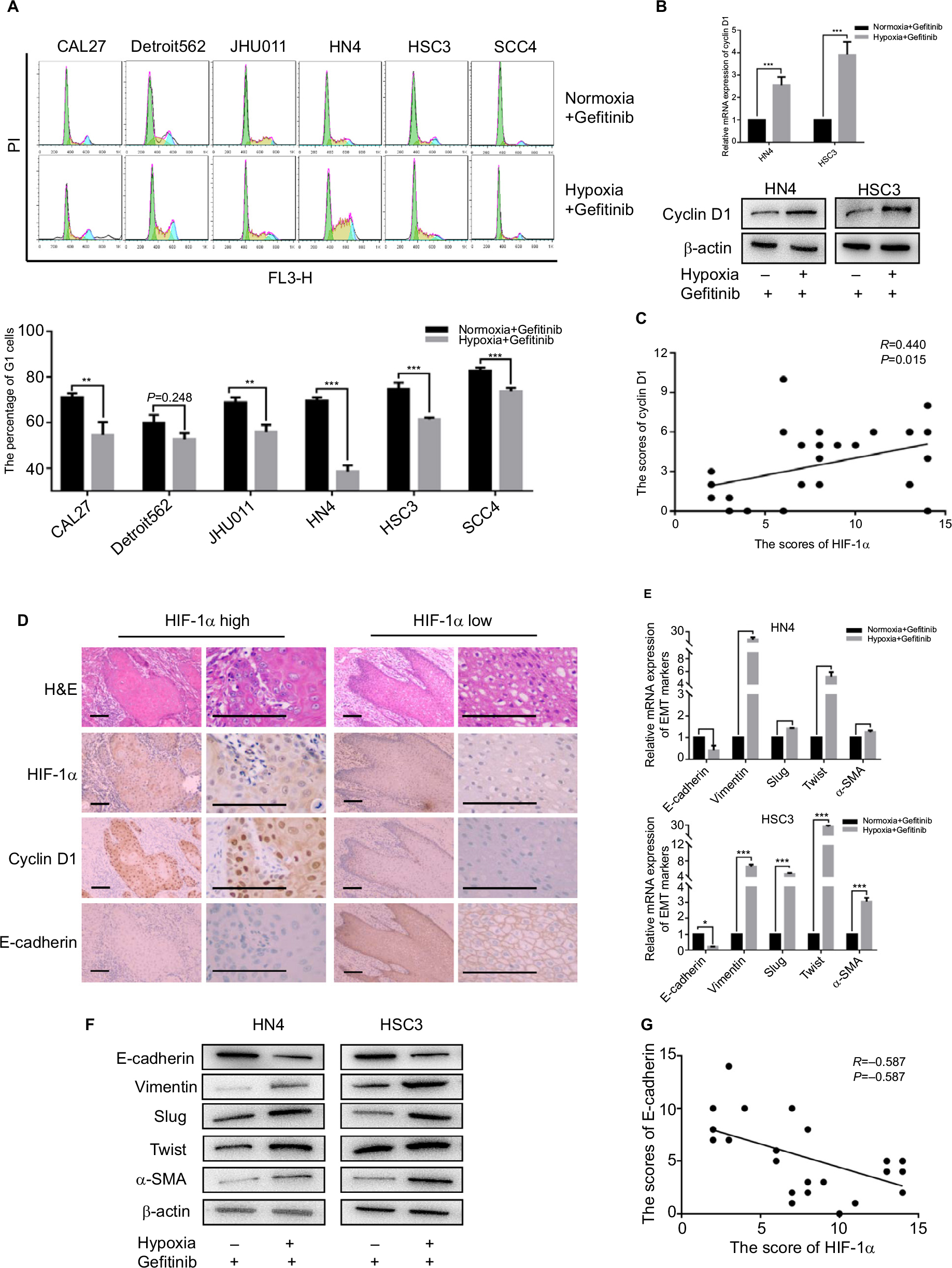 | Figure 2 Hypoxia promoted G1–S cell cycle progression and EMT. Notes: After being treated with 1 µM gefitinib in normoxia or hypoxia for 48 hours, (A) cell cycle was detected by flow cytometry. (B) The expression of cyclin D1 in mRNA and protein level was analyzed by qPCR and Western blot (**P<0.01 and ***P<0.001 compared with normoxia). (C) Relativity between the expression of HIF-1α and cyclin D1 in clinical HNSCC specimens (n=30, R=0.440, P=0.015). (D) Representative images of H&E staining and IHC of HIF-1α, cyclin D1, E-cadherin in HNSCC specimens (scale plate represents 200 µm). (E and F) Expression of EMT markers in mRNA and protein level was analyzed by qPCR and Western blot following treatment with their own IC50 value in normoxia or hypoxia for 48 hours (*P<0.05, **P<0.01, and ***P<0.001 compared with normoxia). (G) Relativity between the expression of HIF-1α and E-cadherin in clinical HNSCC specimens (n=30, R=0.587, P=0.001). Abbreviations: EMT, epithelial–mesenchymal transition; HNSCC, head and neck squamous cell carcinoma; HIF-1α, hypoxic inducible factor-1α; IHC, immunohistochemistry; PI, propidium iodide; qPCR, quantitative PCR; α-SMA, α-smooth muscle actin; PI, propidium iodide. |
Inhibition of cyclin D1 and EMT re-sensitized HNSCC to gefitinib treatment
Since we concluded that HNSCC tumor hypoxia might desensitize cells to gefitinib treatment via regulation of the cell cycle and induction of EMT, it was necessary to explore whether inhibition of such signaling might re-establish the efficacy and effectiveness of gefitinib under hypoxic conditions. Palbociclib, a well-known cyclin D1 inhibitor, was used in this study.26 First, we devaluated the effects of palbociclib on the inhibition of cell proliferation. The results demonstrated that palbociclib alone (0.1 µM) did not induce obvious cell cytotoxicity (Figure 3A). Thus, we chose this concentration for the subsequent experiments. For the same reason, we tested the effect of the EMT inhibitor – LY294002,27 and confirmed that a concentration of 1 µM was appropriate (Figure 3B). Our in vitro experiment confirmed that 0.1 µM palbociclib could significantly change the percentage of cells in the G1 phase during hypoxia in HN4 and HSC3 cells (P<0.05) (Figure 3C), and 1 µM LY294002 reversed tumor cell EMT during hypoxia (Figure 3D). To clearly demonstrate the final treatment effect, combined therapy (gefitinib plus palbociclib or gefitinib plus LY294002) was applied to compare the results to the single agent gefitinib results under normoxic and hypoxic conditions. The results proved that the combined treatment significantly enhanced the percentage of apoptotic cells (Figure 3F), while treatment with palbociclib (0.1 µM) or LY294002 (1 µM) alone did not induce significant cell apoptosis (Figure 3E). These results illustrate the synergistic effect of such agents and emphasize the importance of inhibiting the function of cyclin D1 and EMT reversal.
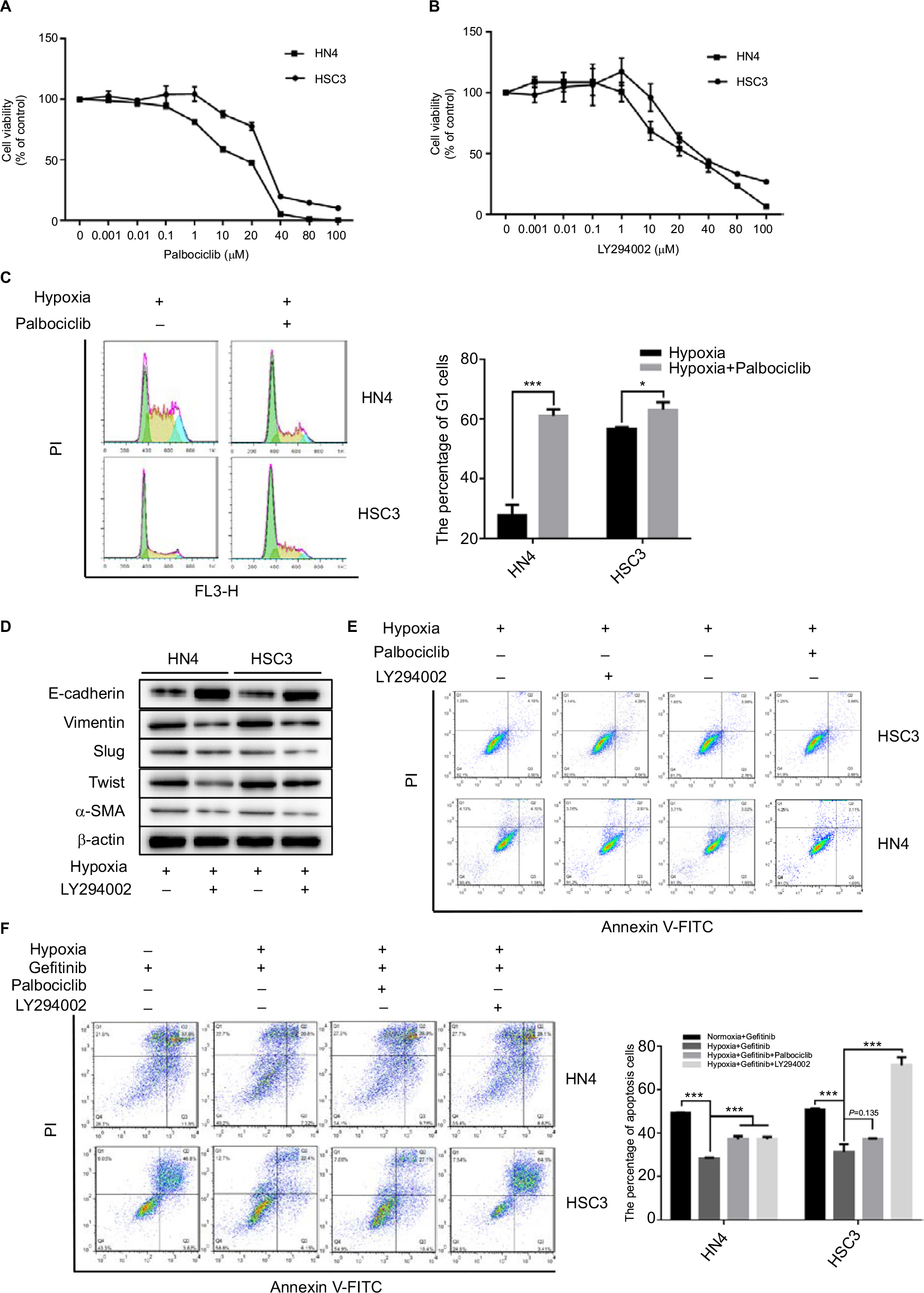 | Figure 3 The inhibitor of cyclin D1 and EMT could re-sensitize HNSCC cells to gefitinib in hypoxia. Notes: (A and B) HN4 and HSC3 cells were treated with the indicated doses of palbociclib or LY294002 for 48 hours. Cell viability was assessed by CCK-8 assay. (C) The cell cycle of HN4 and HSC3 was detected by flow cytometry following treatment with palbociclib (0.1 µM) in hypoxia for 48 hours (*P<0.05 and ***P<0.001 compared with control). (D and E) After treatment with LY294002 (1 µM) in hypoxia for 48 hours, the expression of EMT markers among HN4 and HSC3 cells was analyzed by Western blot; flow cytometry was used to detect the apoptosis condition. (F) The apoptosis condition of HN4 and HSC3 cells was assessed by flow cytometry following treatment with gefitinib (IC50 value) combined with palbociclib (0.1 µM) or LY294002 (1 µM) in hypoxia for 48 hours. Images are representative of three independent experiments (***P<0.001 compared with normoxia). Abbreviations: HNSCC, head and neck squamous cell carcinoma; CCK-8, cell counting kit-8; EMT, epithelial–mesenchymal transition; HNSCC, head and neck squamous cell carcinoma; PI, propidium iodide; α-SMA, α-smooth muscle actin; PI, propidium iodide. |
Metformin re-sensitized cells to hypoxia-induced gefitinib resistance in vitro and in vivo
There have been many reports of the versatile nature of metformin in the inhibition of cancer.28–30 Our previous study confirmed the effects of metformin in the suppression of HIF-1α expression and the subsequent enhancement of the therapeutic effects of cisplatin.23 In addition, metformin is a clinically approved drug which may have great potential for future clinical trials if the capacity to re-sensitize tumors to gefitinib is shown. Our in vitro experiment demonstrated that metformin (100 µM) could significantly decrease the expression of cyclin D1 under hypoxic conditions (Figure 4A) and change the percentage of G1 cells (Figure 4B) when cells were cultured with gefitinib. Meanwhile, metformin also decreased the expression of EMT markers during hypoxia along with the treatment of gefitinib (Figure 4C). More importantly, the combined use of metformin and gefitinib significantly inhibited cell growth and induced apoptosis in all six HNSCC cell lines, compared with metformin and gefitinib alone (Figure 4D and E, P<0.05). It has been reported that gefitinib alone might activate parallel signaling pathways such as PI3K/AKT, which may partially explain the resistance of EGFR TKIs.31 Hence, we explored the activation status of the aforementioned signaling pathway. Western blot clearly demonstrated that gefitinib could partially increase the expression of p-AKT, while no significant change in p-ERK was observed. Encouragingly, the addition of metformin might suppress HIF-1α expression along with p-AKT in both HSC3 and HN4 cells (Figure 4F). These results provide strong evidence that metformin can regulate oncogenic signaling to induce cell cycle arrest and EMT, as well as that gefitinib itself can activate cell signaling pathways such as PI3K/AKT. The combined treatment strategy may increase apoptosis and achieve far better therapeutic effects in vitro.
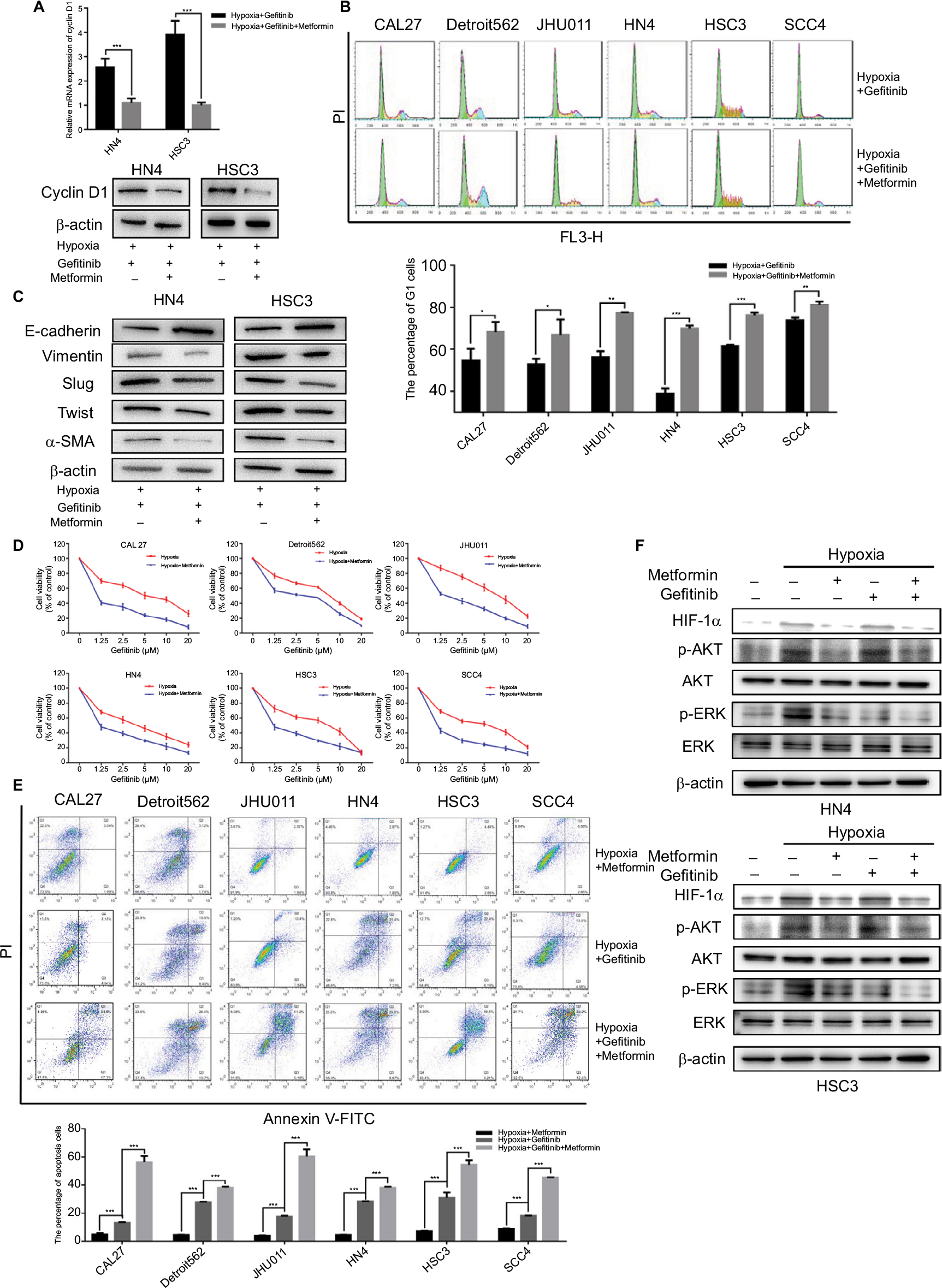 | Figure 4 Metformin sensitized HNSCC cells to gefitinib in hypoxic condition in vitro. Notes: (A and B) HN4 and HSC3 cells were treated with gefitinib (1 µM) alone or combined with metformin (100 µM) in hypoxia for 48 hours. Whole cell protein was immunoblotted with indicated antibodies, and the cell cycle was analyzed by flow cytometry (*P<0.05, **P<0.01, and ***P<0.001 compared with control). (C) The expression of EMT markers among HN4 and HSC3 cells was analyzed by Western blot following treatment with gefitinib (IC50 value) alone or in combination with metformin (100 µM) in hypoxia for 48 hours. (D) After being treated with the indicated doses of gefitinib combined with metformin (100 µM) in normoxia or hypoxia for 48 hours, cell viability was assessed by the CCK-8 assay. (E) The apoptosis condition of HNSCC cell lines was assessed by flow cytometry after treatment with gefitinib (IC50 value), metformin (100 µM), or in combination in hypoxia for 48 hours. Images are representative of three independent experiments (***P<0.001 compared with control). (F) HN4 and HSC3 cells were treated with gefitinib (IC50 value), metformin (100 µM), or in combination in normoxia or hypoxia for 48 hours. Whole cell protein was immunoblotted with indicated antibodies. Abbreviations: HNSCC, head and neck squamous cell carcinoma; CCK-8, cell counting kit-8; EMT, epithelial–mesenchymal transition; HIF-1α, hypoxic inducible factor-1α; HNSCC, head and neck squamous cell carcinoma; PI, propidium iodide α-SMA, α-smooth muscle actin; PI, propidium iodide; HIF-1α, hypoxic inducible factor-1α. |
To further examine the in vivo therapeutic effects of our combined treatment strategy, orthotopic HNSCC animal models were established. Ten days after injection of 30 µL HSC3 (2×107/mL), 24 mice were evenly distributed into four groups to receive PBS, metformin, gefitinib, or metformin plus gefitinib. The dose of gefitinib (60 mg/kg/day) and metformin (200 mg/kg/day) used in this study was selected based on studies of Li et al and Al-Wahab et al.32,33 Following treatment, tumor volume and body weight were measured every 3 days. The mice were killed 3 weeks after the treatment (Figure 5A). Our in vivo study revealed a significant increase in body weight in the combined treatment group compared with the single agent and control groups (Figure 5B), which might explain why the combined treatment could exert better tumor inhibition effect in the tongue, and it allowed the mice to have more food intake. Tumor volume was significantly decreased in the combined treatment group. Interestingly, we also found that metformin alone exerted antitumor effects to some degree when compared with the control group (P<0.05). This may be explained by the effect of metformin on the inhibition of hypoxic signaling (Figure 5C). The IHC results supported our hypothesis that the combined treatment may inhibit the expression of HIF-1α and decrease the expression of cyclin D1 and Ki-67. Reversal of EMT, reflected by increased expression of E-cadherin and significant positive TUNEL staining, was also observed (Figure 5D). All these data strongly indicated that metformin might have a synergistic effect with gefitinib in the treatment of HNSCC.
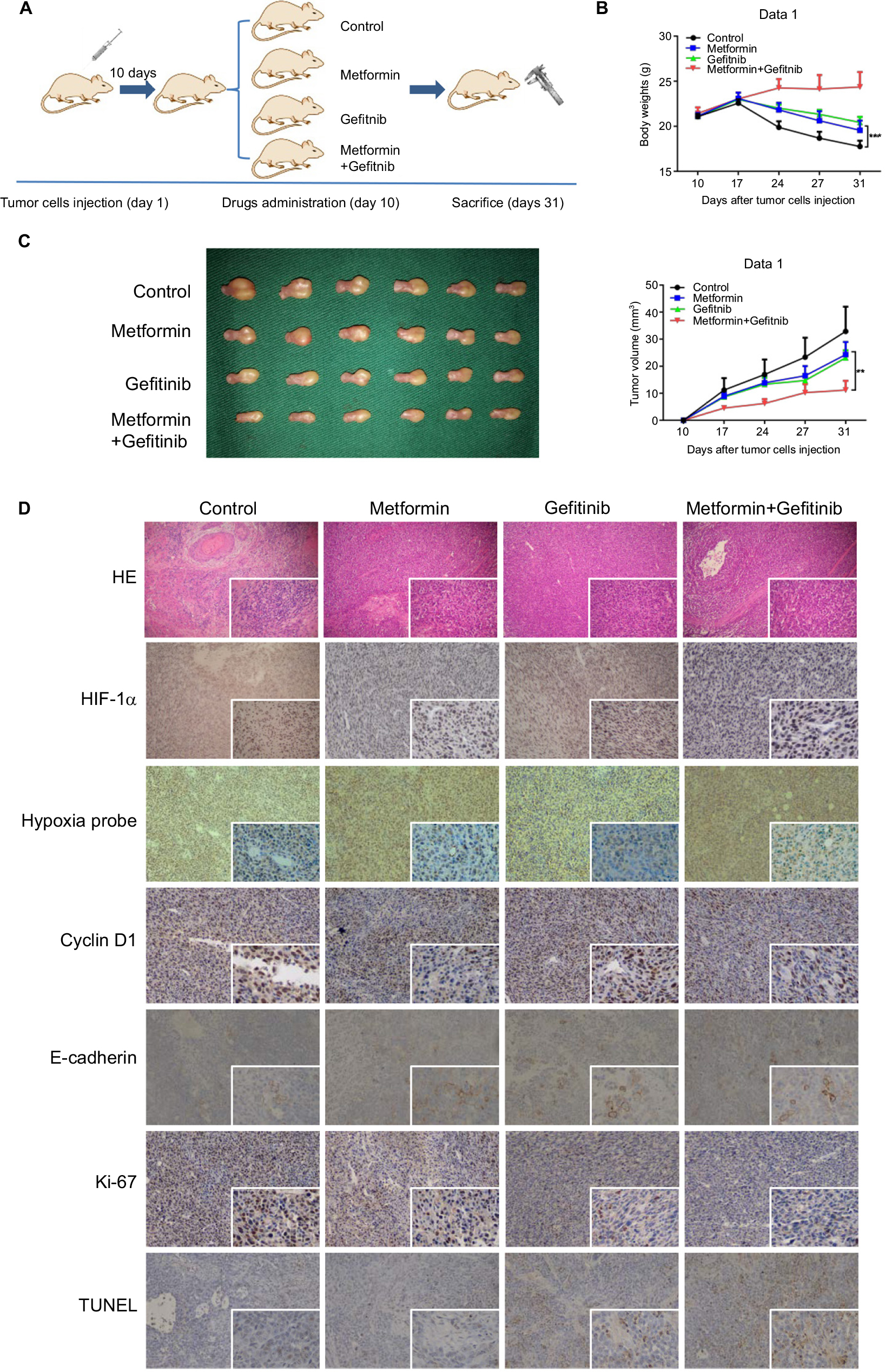 | Figure 5 Metformin enhanced the chemotherapeutic efficacy of gefitinib in vivo. Notes: (A) Schematic figure of the orthotopic xenograft model used in this study. (B) Body weight (g) of mice treated with PBS, metformin (200 mg/kg), gefitinib (60 mg/kg), and metformin plus gefitinib. (C) Macroscopic appearance of the tumors on day 31 after drug administration and tumor volume (mm3) during treatment. (D) Paraffin-embedded sections (4 mm) from tumor tissues were stained for HIF-1α, hypoxia probes, cyclin D1, E-cadherin, Ki67, and TUNEL using IHC (100× and 400×). **P<0.01, and ***P<0.001. Abbreviations: HIF-1α, hypoxic inducible factor-1α; IHC, immunohistochemistry. |
Discussion
Currently, gefitinib is widely regarded as one of the most effective molecular target therapeutic drugs used in lung cancers, especially non-small-cell lung cancer (NSCLC).34 Gefitinib targets EGFR by competitively combining with ATP in the receptor tyrosine kinase of the catalytic center of the Mg-ATP binding site. Consequently, gefitinib inhibits tyrosine kinase activity, blocks intracellular tyrosine kinase phosphorylation, and suspends EGFR signal conduction, thus blocking the growth of tumor cells.35 Despite the effectiveness and efficacy reported in lung cancer, gefitinib has not been suggested as a possible alternative approach in HNSCC treatment guidelines, especially as a first-line drug. Gefitinib is also not yet approved by the FDA, which might be explained by the observed treatment resistance and the limited benefits gained by patients in reported clinical trials.11 Thus, a deeper understanding of the mechanisms behind gefitinib treatment resistance is crucially important. In the present study, HNSCC tumor hypoxia was proven to be one of the major causes of treatment resistance, which is in accordance with previous reports.36–38 An et al reported that hypoxia played an important role in gefitinib adaptive resistance in NSCLC due to the inductive expression of IL-6.39 Murakami et al reported that hypoxia could increase the proportion of cancer stem cells in lung cancer, which in return would generate more gefitinib-resistant cells.17 Our work demonstrated that hypoxia induced HNSCC gefitinib resistance by regulating the cell cycle and inducing EMT.
Due to our earlier findings, overcoming hypoxia-induced gefitinib treatment resistance in HNSCC has been our primary goal. Basic principles to re-sensitize tumor cells to gefitinib treatment include simplicity and practicability in the clinic. Recently, many studies have confirmed the anticancer effects of metformin.40–42 In addition, our previous work has also revealed that metformin inhibited the expression of HIF-1α through interference with the nuclear factor-κB pathway under hypoxic conditions and then sensitized oral squamous cell carcinoma to cisplatin.23 Therefore, we hypothesized that the combination of metformin with gefitinib may lead to a better curative effect during hypoxia in HNSCC. In line with our research, there are clinical trials currently in progress utilizing treatment with metformin and gefitinib (NCT01864681, NCT03071705, and NCT01997775; www.ClinicalTrials.gov) in lung cancer patients. To our knowledge, there have been no similar clinical trials yet in HNSCC. In the present study, we showed that metformin could overcome hypoxia-induced gefitinib resistance, which is favorable evidence for possible future application in HNSCC.
Although metformin is a commonly used hypoglycemic drug, the safety and possible systemic toxicity are still of major concern. In our study, the dose of metformin (100 μmol/L) is higher than that used in diabetic patients. Moreover, Iliopoulos et al proved that metformin tends to accumulate in tissues rather than blood; thus, a range of drug concentrations from 1 to 10 mmol/L would not affect normal blood sugar metabolism.43 In addition, Morgillo et al validated the safety of 1,500 mg metformin per day in nondiabetic patients. This dose corresponds to ~308 mg/kg in the mouse, which is higher than what we used in our study.44,45 Thus, we have provided strong evidence for the effectiveness and safety of metformin in our study.
Despite the encouraging outcomes obtained, challenges remain. Drug resistance is a consequence of multiple driving factors. It is difficult to determine the contribution of the microenvironment in this process, especially tumor hypoxia. Therefore, the strategy of inhibiting hypoxia signaling to overcome barriers is unrealistic, and more trials of combined therapy are desperately needed. In addition, although metformin is reported to have anticancer effects, it cannot be included as a first-line therapeutic drug. Hence, it is necessary to further explore the mechanisms of metformin and more novel treatment strategies in the field of HNSCC molecular target therapy.
Conclusion
This study demonstrates that metformin has the potential to synergize with gefitinib to overcome hypoxia-induced treatment resistance in HNSCC via cell cycle regulation and EMT reversal. Thus, a combination of metformin with EGFR TKIs is implicated as a promising therapeutic strategy for HNSCC.
Acknowledgments
This study was supported by Jiangsu Provincial Key Research and Development Program (BE2017741), Nanjing Foundation for Development of Science and Technology (2017sc512031 and 201605042), Jiangsu Provincial Natural Science Foundation (BK20150089 and BK 20160051), the Distinguished Young Investigator Project of Nanjing (JQX14010), and Medical Science and Technology Development Foundation (YKK15116). We would like to thank LetPub (www.letpub.com) for providing linguistic assistance during the preparation of this manuscript.
Author contributions
WH and ZTL conceived and designed the study. XTY, ZW, JQX, CHS, CCT, WGX and YFW performed the experiments and analyzed the data. XTY and ZW wrote the paper; WH and ZTL reviewed and edited the manuscript. All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66(4):271–289. | ||
Marur S, Forastiere AA. Head and Neck Squamous Cell Carcinoma: Update on Epidemiology, Diagnosis, and Treatment. Mayo Clin Proc. 2016;91(3):386–396. | ||
Metro G. EGFR targeted therapy for lung cancer: are we almost there? Transl Lung Cancer Res. 2018;7(Suppl 2):S142–S145. | ||
Bhatti M, Ippolito T, Mavis C, et al. Pre-clinical activity of targeting the PI3K/Akt/mTOR pathway in Burkitt lymphoma. Oncotarget. 2018;9(31):21820–21830. | ||
Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front Immunol. 2018;9:978. | ||
Toschi L, Rossi S, Finocchiaro G, Santoro A. Non-small cell lung cancer treatment (r)evolution: ten years of advances and more to come. Ecancermedicalscience. 2017;11:787. | ||
Kalyankrishna S, Grandis JR. Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol. 2006;24(17):2666–2672. | ||
Soulieres D, Senzer NN, Vokes EE, Hidalgo M, Agarwala SS, Siu LL. Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neck. J Clin Oncol. 2004;22(1):77–85. | ||
Magnes T, Egle A, Greil R, Melchardt T. Update on squamous cell carcinoma of the head and neck: ASCO annual meeting 2017. Memo. 2017;10(4):220–223. | ||
Cohen EE, Rosen F, Stadler WM, et al. Phase II trial of ZD1839 in recurrent or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol. 2003;21(10):1980–1987. | ||
Maseki S, Ijichi K, Tanaka H, et al. Acquisition of EMT phenotype in the gefitinib-resistant cells of a head and neck squamous cell carcinoma cell line through Akt/GSK-3β/snail signalling pathway. Br J Cancer. 2012;106(6):1196–1204. | ||
Jameson MJ, Beckler AD, Taniguchi LE, et al. Activation of the insulin-like growth factor-1 receptor induces resistance to epidermal growth factor receptor antagonism in head and neck squamous carcinoma cells. Mol Cancer Ther. 2011;10(11):2124–2134. | ||
Jia Y, Yun CH, Park E, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534(7605):129–132. | ||
Ocaña MC, Martínez-Poveda B, Quesada AR, Medina MÁ. Metabolism within the tumor microenvironment and its implication on cancer progression: An ongoing therapeutic target. Med Res Rev. Epub 2018 May 22. | ||
Ma S, Pradeep S, Hu W, Zhang D, Coleman R, Sood A. The role of tumor microenvironment in resistance to anti-angiogenic therapy. F1000Res. 2018;7:326. | ||
Murakami A, Takahashi F, Nurwidya F, et al. Hypoxia increases gefitinib-resistant lung cancer stem cells through the activation of insulin-like growth factor 1 receptor. PLoS One. 2014;9(1):e86459. | ||
Minakata K, Takahashi F, Nara T, et al. Hypoxia induces gefitinib resistance in non-small-cell lung cancer with both mutant and wild-type epidermal growth factor receptors. Cancer Sci. 2012;103(11):1946–1954. | ||
Han S, Xu W, Wang Z, et al. Crosstalk between the HIF-1 and Toll-like receptor/nuclear factor-κB pathways in the oral squamous cell carcinoma microenvironment. Oncotarget. 2016;7(25):37773–37789. | ||
Wahdan-Alaswad RS, Edgerton SM, Salem HS, Thor AD. Metformin Targets Glucose Metabolism in Triple Negative Breast Cancer. J Oncol Transl Res. 2018;4(1):pii:129. | ||
Tseng CH. Metformin and risk of developing nasopharyngeal cancer in patients with type 2 diabetes mellitus. Metabolism. 2018;85:223–226. | ||
Woo SH, Yang LP, Chuang HC, et al. Down-regulation of malic enzyme 1 and 2: Sensitizing head and neck squamous cell carcinoma cells to therapy-induced senescence. Head Neck. 2016;38(Suppl 1):E934–E940. | ||
Qi X, Xu W, Xie J, et al. Metformin sensitizes the response of oral squamous cell carcinoma to cisplatin treatment through inhibition of NF-κB/HIF-1α signal axis. Sci Rep. 2016;6:35788. | ||
Shien K, Toyooka S, Yamamoto H, et al. Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell-like properties in cancer cells. Cancer Res. 2013;73(10):3051–3061. | ||
Kohnoh T, Hashimoto N, Ando A, et al. Hypoxia-induced modulation of PTEN activity and EMT phenotypes in lung cancers. Cancer Cell Int. 2016;16:33. | ||
Fry DW, Harvey PJ, Keller PR, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3(11):1427–1438. | ||
Reka AK, Kuick R, Kurapati H, Standiford TJ, Omenn GS, Keshamouni VG. Identifying inhibitors of epithelial–mesenchymal transition by connectivity map-based systems approach. J Thorac Oncol. 2011;6(11):1784–1792. | ||
Camacho L, Dasgupta A, Jiralerspong S. Metformin in breast cancer - an evolving mystery. Breast Cancer Res. 2015;17:88. | ||
You A, Cao M, Guo Z, et al. Metformin sensitizes sorafenib to inhibit postoperative recurrence and metastasis of hepatocellular carcinoma in orthotopic mouse models. J Hematol Oncol. 2016;9:20. | ||
El-Arabey AA. New insight for metformin against bladder cancer. Genes Environ. 2017;39:13. | ||
Janmaat ML, Kruyt FA, Rodriguez JA, Giaccone G. Response to epidermal growth factor receptor inhibitors in non-small cell lung cancer cells: limited antiproliferative effects and absence of apoptosis associated with persistent activity of extracellular signal-regulated kinase or Akt kinase pathways. Clin Cancer Res. 2003;9(6):2316–2326. | ||
Al-Wahab Z, Mert I, Tebbe C, et al. Metformin prevents aggressive ovarian cancer growth driven by high-energy diet: similarity with calorie restriction. Oncotarget. 2015;6(13):10908–10923. | ||
Li L, Han R, Xiao H, et al. Metformin sensitizes EGFR-TKI-resistant human lung cancer cells in vitro and in vivo through inhibition of IL-6 signaling and EMT reversal. Clin Cancer Res. 2014;20(10):2714–2726. | ||
Onn A, Tsuboi M, Thatcher N. Treatment of non-small-cell lung cancer: a perspective on the recent advances and the experience with gefitinib. Br J Cancer. 2004;91(Suppl 2):S11–S17. | ||
Tetsu O, Hangauer MJ, Phuchareon J, Eisele DW, Mccormick F. Drug Resistance to EGFR Inhibitors in Lung Cancer. Chemotherapy. 2016;61(5):223–235. | ||
Stegeman H, Kaanders JH, Wheeler DL, et al. Activation of AKT by hypoxia: a potential target for hypoxic tumors of the head and neck. BMC Cancer. 2012;12:463. | ||
Vigneswaran N, Wu J, Song A, Annapragada A, Zacharias W. Hypoxia-induced autophagic response is associated with aggressive phenotype and elevated incidence of metastasis in orthotopic immunocompetent murine models of head and neck squamous cell carcinomas (HNSCC). Exp Mol Pathol. 2011;90(2):215–225. | ||
Löck S, Perrin R, Seidlitz A, et al. Residual tumour hypoxia in head-and-neck cancer patients undergoing primary radiochemotherapy, final results of a prospective trial on repeat FMISO-PET imaging. Radiother Oncol. 2017;124(3):533–540. | ||
An SM, Lei HM, Ding XP, et al. Interleukin-6 identified as an important factor in hypoxia- and aldehyde dehydrogenase-based gefitinib adaptive resistance in non-small cell lung cancer cells. Oncol Lett. 2017;14(3):3445–3454. | ||
Xu S, Yang Z, Jin P, et al. Metformin suppresses tumor progression by inactivating stromal fibroblasts in ovarian cancer. Mol Cancer Ther. 2018;17(6):1291–1302. | ||
Sui X, Xu Y, Wang X, et al. Metformin: A Novel but Controversial Drug in Cancer Prevention and Treatment. Mol Pharm. 2015;12(11):3783–3791. | ||
Sivalingam V, Mcvey R, Gilmour K, et al. A presurgical window-of-opportunity study of metformin in obesity-driven endometrial cancer. Lancet. 2015;385(Suppl 1):S90. | ||
Iliopoulos D, Hirsch HA, Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011;71(9):3196–3201. | ||
Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22(3):659–661. | ||
Morgillo F, Fasano M, della Corte CM, et al. Results of the safety run-in part of the METAL (METformin in Advanced Lung cancer) study: a multicentre, open-label phase I-II study of metformin with erlotinib in second-line therapy of patients with stage IV non-small-cell lung cancer. ESMO Open. 2017;2(2):e000132. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.