")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 15
Metformin Inhibits HaCaT Cell Proliferation Under Hyperlipidemia Through Reducing Reactive Oxygen Species via FOXO3 Activation
Authors Zhang L, Liu X, Huang M, Wang R, Zhu W, Li Y, Shen L, Li C
Received 2 April 2022
Accepted for publication 11 July 2022
Published 22 July 2022 Volume 2022:15 Pages 1403—1413
DOI https://doi.org/10.2147/CCID.S368845
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Jeffrey Weinberg
Li Zhang,1 Xiaoling Liu,1 Min Huang,1 Rui Wang,1 Wenwei Zhu,1 Yu Li,2 Lin Shen,1 Chengxin Li1
1Department of Dermatology, The First Medical Center of Chinese PLA General Hospital, Beijing, 100853, People’s Republic of China; 2Department of Dermatology, The Fifth Medical Center of Chinese PLA General Hospital, Beijing, 100853, People’s Republic of China
Correspondence: Chengxin Li, Department of Dermatology, The First Medical Center of Chinese PLA General Hospital, Beijing, 100853, People’s Republic of China, Tel +86 15210866656, Email [email protected]
Purpose: Metformin (MET) has been proved to be effective for the treatment of psoriasis. The mechanisms of its action under the hyperlipidemia have yet to be fully elucidated. Here, we investigated the effect of metformin on the cell proliferation induced by hyperlipidemia and the underlying mechanism in immortalized human keratinocyte cell line (HaCat).
Methods: Wild-type or FOXO3 knockdown HaCat cells were treated with free fatty acids (FFA) for 10 days and then co-treated with metformin for another 4 days. Triglyceride (TG) level, cell viability, proliferation, apoptosis, antioxidant enzymes, reactive oxygen species (ROS) levels, as well as the transcription activity of FOXO3 were analyzed.
Results: Metformin decreased HaCaT cell proliferation and induced cell apoptosis after FFA treatment. Metformin was found to significantly increase the expressions and the activities of superoxide dismutase (SOD) as well as catalase (CAT), and reduced the reactive oxygen species (ROS) level. Metformin significantly promoted the autophagy and increase FOXO3 protein level in the nucleus under hyperlipidemia. However, all of the effects from metformin were partially blocked by FOXO3 knockdown.
Conclusion: This study demonstrated that under the hyperlipidemia, metformin has significant antiproliferation and proapoptosis effects by reducing ROS level as well as increasing autophagy. All of these effects from metformin were through FOXO3-dependent pathway.
Keywords: reactive oxygen species, ROS, apoptosis, autophagy, forkhead box O3, FOXO3
Introduction
Psoriasis is a kind of inflammatory skin disease that influences approximately 3% of the population worldwide. The characteristic phenotypes of psoriasis are the progressive erythematous papules as well as overlying scaly plaques.1 The pathomechanism underlying psoriasis is thought to involve the acceleration of cell proliferation and the rapid migration of keratinocytes from the basal layer to the granular layer.2 It is reported that various factors, such as hereditary factors, environmental factors, immune responses, and metabolic disturbance, are involved in the pathological progress of psoriasis.3–5 More and more clinical studies report that metabolic syndrome contributes to the high morbidity of psoriasis.6–9 A retrospective study has shown that in psoriasis patients, there is a high incidence of metabolic syndrome. The treatment to metabolic syndrome was useful to psoriasis.9 Clinical studies report that patients with psoriasis show significant dyslipidemia.10 Further studies found that elevated cholesterol, triglyceride, and low-density lipoprotein (LDL) promote the adhesion and aggregation of platelets. Activated platelets could help foster an inflammatory environment by the release of biologically active molecules to aggravate psoriasis.11–13 These studies illustrate that dyslipidemia might be one of the pathogenic factors for the psoriasis. Further understanding the mechanism of dyslipidemia triggering this chronic skin development would prompt the new methods and drugs for the psoriasis.
Excessive and long-term lipid accumulation could induce oxidative stress and inflammation in the cell. It has been demonstrated oxidative stress contributed to the development of psoriasis.14,15 Impaired antioxidant system, together with excessive reactive oxygen species (ROS) production, is involved in the pathogenesis of psoriasis. Exogenous harmful factors including cigarette smoking, ethyl alcohol, air pollution, as well as pathogenic microorganisms (virus, bacteria, etc) could damage keratinocytes by excessive ROS.16 Several studies have found there was an raised ROS level in psoriatic skin.14 The strategy to reduce the oxidative stress was effective for the treatment of psoriasis.17 It has been widely accepted that lipid accumulation increases the level of reactive oxygen species (ROS). Furthermore, it has been demonstrated that hydrogen peroxide (H2O2) could promote cell proliferation in the tissue regeneration as well as embryonic development.18 Therefore, we hypothesize that a mild increase of ROS induced by hyperlipidemia in keratinocytes stimulates the cell proliferation. A series of antioxidative enzymes, including SOD and CAT, comprise the antioxidant defense system, which scavenge excessive ROS to maintain the normal function of the cell. We speculated that in psoriatic skin, the expressions of several antioxidative enzymes decreased, which would be responsible for the increased ROS level. As a consequence, increased ROS might aggravate the psoriasis through stimulating keratinocytes proliferation.
It was well accepted that long-term hyperlipidemia inhibited the autophagy in a series of metabolic diseases. Autophagy is a self-digestion process in the cells that play a crucial role to balance the catabolism and anabolism. Basal level of autophagy in a cell helps to maintain its homeostatic state and normal function under stressful conditions. During the early stage of nonalcoholic fatty liver disease (NAFLD), lipid would trigger autophagy upregulation to decrease the lipid accumulation. However, excessive lipid accumulation in the liver for a long time would block the autophagy process.19 It has been well established that many therapeutic strategies induce apoptosis through promoting autophagy in tumor cells. Through inducing autophagy, rapamycin ameliorated skin inflammation in a psoriatic mouse model.20 Therefore, we speculated that increasing autophagy in metformin-treated keratinocytes under hyperlipidemia would induce apoptosis and ameliorate psoriasis effectively.
Psoriasis and psoriatic arthritis are multifactorial chronic disorders whose etiopathogenesis essentially derives from the alteration of several signaling pathways and the co-occurrence of genetic, epigenetic and non-genetic susceptibility factors that altogether affect the functional and structural property of the skin.21 As a crucial transcription factor, FOXO3 regulates a wide spectrum of genes, which are involved in various biological processes, including proliferation, differentiation, apoptosis, immunoregulation as well as metabolism.22 All of these might contribute to the psoriasis. FOXO3 initiates the gene expression of some cell cycle kinase inhibitors such as p16, p21 and p27 to inhibit cell proliferation. In addition, under more oxidizing conditions, to reduce the level of ROS, FOXO3 regulates a series of antioxidant genes, including CAT and SOD2.23 It is reported that FOXO3 is involved in several autophagy pathways to repress cell proliferation. In response to the accumulation of stress, FOXO3 may activate both the ubiquitin-proteasome pathway and the transcription of autophagy-related genes such as those encoding LC3 and BNIP3, inducing the formation of autophagy.24 As these, we speculated that FOXO3 would be a drug target for regulating cell proliferation and apoptosis in psoriasis.
Metformin is utilized as a first-line antidiabetic agent for the treatment of type 2 diabetes mellitus (T2DM), as it has been shown to improve metabolic homeostasis including hyperglycemia and dyslipidemia. FOXO3 pathway plays a role in the effects of metformin. Metformin inhibits hepatocellular carcinoma development by inducing apoptosis through regulating FOXO3.25 In immune cells, metformin was shown to attenuate ROS via FOXO3 activation.26 Previous studies have demonstrated that the long-term use of metformin was associated with a reduced risk of psoriasis.27,28 However, further studies are required to identify the potential mechanism.
In our current study, we demonstrated for the first time that under hyperlipidemia conditions, metformin suppressed cell proliferation and induced apoptosis in free fatty acids (FFA)-treated human keratinocyte via FOXO3-dependent pathway. Metformin activated FOXO3 to upregulate expressions of antioxidative enzymes SOD2 and CAT to reduce ROS. Consequently, it would block the proliferation caused by ROS. Meanwhile, FOXO3 activation induced the apoptosis by increasing autophagy. Both of these were responsible for the alleviation of psoriasis under hyperlipidemia.
Materials and Methods
Cell Culture
HaCaT cells purchased from Cell Culture Unit of Shanghai Science Academy (Shanghai, China) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) (Hyclone, UT, USA) with 10% fetal bovine serum (FBS) (Gibco, Invitrogen, NY, USA) and 1% penicillin/streptomycin (Gibco, Invitrogen, NY, USA) in a 5% CO2 atmosphere at 37 °C. FFA was the mixture of oleate (Sigma-Aldrich, MO, USA) and palmitate (Sigma-Aldrich, MO, USA). FFA was used to make the cell model of lipid accumulation.29 Oleate and palmitate were dissolved in 0.5 M BSA to make a 4 mM total FFA mixture (Oleate:palmitate, 2:1 molar ratio). The final concentration of FFA in the working solution was 400 μM. The cells were treated with FFA for 10 days, and then the metformin was added to co-treat the cells for additional 4 days.
Cell Viability Assay
HaCaT cells were treated with FFA and metformin. Before the measurement, cells were seeded into 96‐well plates and incubated for 24 hours, and then 10 μL of Cell Counting Kit‐8 (CCK8) (Topscience, Shanghai, China) solution was added and incubated for 1 hour. Absorbance (450 nm) was measured using a quantitative automatic microplate reader.
Apoptosis Assay
Apoptosis was measured with a apoptosis kit (Beyotime Biotechnology, Shanghai, China). The experimental procedures were strictly according to the instruction from the vendor. In brief, HaCaT cells were detached and washed with PBS for 3 times. Cells were stained with binding buffer containing Annexin V for 15 min and the PI for 5 min at room temperature. Cells were analyzed using BD Accuri C6 (BD biosciences, CA, USA).
SOD Activity and CAT Activity Assay
Activities of SOD and CAT were measured by Total Superoxide Dismutase Assay Kit and Catalase Assay Kit, respectively (Beyotime Biotechnology, Shanghai, China). The experimental procedures were strictly according to the instruction from the vendor.
Western Blotting
The Western blot was performed using specific primary antibodies. SOD2 (Cat# 13141) and CAT (Cat#12980) antibodies were from Cell Signaling Technology (Beverly, MA, USA). Anti-LC3 (NB600-1384) antibody was from Novus Biologicals (Centennial, CO, USA). P62 (ab240635) antibody was from Abcam (Abcam, Cambridge, UK). Tubulin (MB0009) antibody was from Bioworld Technology (Louis Park, MN, USA). RIPA lysis buffer was used to prepare rat tissue and cell lysates. Ten to twenty micrograms of protein was loaded and separated on SDS-PAGE gels. Fractionated proteins were then transferred to nitrocellulose membranes, blocked in 5% nonfat milk for 2 h, and probed overnight with primary antibodies. Immunoblots were washed three times (5 min each) with TBS containing 0.1% Tween 20 and then incubated with horseradish peroxidase conjugated secondary antibody for 2 h. Blots were washed four times (5 min each) with TBS containing 0.1% Tween 20, developed in enhanced chemiluminescent reagent (Cat. No. WBKLS0500, MilliporeSigma), and visualized with an image analyzer Quantity One System (Bio-Rad).
Quantitative RT-PCR
Total RNA was isolated from cells using FastPure Cell/Tissue Total RNA Isolation Kit (Vazyme, Nanjing, China). Then, 1μg of RNA was reverse-transcribed with HiScript III First Strand cDNA Synthesis Kit (Vazyme, Nanjing, China). The following PCR primers were used: human CDKN2A (p16), 5͑ʹ-GACCTGGCTGAGGAGCTG-3ʹ (forward) and 5ʹ͑-GCATGGTTACTGCCTCTGGT-3ʹ (reverse); human CDKN1A (p21), 5ʹ-CATTTGGTGGACCCAAAGAC-3ʹ (forward) and 5ʹ-TTGCAGGTCGCTTCCTTATT-3ʹ (reverse); human actin, 5ʹ-GAGCGCGGCTACAGCTT-3ʹ (forward) and 5ʹ-TCCTTAATGTCACGCACGATTT-3ʹ (reverse). Quantitative RT-PCR analysis was performed with the ChamQ SYBR qPCR Master Mix (Vazyme, Nanjing, China) and the CFX Connect Realtime System (BioRad, CA, USA). Relative gene expression was obtained after normalization to actin expression. Fold differences in comparisons were expressed as relative mRNA levels using the 2−ΔΔCt method.
Intracellular ROS Measurement
Intracellular ROS was measured by flow cytometry using DCFH-DA (Sigma-Aldrich, MO, USA) as the fluorescent probe. Briefly, after the treatment, the cells were washed once with PBS and then incubated with 10μM DCFH-DA in fresh medium for 40 min in the cell culture incubator. After the incubation, the cells were trypsinized and collected, and then washed three times with PBS before flow cytometry analysis (Excitation 488 nm/Emission 525 nm for DCFH-DA).
Autophagic Flux Quantification
RFP-GFP-LC3 adeno virus (Orbitalgene, Xi`an, China) was used to perform the autophagic flux.30 Briefly, 48 hours before the measurement, HaCaT cells were infected with RFP-GFP-LC3 adeno virus. As reported, the red only puncta were autolysosome, yellow was early autophagosome (red and green = yellow). In acidic pH condition, the GFP fluorescence decreased while RFP was stable The readout for autophagic flux was set as the conversion of yellow puncta to red puncta. The puncta were analyzed with a confocal laser scanning microscope (LSM800, Carl Zeiss), using a 40× oil immersion objective. The fluorescence of the puncta was quantified with the Image J program.
shRNA Lentivirus Knockdown
The FOXO3 shRNA and scramble shRNA lentiviruses were obtained from Orbitalgene Co. Ltd (Orbitalgene, Xi`an, China). The shRNA targeting the FOXO3 coding sequence was as follows: FOXO3-shRNA (NM_001455.4): 5′- CCGGCACCATGAATCTGAATG −3′, Scramble-shRNA: CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG. Scramble shRNA was treated as a negative control group. HaCat cells were infected with FOXO3 shRNA and scramble shRNA lentivirus for 48h, and the cells were selected with 1 µg/mL puromycin for 48h according to the manufacturer’s protocol. Stably FOXO3 knockdown cell line was confirmed by Western blotting.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism software (version 6.01). Data are presented as mean ± SEM. One-way or two-way ANOVA were performed and significance was accepted at P<0.05.
Results
Metformin Inhibited FFA-Treated Human Keratinocyte Proliferation
Human HaCaT cells were treated with FFA for 10 days and then cotreated with different concentrations of metformin for another 4 days. FFA treatment increased the TG level in HaCaT cells, which suggested that excessive lipid accumulation. Metformin significantly reduced the TG in the cell in a dose-dependent manner (Figure 1A, *p < 0.05). Data from CCK8 assay showed that FFA significantly increased cell viability, while metformin hindered cell growth (Figure 1B, *p < 0.05). qPCR data showed that metformin upregulated expressions of p16 (Figure 1C, *p < 0.05) and p21 (Figure 1D, *p < 0.05) which could arrest the cell cycle in the G1 phase and suppress cell proliferation, which were repressed by FFA. Apoptosis was assessed by flow cytometry. Our data showed that FFA treatment significantly promoted cell proliferation and inhibited cell apoptosis, while it was reversed by metformin (Figure 1E, *p < 0.05).
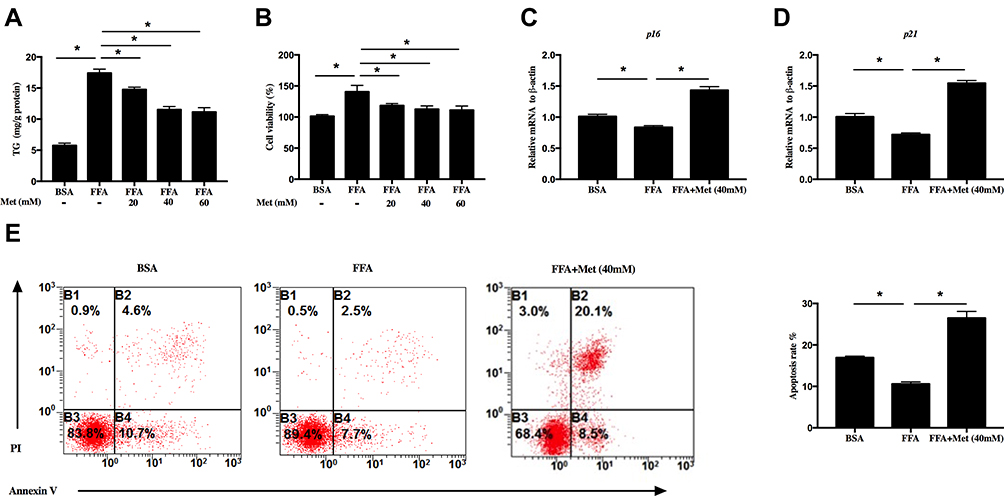 |
Figure 1 Metformin prohibits free fatty acids (FFA)-treated human keratinocyte proliferation. HaCat cells were treated with 400 μM FFA for 10 days, and then the intervention groups were co-treated with metformin at different concentrations and FFA for additional 4 days. (A) Metformin inhibited triglyceride (TG) level of HaCaT cells. (B) Metformin inhibited HaCaT cell proliferation analyzed by CCK8. Metformin (40mM) increased the mRNA levels of p16 (C) and p21 (D). (E) Metformin (40mM) promoted HaCaT cells apoptosis. Representative FACS analysis (left panel) and the ratios (right panel) of Annexin V+ PI− HaCaT cells. Data are presented as the mean ± SD and are representative of three independent experiments. *p < 0.05. |
Metformin Alleviated Oxidative Stress in FFA-Treated Human Keratinocyte
SOD and CAT are important antioxidative enzymes that comprise the antioxidative defense system. FFA treatment significantly decreased the SOD2 and CAT protein levels (Figure 2A, *p < 0.05) as well as enzyme activities of total SOD (Figure 2B, *p < 0.05) and CAT (Figure 2C, *p < 0.05). Here, metformin (at 40 mM) upregulated the protein levels as well as activities of these antioxidant enzymes. Furthermore, flow cytometry of DCFH-DA revealed that the intracellular ROS level in FFA-treated HaCaT cells was significantly increased, and this was effectively reversed by metformin at 40 mM (Figure 2D, *p < 0.05). All of these data suggested that decreasing intracellular ROS level could be an important mechanism for metformin inhibiting proliferation in FFA-treated HaCaT cells.
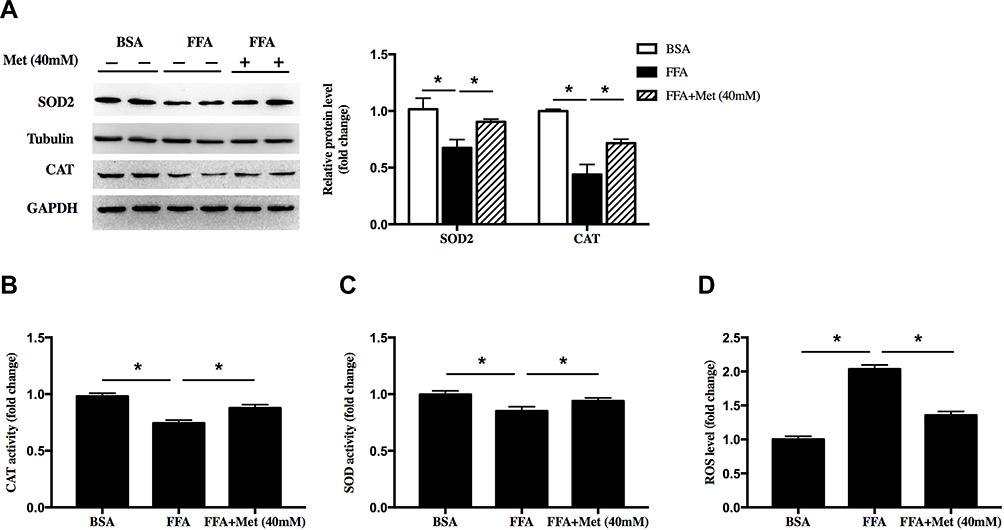 |
Figure 2 Metformin alleviated oxidative stress in FFA-treated human keratinocyte. HaCat cells were treated with 400 μM FFA for 10 days, and then the intervention groups were co-treated with metformin (40mM) and FFA for additional 4 days. (A) Immunoblot of superoxide dismutase 2 (SOD2) and catalase (CAT) in HaCaT cells. Total (B) CAT activity and (C) SOD activity in HaCat cells. (D) Total ROS levels in the HaCat cells. Data are presented as the mean ± SD and are representative of three independent experiments. *p < 0.05. |
Metformin Promoted Autophagy in FFA-Treated Human Keratinocyte
Next, we sought to determine whether metformin promoted keratinocyte autophagy. Western blot showed that FFA treatment significantly reduced the levels of LC3II/LC3I, increased the P62, suggesting FFA inhibited the autophagy in the cell. However, the inhibition of autophagy by FFA was reversed by metformin at 40mM concentration (Figure 3A, *p < 0.05). To confirm this, the autophagy flux was detected in FFA-treated cells that were transfected with a tandem fluorescent tagged RFP-GFP-LC3 adenovirus. Data showed more early autophagosome (yellow puncta on colocalization) and less auto-lysosome (red only puncta) were in FFA-treated cells, while metformin reversed this (Figure 3B, *p < 0.05). The above data indicated that metformin could attenuate the FFA-caused suppression of autophagy.
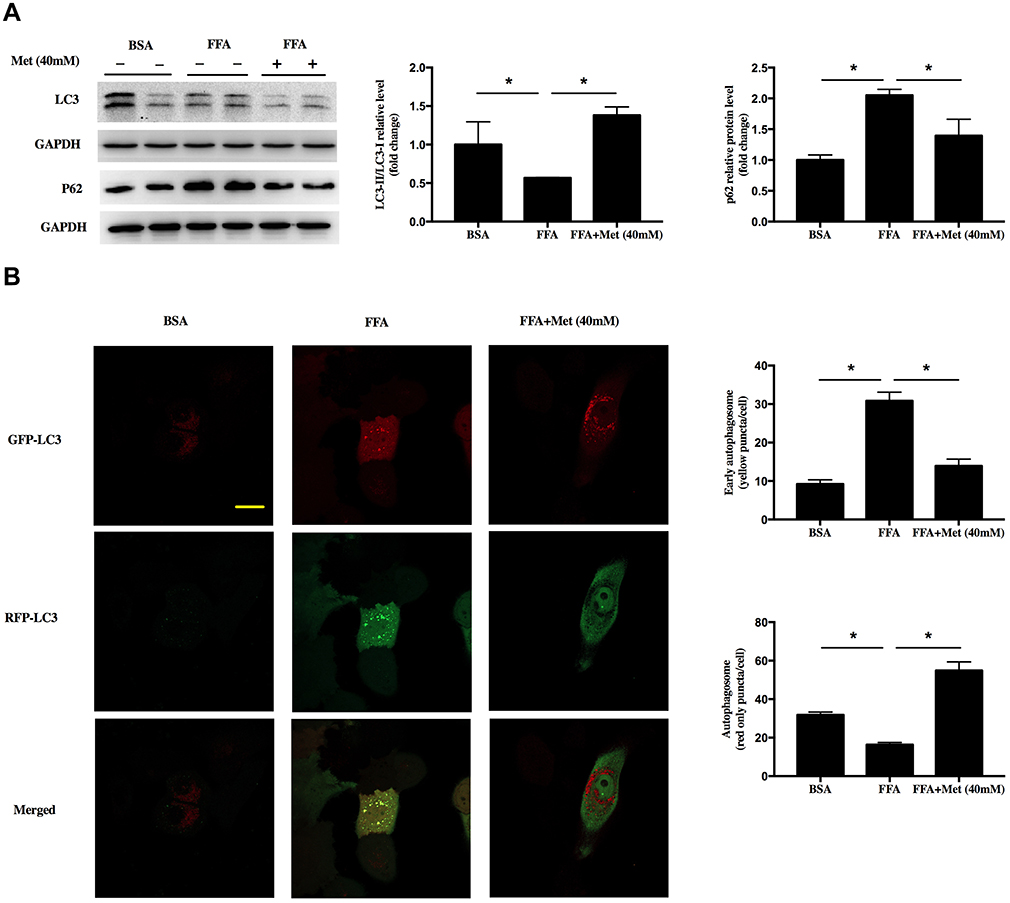 |
Figure 3 Metformin promotes autophagy in FFA-treated human keratinocyte. HaCat cells were treated with 400 μM FFA for 10 days, and then the intervention groups were co-treated with metformin (40mM) and FFA for additional 4 days. (A) Immunoblot of LC3 and P62 in HaCaT cells. (B) Representative confocal images of HaCat cells expressing GFP-RFP- LC3 and quantitation of early autophagosome puncta and autolysosome puncta following FFA and metformin treatment. Yellow showed co-localization of GFP and RFP, indicating early autophagosomes. Red only showed autolysosomes, scale: 20 μm. Data are presented as the mean ± SD and are representative of three independent experiments. *p < 0.05. |
Activation of FOXO3 Contributed to the Antiproliferative Effects of Metformin Through Reducing ROS in FFA-Treated Human Keratinocyte
In this study, FFA treatment inhibited the nuclear translocation of FOXO3. However, metformin remarkably reversed this effect, which mean metformin could increase the activity of FOXO3 (Figure 4A, *p < 0.05). To further confirm whether FOXO3 mediated the antiproliferative roles of metformin in FFA-treated human keratinocyte, we speculated FOXO3 shRNA lentivirus to knockdown FOXO3 in HaCat cells as confirmed by Western blot (Figure 4B, *p < 0.05). In the scramble control group, consistent with above data, metformin inhibited cell viability (Figure 4C, *p < 0.05), upregulated p16 (Figure 4D, *p < 0.05) and p21 gene (Figure 4E, *p < 0.05) expressions, whereas FOXO3 knockdown significantly attenuated these effects. To analyze the role of FOXO3 in FFA-induced ROS generation, we firstly detected the cellular ROS level. The level of ROS was significantly reduced by metformin, while FOXO3 knockdown blocked this effect (Figure 4F, *p < 0.05). Then, we measured the activities of antioxidant enzyme SOD and CAT as well as the expressions of SOD2 and CAT after FOXO3 knockdown. Results showed FOXO3 knockdown reversed the effects of metformin on increasing activities (Figure 4G, h, *p < 0.05) and protein levels (Figure 4I, *p < 0.05) of these two antioxidant enzymes. All of above implied that in FFA-treated human keratinocyte, metformin inhibited proliferation potentially through promoting FOXO3 activity to reduce ROS level.
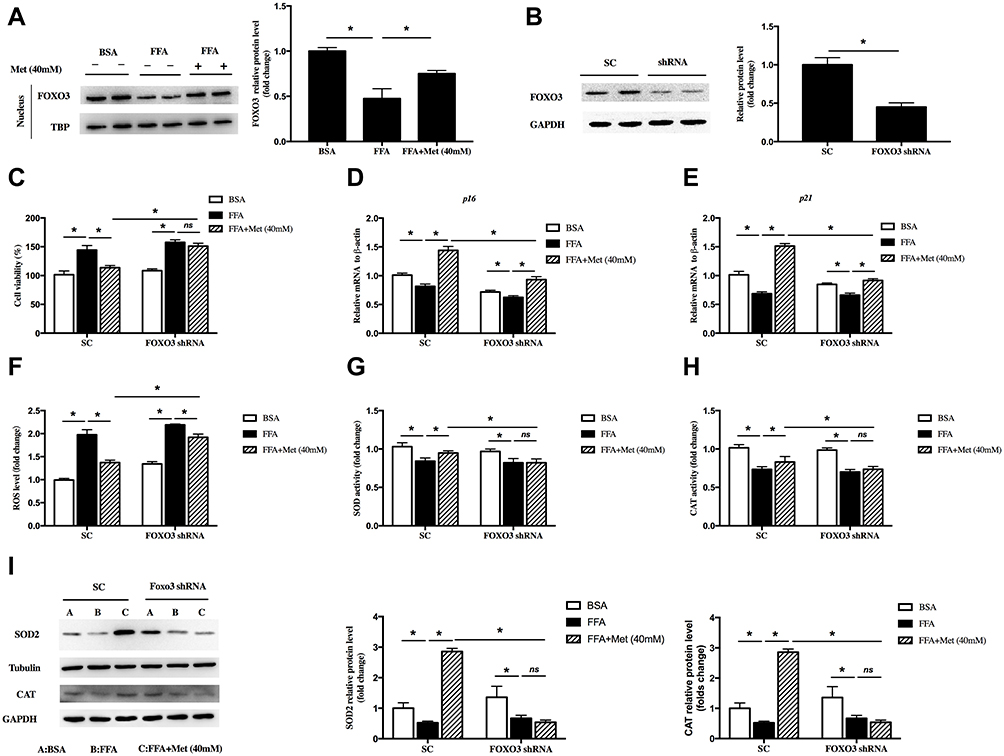 |
Figure 4 Forkhead box O 3 (FOXO3) was involved in the reduction of reactive oxygen species (ROS) by metformin in FFA-treated human keratinocyte. HaCat cells were treated with 400 μM FFA for 10 days, and then the intervention groups were co-treated with metformin (40mM) and FFA for additional 4 days. (A) Immunoblot of FOXO3 in the nucleus of HaCaT cells. HaCat cells were transfected with FOXO3 shRNA or scramble control (SC) by lentivirus. (B) Immunoblot of FOXO3 in the HaCaT cells. HaCat cells were pretransfected with FOXO3 shRNA or scramble control (SC) by lentivirus, then treated with 400 μM FFA for 10 days. After that, the intervention groups were co-treated with metformin (40mM) and FFA for additional 4 days. (C) Proliferation of the HaCaT cells detected by CCK8. mRNA levels of p16 (D) and p21 (E) in the HaCaT cells. ROS levels (F), total SOD activities (G) and CAT activities (H) in HaCat cells. (I) Immunoblot of SOD2 and CAT in HaCaT cells. A: BSA; B: FFA; C: FFA+Metformin (40mM). Data are presented as the mean ± SD and are representative of three independent experiments. *p < 0.05. |
FOXO3 Mediates the Proapoptosis of Metformin Through Inducing Autophagy in FFA-Treated Human Keratinocyte
We determined if FOXO3 was involved in the effects of metformin on proapoptosis in FFA-treated human keratinocyte. In the control group, metformin induced apoptosis in FFA-treated HaCat cells, while this effect was blocked by FOXO3 knockdown (Figure 5A, *p < 0.05). In addition, metformin induced the autophagy in FFA-treated HaCat cells, assessed by protein levels of LC3II/LC3I and P62. However, these effects were also weakened by FOXO3 knockdown (Figure 5B, *p < 0.05). These results suggested that metformin increased FOXO3 activity to promote apoptosis through inducing autophagy in FFA-treated keratinocyte, which might contribute to its inhibitory role in psoriasis under hyperlipidemia.
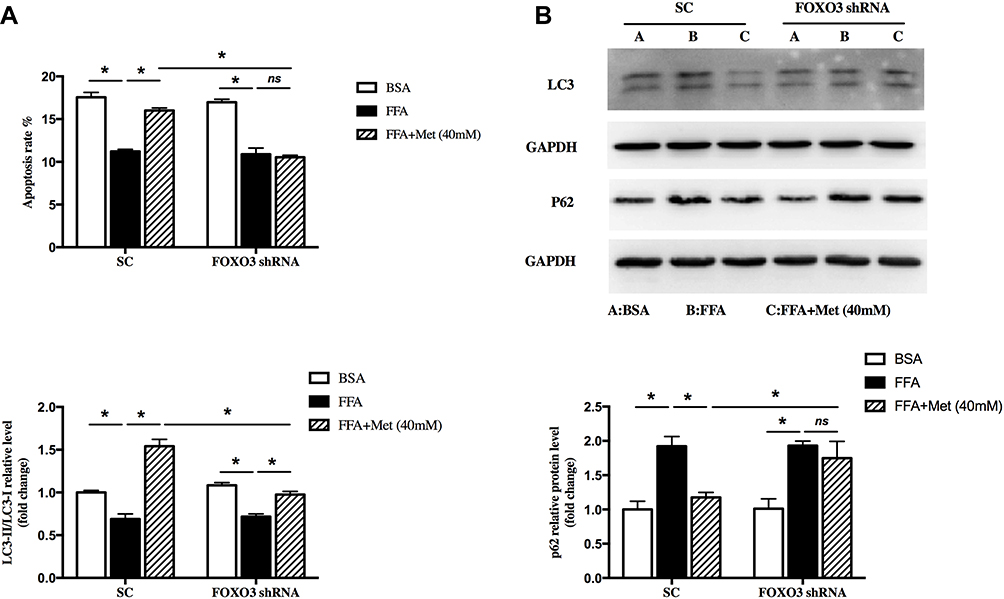 |
Figure 5 FOXO3 mediated the effect of pro-autophagy by metformin in FFA-treated human keratinocyte. HaCat cells were pretransfected with FOXO3 shRNA or empty vector by lentivirus, then treated with 400 μM FFA for 10 days. After that, the intervention groups were co-treated with metformin (40mM) and FFA for additional 4 days. (A) Cell apoptosis was detected by FACS analysis. (B) Immunoblot of LC3 and P62 in HaCaT cells. A: BSA; B: FFA; C: FFA+Metformin (40mM). Data are presented as the mean ± SD and are representative of three independent experiments. *p < 0.05. |
Discussion
Skin has been considered as a mirror of underlying metabolic sinister. Various dermatological conditions like psoriasis, acanthosis nigricans, lichen planus, acne vulgaris, acrochordons, atopic dermatitis, etc have been reported to be associated with metabolic syndromes.8 Dyslipidemias are one of the common metabolic disorders. A link between dyslipidemia and dermatological disorders such as psoriasis has been established in the recent past.31
Psoriasis is a multifactorial skin disease that inconveniences many patients. As abnormally excessive and rapid proliferation of the keratinocytes accounts for the skin lesion of psoriasis, inhibition of the cell proliferation as well as induction of the apoptosis are the primary method of treating psoriasis.32 Normally, skin cells are replaced every 28 to 30 days; however, in patients with psoriasis, skin cells are replaced every 3 to 5 days. These alterations are believed to originate from the excessive proliferation of keratinocytes.33 Despite remarkable advances in understanding the pathogenesis of psoriasis, the chronological order of keratinocyte hyperproliferation has not been completely elucidated. Therefore, the strategies for proliferation inhibition would be effective treatment of psoriasis.
Several studies have demonstrated a linear correlation between body mass index (BMI) and risk of incident psoriasis existed.11,34 The results of studies that investigated FFA levels in psoriatic patients revealed that significant elevation. Other clinical research had also demonstrated the existence of lipid metabolism disorders in patients with psoriasis, which is more likely to be complicated with fatty liver.35,36 Hence, dyslipidemia could be the important risk factor for the development of psoriasis. However, the underlying mechanism of lipid-induced psoriasis is still unknown.37,38 In the present study, we treated the HaCat cells with FFA to create a hyperlipidemia microenvironment in vitro. We found that ROS, induced by excessive lipid accumulation, could be the key factor to promote the proliferation of keratinocytes in psoriasis. Currently, several studies have reported that ROS is a key factor for cell proliferation in different cell and animal models.39 Therefore, understanding the mechanism of regulating ROS generation in psoriasis under hyperlipidemia conditions may be an important hint to cure this intricate disease.
Many studies have demonstrated that metformin could ameliorate psoriasis through suppressing inflammation via different pathways in the keratinocytes. In our study, we have shown for the first time that metformin significantly decreased ROS level and promoted autophagy in FFA-treated keratinocytes, which contributed to the antiproliferation. Increased ROS production and decreased antioxidant system functions have been demonstrated to be associated with the pathogenesis of psoriasis.14,40 One study found high levels of ROS promoted the function of Treg and so prevent the psoriatic dermatitis induced by imiquimod.41 Another study has shown that metformin could increase ROS production and then promote apoptosis.42 In all of these studies, ROS was considered as the factor to aggravate inflammation. In contrast, in the present study, metformin reduced ROS level to inhibit proliferation of HaCaT cells, as ROS could be the key signal factor to promote cell proliferation. Like the dual function in cancer, ROS can also act as a regulator or a suppressor of immune-mediated diseases. At high level, ROS would induce apoptosis and inhibit proliferation, while at low level, ROS would only be as a regulator to induce proliferation. FFA treatment was a mild stress, which stimulated a low level of ROS production. Under this condition, metformin played a major role in repressing ROS to inhibit proliferation.
In addition, we found autophagy was suppressed in the HaCaT cells treated with FFA. Many studies have shown that autophagy was involved in various types of physiological activities. Deficiency of autophagy induced inflammatory cytokine generation and cell proliferation in keratinocytes, thus aggravated the psoriasis. PSORI-CM02 suppressed the proliferation of TNF-α-stimulated HaCaT cells via induction of autophagy. The classical treatments for psoriasis such as retinoids, vitamin D analogues and ultraviolet B therapy can induce autophagy, which suggests that the clinical benefits of these drugs may be related to autophagy activation.43,44 Excessive lipid accumulation could inhibit autophagy in different cells. Consistent with these studies, in the present study, metformin promoted the autophagy to induce apoptosis in FFA-treated HaCaT cells.
FOXO transcription factor family is a central player in cell proliferation and antioxidant defense. Research have shown that the nuclear translocation of FOXO proteins can regulate cell survival. Some of the target genes like p16, p21, and p27 have important roles in the suppressing cell proliferation.45 Activation of FOXO3 blocks cellular proliferation and drives cells into a quiescent state.22 FOXO3 also triggered the expression of several antioxidant enzymes, such as SOD2 and CAT, to cope with oxidative stress.46 Studies investigated the relationship between FOXO3 and the proliferation of psoriatic keratinocyte, and found out that the gene expression and the activity of FOXO3 are both significantly decreased in psoriatic lesions compared with that uninvolved psoriatic lesions and normal skin.47 Consistent with these findings, we found FFA treatment significantly decreased the FOXO3 level in nuclease, while metformin upregulated its activity, increased the expression of p21, SOD2 as well as SOD. FOXO3 knockdown partially blocked these effects from metformin. It suggested under hyperlipidemia conditions, metformin induced autophagy and reduced ROS to suppress keratinocyte proliferation via promoting FOXO3 activity.
Conclusion
In summary, this study for the first time demonstrated that metformin inhibited human keratinocyte proliferation by reducing ROS level under hyperlipidemia. Furthermore, metformin induced the apoptosis by promoting autophagy. All these effects of metformin were through FOXO3-dependent pathways.
Abbreviations
FOXO3, forkhead box O3; FFA, free fatty acids; ROS, reactive oxygen species; DCFH-DA, 2,7-dichlorofluorescein-diacetate; SOD, superoxide dismutase; CAT, catalase; LDL, low-density lipoprotein; H2O2, hydrogen peroxide; NAFLD, nonalcoholic fatty liver disease; T2DM, type 2 diabetes mellitus; CCK8, cell counting kit‐8; TG, triglyceride; qPCR, quantitative RT-PCR; BMI, body mass index.
Data Sharing Statement
The data collected for the study are available from the corresponding author upon request.
Funding
This work was funded by the Project funded by China Postdoctoral Science Foundation (Grant Nos. 2019M653939 and 2019M664000), the National Natural Science Foundation of China (Grant No. 81972936), and the Capital Characteristic Clinical Application Research and Achievement Promotion Project of China (Z181100001718038).
Disclosure
The authors declare that they have no conflicts of interest in relation to this work.
References
1. Korman NJ. Management of psoriasis as a systemic disease: what is the evidence? Br J Dermatol. 2020;182(4):840–848. doi:10.1111/bjd.18245
2. Ogawa E, Sato Y, Minagawa A, Okuyama R. Pathogenesis of psoriasis and development of treatment. J Dermatol. 2018;45(3):264–272. doi:10.1111/1346-8138.14139
3. Rendon A, Schakel K. Psoriasis pathogenesis and treatment. Int J Mol Sci. 2019;20(6). doi:10.3390/ijms20061475
4. Choi MR, Kim HD, Cho S, et al. Anoctamin1 induces hyperproliferation of HaCaT keratinocytes and triggers imiquimod-induced psoriasis-like skin injury in mice. Int J Mol Sci. 2021;22(13):7145.
5. Campione E, Cosio T, Lanna C, et al. Predictive role of vitamin A serum concentration in psoriatic patients treated with IL-17 inhibitors to prevent skin and systemic fungal infections. J Pharmacol Sci. 2020;144(1):52–56. doi:10.1016/j.jphs.2020.06.003
6. Gisondi P, Fostini AC, Fossa I, Girolomoni G, Targher G. Psoriasis and the metabolic syndrome. Clin Dermatol. 2018;36(1):21–28. doi:10.1016/j.clindermatol.2017.09.005
7. Gisondi P, Tessari G, Conti A, et al. Prevalence of metabolic syndrome in patients with psoriasis: a hospital-based case-control study. Br J Dermatol. 2007;157(1):68–73. doi:10.1111/j.1365-2133.2007.07986.x
8. Fatima F, Das A, Kumar P, Datta D. Skin and metabolic syndrome: an evidence based comprehensive review. Indian J Dermatol. 2021;66(3):302–307. doi:10.4103/ijd.IJD_728_20
9. Mazzilli S, Lanna C, Chiaramonte C, et al. Real life experience of apremilast in psoriasis and arthritis psoriatic patients: preliminary results on metabolic biomarkers. J Dermatol. 2020;47(6):578–582. doi:10.1111/1346-8138.15293
10. Raychaudhuri SK, Chatterjee S, Nguyen C, Kaur M, Jialal I, Raychaudhuri SP. Increased prevalence of the metabolic syndrome in patients with psoriatic arthritis. Metab Syndr Relat Disord. 2010;8(4):331–334. doi:10.1089/met.2009.0124
11. Miao C, Li J, Li Y, Zhang X. Obesity and dyslipidemia in patients with psoriasis: a case-control study. Medicine. 2019;98(31):e16323. doi:10.1097/MD.0000000000016323
12. Pietrzak A, Chabros P, Grywalska E, et al. Serum lipid metabolism in psoriasis and psoriatic arthritis - an update. Arch Med Sci. 2019;15(2):369–375. doi:10.5114/aoms.2018.74021
13. Fan Z, Wang L, Jiang H, Lin Y, Wang Z. Platelet dysfunction and its role in the pathogenesis of psoriasis. Dermatology. 2021;237(1):56–65. doi:10.1159/000505536
14. Cannavo SP, Riso G, Casciaro M, Di Salvo E, Gangemi S. Oxidative stress involvement in psoriasis: a systematic review. Free Radic Res. 2019;53(8):829–840. doi:10.1080/10715762.2019.1648800
15. Ko W, Kim N, Lee H, et al. Anti-inflammatory effects of compounds from cudrania tricuspidata in HaCaT human keratinocytes. Int J Mol Sci. 2021;22(14):7472. doi:10.3390/ijms22147472
16. Sreedhar A, Aguilera-Aguirre L, Singh KK. Mitochondria in skin health, aging, and disease. Cell Death Dis. 2020;11(6):444. doi:10.1038/s41419-020-2649-z
17. Campione E, Mazzilli S, Di Prete M, et al. The role of Glutathione-S transferase in psoriasis and associated comorbidities and the effect of dimethyl fumarate in this pathway. Front Med. 2022;9:760852. doi:10.3389/fmed.2022.760852
18. Bai H, Zhang W, Qin XJ, et al. Hydrogen peroxide modulates the proliferation/quiescence switch in the liver during embryonic development and posthepatectomy regeneration. Antioxid Redox Signal. 2015;22(11):921–937. doi:10.1089/ars.2014.5960
19. Liu C, Liao JZ, Li PY. Traditional Chinese herbal extracts inducing autophagy as a novel approach in therapy of nonalcoholic fatty liver disease. World J Gastroenterol. 2017;23(11):1964–1973. doi:10.3748/wjg.v23.i11.1964
20. Kim HR, Kim JC, Kang SY, Kim HO, Park CW, Chung BY. Rapamycin alleviates 2,3,7,8-Tetrachlorodibenzo-p-dioxin-Induced aggravated dermatitis in mice with imiquimod-induced psoriasis-like dermatitis by inducing autophagy. Int J Mol Sci. 2021;22(8):3968.
21. Caputo V, Strafella C, Termine A, et al. Overview of the molecular determinants contributing to the expression of Psoriasis And Psoriatic Arthritis phenotypes. J Cell Mol Med. 2020;24(23):13554–13563. doi:10.1111/jcmm.15742
22. Morris BJ, Willcox DC, Donlon TA, Willcox BJ. FOXO3: a major gene for human Longevity–A Mini-Review. Gerontology. 2015;61(6):515–525. doi:10.1159/000375235
23. Tsitsipatis D, Klotz LO, Steinbrenner H. Multifaceted functions of the forkhead box transcription factors FoxO1 and FoxO3 in skin. Biochim Biophys Acta Gen Subj. 2017;1861(5 Pt A):1057–1064. doi:10.1016/j.bbagen.2017.02.027
24. Lin F. Molecular regulation and function of FoxO3 in chronic kidney disease. Curr Opin Nephrol Hypertens. 2020;29(4):439–445. doi:10.1097/MNH.0000000000000616
25. Shen Z, Zhou H, Li A, et al. Metformin inhibits hepatocellular carcinoma development by inducing apoptosis and pyroptosis through regulating FOXO3. Aging. 2021;13(18):22120–22133. doi:10.18632/aging.203464
26. Hartwig J, Loebel M, Steiner S, et al. Metformin attenuates ROS via FOXO3 activation in immune cells. Front Immunol. 2021;12:581799. doi:10.3389/fimmu.2021.581799
27. Wu CY, Shieh JJ, Shen JL, Liu YY, Chang YT, Chen YJ. Association between antidiabetic drugs and psoriasis risk in diabetic patients: results from a nationwide nested case-control study in Taiwan. J Am Acad Dermatol. 2015;72(1):123–130. doi:10.1016/j.jaad.2014.08.042
28. Chang JE, Choi MS. A molecular perspective on the potential benefits of metformin for the treatment of inflammatory skin disorders. Int J Mol Sci. 2020;21(23):8960. doi:10.3390/ijms21238960
29. Gao Y, Zhang W, Zeng LQ, et al. Exercise and dietary intervention ameliorate high-fat diet-induced NAFLD and liver aging by inducing lipophagy. Redox Biol. 2020;36:101635. doi:10.1016/j.redox.2020.101635
30. Proenca CC, Stoehr N, Bernhard M, et al. Atg4b-dependent autophagic flux alleviates Huntington’s disease progression. PLoS One. 2013;8(7):e68357. doi:10.1371/journal.pone.0068357
31. Shenoy C, Shenoy MM, Rao GK. Dyslipidemia in dermatological disorders. N Am J Med Sci. 2015;7(10):421–428. doi:10.4103/1947-2714.168657
32. Armstrong AW, Read C. Pathophysiology, clinical presentation, and treatment of psoriasis: a review. JAMA. 2020;323(19):1945–1960. doi:10.1001/jama.2020.4006
33. Ba W, Xu Y, Yin G, et al. Metformin inhibits pro-inflammatory responses via targeting nuclear factor-kappaB in HaCaT cells. Cell Biochem Funct. 2019;37(1):4–10. doi:10.1002/cbf.3367
34. Setty AR, Curhan G, Choi HK. Obesity, waist circumference, weight change, and the risk of psoriasis in women: Nurses’ Health Study II. Arch Intern Med. 2007;167(15):1670–1675. doi:10.1001/archinte.167.15.1670
35. Moudgil S, Mahajan R, Narang T, Sachdeva N, Dayal D, Dogra S. Central obesity and dyslipidemia in pediatric patients with psoriasis: an observational study from India. J Am Acad Dermatol. 2021;85(6):1655–1657. doi:10.1016/j.jaad.2020.12.072
36. Gutmark-Little I, Shah KN. Obesity and the metabolic syndrome in pediatric psoriasis. Clin Dermatol. 2015;33(3):305–315. doi:10.1016/j.clindermatol.2014.12.006
37. Rodriguez-Cerdeira C, Cordeiro-Rodriguez M, Carnero-Gregorio M, et al. Biomarkers of inflammation in obesity-psoriatic patients. Mediators Inflamm. 2019;2019:7353420. doi:10.1155/2019/7353420
38. Carrascosa JM, Bonanad C, Dauden E, Botella R, Olveira-Martin A; en nombre del Grupo de Trabajo en Inflamacion Sistemica en P. Psoriasis and nonalcoholic fatty liver disease. Actas Dermosifiliogr. 2017;108(6):506–514. doi:10.1016/j.ad.2016.12.017
39. Checa J, Aran JM. Reactive oxygen species: drivers of physiological and pathological processes. J Inflamm Res. 2020;13:1057–1073. doi:10.2147/JIR.S275595
40. Plenkowska J, Gabig-Ciminska M, Mozolewski P. Oxidative stress as an important contributor to the pathogenesis of psoriasis. Int J Mol Sci. 2020;21(17):6206. doi:10.3390/ijms21176206
41. Kim HR, Lee A, Choi EJ, et al. Reactive oxygen species prevent imiquimod-induced psoriatic dermatitis through enhancing regulatory T cell function. PLoS One. 2014;9(3):e91146. doi:10.1371/journal.pone.0091146
42. Li B, Zhou P, Xu K, et al. Metformin induces cell cycle arrest, apoptosis and autophagy through ROS/JNK signaling pathway in human osteosarcoma. Int J Biol Sci. 2020;16(1):74–84. doi:10.7150/ijbs.33787
43. Yue L, Ailin W, Jinwei Z, et al. PSORI-CM02 ameliorates psoriasis in vivo and in vitro by inducing autophagy via inhibition of the PI3K/Akt/mTOR pathway. Phytomedicine. 2019;64:153054. doi:10.1016/j.phymed.2019.153054
44. Yin H, Wu H, Chen Y, et al. The therapeutic and pathogenic role of autophagy in autoimmune diseases. Front Immunol. 2018;9:1512. doi:10.3389/fimmu.2018.01512
45. Link W. Introduction to FOXO Biology. Methods Mol Biol. 2019;1890:1–9.
46. de Keizer PL, Burgering BM, Dansen TB. Forkhead box o as a sensor, mediator, and regulator of redox signaling. Antioxid Redox Signal. 2011;14(6):1093–1106. doi:10.1089/ars.2010.3403
47. Zhang M, Zhang X. The role of PI3K/AKT/FOXO signaling in psoriasis. Arch Dermatol Res. 2019;311(2):83–91. doi:10.1007/s00403-018-1879-8
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.