")
Back to Journals » OncoTargets and Therapy » Volume 11
Metformin induces miR-378 to downregulate the CDK1, leading to suppression of cell proliferation in hepatocellular carcinoma
Authors Zhou J, Han S, Qian W, Gu Y, Li X, Yang K
Received 8 March 2018
Accepted for publication 12 May 2018
Published 31 July 2018 Volume 2018:11 Pages 4451—4459
DOI https://doi.org/10.2147/OTT.S167614
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Jin Zhou,1,* Sheng Han,2,3,* Weichun Qian,4 Yuanyuan Gu,1 Xiangcheng Li,2,3 Kunxing Yang1
1Department of General Surgery, Nanjing First Hospital, Nanjing Medical University, Nanjing, China; 2Hepatobiliary Center, The First Affiliated Hospital of Nanjing Medical University, Nanjing, China; 3Key Laboratory of Liver Transplantation, Chinese Academy of Medical Science, Beijing, China; 4Division of Cardiology, Nanjing First Hospital, Nanjing Medical University, Nanjing, China
*These authors contributed equally to this work
Abstract: Metformin is one of the extensively and most commonly used oral antihyperglycemic agents, but it has been shown to exert antineoplastic effects in many cancer cells. Recent studies have confirmed that metformin has an antitumor effect on hepatocellular carcinoma (HCC). However, the molecular mechanism underlying this effect needs to be further studied.
Materials and methods: CDK1 and miR-378 expression was analyzed by western blotting and real-time PCR assays. We confirmed the association between miR-378 and CDK1 by dual luciferase reporter assay. The role of the miR-378/CDK1 pathway in proliferation, cell cycle and apoptosis was examined in vitro. The effect of miR-378 on HCC tumor growth was evaluated in nude xenograft mouse model.
Results: Our study found that metformin significantly inhibited the HCC cell proliferation via inducing G2/M arrest. At the same time, metformin efficiently decreased CDK1 expression and elevated miR-378 level. Moreover, the upregulation of miR-378 also repressed HCC cell proliferation by causing G2/M arrest and inhibited tumor growth. Additionally, we demonstrated that miR-378 directly targeted CDK1 3'UTR and downregulated CDK1 mRNA and protein levels. Furthermore, metformin treatment could not decrease CDK1 expression, suppress HCC cell proliferation, and induce G2/M cell cycle arrest.
Discussion: Metformin-suppressed HCC cell proliferation was dependent on the inhibitory effect of miR-378 on CDK1 expression. Taken together, we concluded that metformin inhibited HCC cell proliferation via modulating miR-378/CDK1 axis.
Conclusion: Collectively, the current results provide the first evidence, to our knowledge, that miR-378/CDK1 axis is involved in metformin modulating the proliferation of HCC cells, which suggests a novel molecular mechanism underlying the therapeutic effect of metformin on HCC.
Keywords: cell cycle, apoptosis, metformin, cancer, liver
Introduction
Hepatocellular carcinoma (HCC) is the third most frequent cause of tumor-related deaths in People’s Republic of China.1,2 Due to the high metastatic potential of HCC,3,4 progression is found in ~70% of patients within 1 year of diagnosis, and it is difficult to treat patients with advanced HCC.5,6 Metformin (1,1-dimethylbiguanide), which is commonly used as an oral antihyperglycemic agent of the biguanide family, may reduce cancer risk and improve prognosis.7 Previous studies have demonstrated that metformin could inhibit HCC angiogenesis and invasion and that it enhances the chemosensitivity to chemotherapeutics.8–11 However, the underlying mechanisms have not been elucidated until now.
MicroRNAs (miRNAs) are a novel class of short, noncoding RNAs involved in posttranscriptional gene regulation by binding to the target site in the 3′-untranslated region (3′-UTR) of target mRNAs.12 The role for miRNAs in carcinogenesis, development, and progression of HCC has been well established. MiR-378 has been reported to be related to cell survival, tumor growth, and angiogenesis.13,14 miR-378 inhibits hepatocyte proliferation during liver regeneration.15 Furthermore, metformin affected miR-378 expression in HCC.16 However, whether metformin has antitumor effects on HCC cells through regulating miR-378 expression remains unknown and needs to be studied.
In this study, we explored the molecular mechanisms underlying the antitumor function of metformin on HCC. Interestingly, we found that metformin significantly inhibited the proliferation of HCC cells by upregulating miR-378 and downregulating CDK1 expression, which indicated that miR-378/CDK1 axis may be a potential target for HCC therapy.
Materials and methods
Cell culture and stimulation with metformin
Human HCC cell lines HepG2 and Hep3B were purchased from the American Type Culture Collection (Shanghai, People’s Republic of China). The cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Invitrogen) and antibiotics. Cultured cells were maintained in a humidified incubator containing 5% CO2 at 37°C. Cells were stimulated with metformin (Sigma-Aldrich, St Louis, MO, USA), dissolved in dimethyl sulfoxide (DMSO) at a final concentration of 0.5, 5, 50, and 500 mM. As a control, cultured cells were incubated in complete DMEM medium containing DMSO at a final concentration of <0.1%.
Cell transfection
miR-378 mimics and negative control (NC) were obtained from Shanghai GenePharma (Shanghai, People’s Republic of China). To evaluate the functions of miR-378 in HepG2 and Hep3B cells, cells were transfected with miR-378 mimics or NC using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s protocol. Subsequent to transfection at 37°C for 4 h, cell culture medium was replaced with DMEM medium containing 10% fetal bovine serum. The in vitro transfection efficiency (~75%) was measured by using carboxyfluorescein-labeled NC microRNA (data not shown).
CCK-8 assay
Cell proliferation was performed with Cell Counting Kit-8 (Dojindo, Tokyo, Japan). According to the instructions, CCK-8 reagent was added at 0, 24, 48, and 72 h, respectively, after seeding 4×103 cells per well into 96-well plates and transfecting with miR-378 control or mimics, and incubating at 37°C for 2 h. The OD 450 nm value was detected by using a microplate reader (Bio-Rad, Hercules, CA, USA).
Cell cycle assay
For the cell cycle study, cells were stimulated with metformin at a final concentration of 50 mM for 24 h. Cells were stained by using BD Pharmingen™ Cell Cycle Kit (BD Biosciences, San Jose, CA, USA) according to manufacture-supplied protocols (propidium iodide [PI]/RNase). Flow cytometry was performed using a BD FACSCalibur™ flow cytometer (BD Biosciences) and data were analyzed using MODIFIT software (Becton Dickinson). The experiment was performed in triplicate, and a representative experiment is shown.
Apoptosis
Apoptosis was assayed in control and treated cells with an Annexin V assay kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Briefly, 2×105 cells were harvested after treatment. Annexin V-FITC and PI were added. The reaction was stopped by adding 300 μL of 1× binding buffer, and labeled cells were analyzed by flow cytometry with a FACSCalibur (BD Bioscience).
Real-time PCR
Total RNA was extracted from HCC cell lines HepG2 and Hep3B using the TRIzol method (Invitrogen). Then, reverse transcription was performed by using the reverse-transcriptase kit (Takara, Tokyo, Japan) according to the manufacturer's protocol. RT-PCR was performed using a SYBR premix Ex Taq kit (Takara) on a 7300 Real-time PCR system (Thermo Fisher Scientific) according to the manufacturer’s protocol. The miR-378 and U6 primer sequences used were obtained from Shanghai GenePharma. CDK1 forward, 5′-GGTTCCTAGTACTGCAATTCG-3′ and reverse, 5′-TTTGCCAGAAATTCGTTTGG-3′; and GAPDH forward, 5′-TGACTTCAACAGCGACACCCA-3′ and reverse, 5′-CACCCTGTTGCTGTAGCCAAA-3′. The relative expression level was calculated using the 2−ΔΔCt method. U6 small nuclear RNA and GAPDH were used as an internal control. Each sample was analyzed in triplicate.
Western blotting analysis
The protein contents of CDK1 and β-actin in HCC cells were detected by performing immunoblotting. We lysed cultured or transfected cells in RIPA buffer with 1% phenylmethylsulfonyl fluoride and loaded the protein onto an SDS-PAGE and transferred them onto polyvinylidene difluoride membrane. After probing with the antibodies for CDK1 (Cat# A17, Abcam, Cambridge, MA, USA) and β-actin (#4967, CST, Danvers, MA, USA) at 4°C overnight, the blots were then incubated with horseradish peroxidase-conjugated secondary antibody (1:5,000). Enhanced chemiluminescence substrates were used to visualize signals (Thermo Fisher Scientific). β-actin was used as an endogenous protein for normalization.
Nude mice model
The animal experiments were approved by the Committee on Animal Care of the First Affiliated Hospital of Nanjing Medical University and were conducted according to NIH Guidelines for the Care and Use of Laboratory Animals. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals. Nude mice were purchased from the Vital River Laboratories (Beijing, People’s Republic of China). A total of 6×106 HepG2 cells were transfected with miR-378 mimics or NC. Cells were cultured for 24 h before harvest for transplantation into the animal. Before injection, cells were pooled and mixed with Matrigel (BD Biosciences). A total of 3×106 cells were injected subcutaneously into the left flank of nude mice. Tumor volumes were calculated using the formula V = length × width ^2/2. The animals were sacrificed 4 weeks after injection. Pictures were recorded with a Nikon d800 digital camera (Nikon, Tokyo, Japan).
Dual-luciferase reporter assay
A Dual-Luciferase Reporter assay was performed to explore whether CDK1 was a direct target of miR-378. Cells were transfected with miR-378 mimics or NC, and cotransfected with pGL3-CDK1-3′UTR wild-type or pGL3-CDK1-3′UTR mutant using Lipofectamine 2000. Following incubation at 37°C for 24 h, a Dual-Luciferase Reporter assay (Promega Corporation, Madison, WI, USA) was performed to detect Firefly and Renilla luciferase activity according to the manufacturer’s protocol. Renilla luciferase activity was measured as an internal control. Each experiment was repeated at least 3 times.
Statistical analysis
Data in the bar graphs are presented as mean ± SD. All the statistical analyses were performed using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA, USA). All the data were analyzed by unpaired Student’s t-test and P<0.05 was considered to be statistically significant.
Results
Metformin inhibited HCC cell proliferation accompanied with the alteration of CDK1 and miR-378 expression
To determine the effects of metformin on the proliferation of HCC cells, CCK-8 assay was performed on HCC cells HepG2 and Hep3B with metformin (0.5, 5, 50, and 500) treatment. The results showed that the proliferation of HCC cells was significantly inhibited by metformin at 50 and 500 mM (Figure 1A). Therefore, we treated HCC cells with metformin at 50 mM in our study. To investigate the mechanism responsible for the growth inhibition caused by metformin, cell cycle distribution was determined by flow cytometry analysis. After metformin treatment for 24 h, the percentage of HCC cells in the G2/M phase increased (9.3%–22.8%; 6.5%–17.3%) compared to the control (Figure 1B). This cell cycle delay may be responsible for suppression of cell proliferation. Moreover, we found that the expression of CDK1 was obviously downregulated when the HCC cells were treated with metformin (Figure 1C), while the expression of miR-378 was substantially increased (Figure 1D). To examine the cell apoptosis caused by metformin, cell apoptosis was determined by Annexin V/PI staining and flow cytometry assays. After metformin treatment for 24 h, the apoptosis rate of HCC cells was increased compared to the control (Figure 1E).
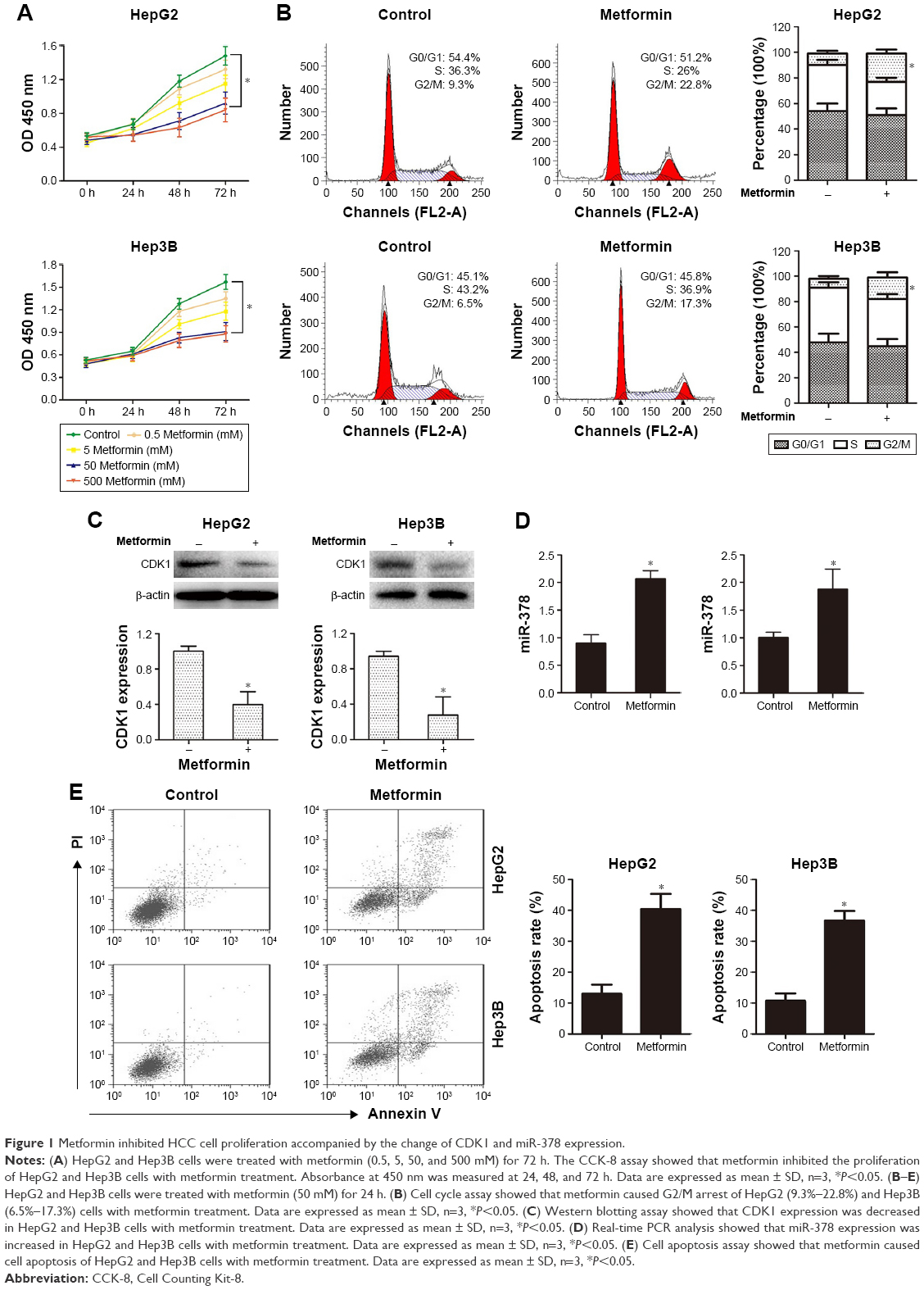 | Figure 1 Metformin inhibited HCC cell proliferation accompanied by the change of CDK1 and miR-378 expression. |
Overexpression of miR-378 inhibited HCC cell proliferation
To explore the function of miR-378 in the regulation of cell proliferation, miR-378 mimics was transfected into human HCC cell lines HepG2 and Hep3B. MiR-378 expression was upregulated with miR-378 mimic transfection at 24, 48, and 72 h compared to the NC, determined by RT-PCR (Figure 2A). The effect of miR-378 expression on cell proliferation was evaluated by CCK-8 assay. Overexpression of miR-378 by miR-378 mimic transfection reduced the proliferation capacity of HepG2 and Hep3B cells, as compared with those treated with NC (Figure 2B). To investigate the mechanism responsible for the growth inhibition caused by miR-378, cell cycle distribution was determined by flow cytometry analysis. After miR-378 mimic transfection for 24 h, the percentage of HCC cells in the G2/M phase increased (8.7%–20.4%; 6.5%–17.9%) compared to the control (Figure 2C). The repression of cell proliferation may also due to cell cycle arrest.
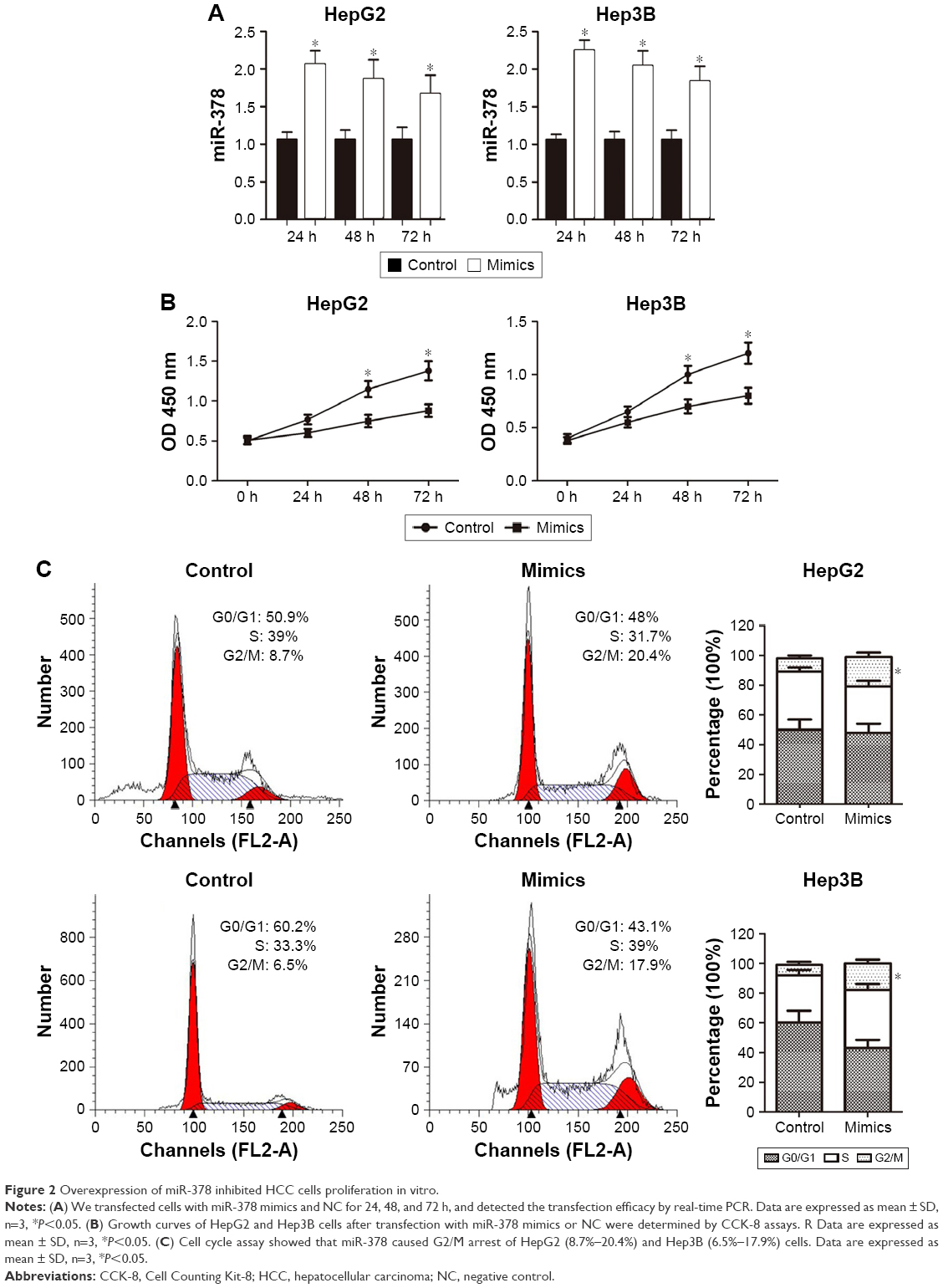 | Figure 2 Overexpression of miR-378 inhibited HCC cells proliferation in vitro. |
The CDK1 3′-UTR is a direct target for miR-378
A target miRNA prediction website (http://www.mirbase.org) and the luciferase reporter assay showed that there was a binding site between miR-378 and CDK1 (Figure 3A and B). RT-PCR analysis showed that miR-378 mimic transfection reduced CDK1 mRNA expression compared with transfection with the NC in HepG2 and Hep3B cells (Figure 3C). Western blotting analysis showed that CDK1 protein expression decreased in HepG2 and Hep3B cells after miR-378 mimic transfection compared with NC (Figure 3D). Taken together, these results suggested that CDK1 was the target gene of miR-378 and the expression of CDK1 was inhibited by miR-378 in HCC cells.
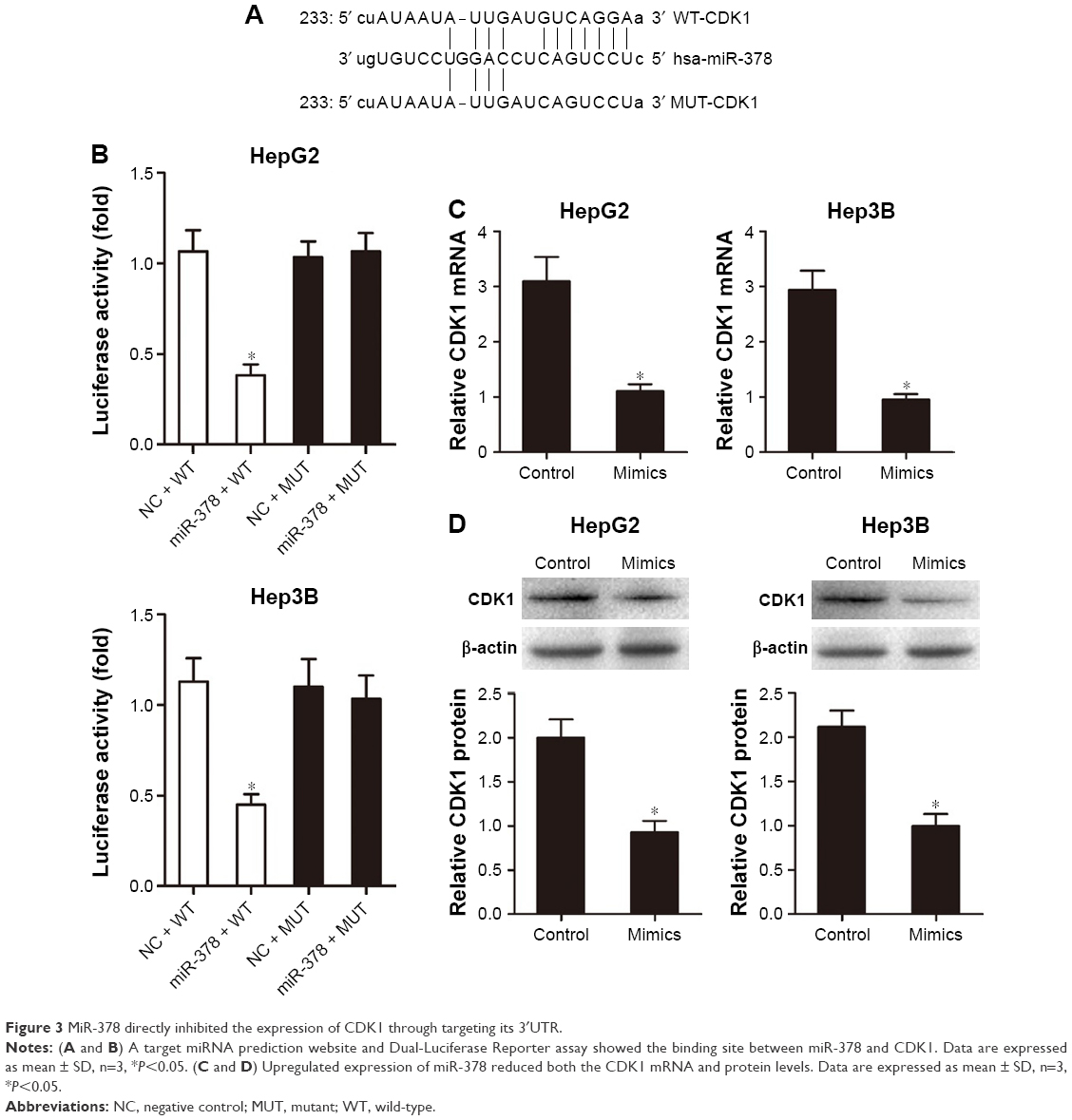 | Figure 3 MiR-378 directly inhibited the expression of CDK1 through targeting its 3′UTR. |
Overexpression of miR-378 inhibited tumor growth in xenograft models of HCC
For further verification, we next evaluated tumor growth of xenografts derived from HepG2 cells that were transfected with miR-378 mimics or NC by subcutaneous injection into nude mice. We found that tumor growth suppression was abrogated in HCC cells xenografts where miR-378 was overexpressed (Figure 4A and B). RT-PCR analysis showed that miR-378 expression was increased in the tumor tissues of miR-378 mimic group compared with that of the control group (Figure 4C). Western blotting analysis showed that CDK1 protein expression was decreased in the tumor tissues of miR-378 mimic group compared with that of control group (Figure 4D). Taken together, we concluded that miR-378 inhibited HCC cell proliferation in vitro and in vivo.
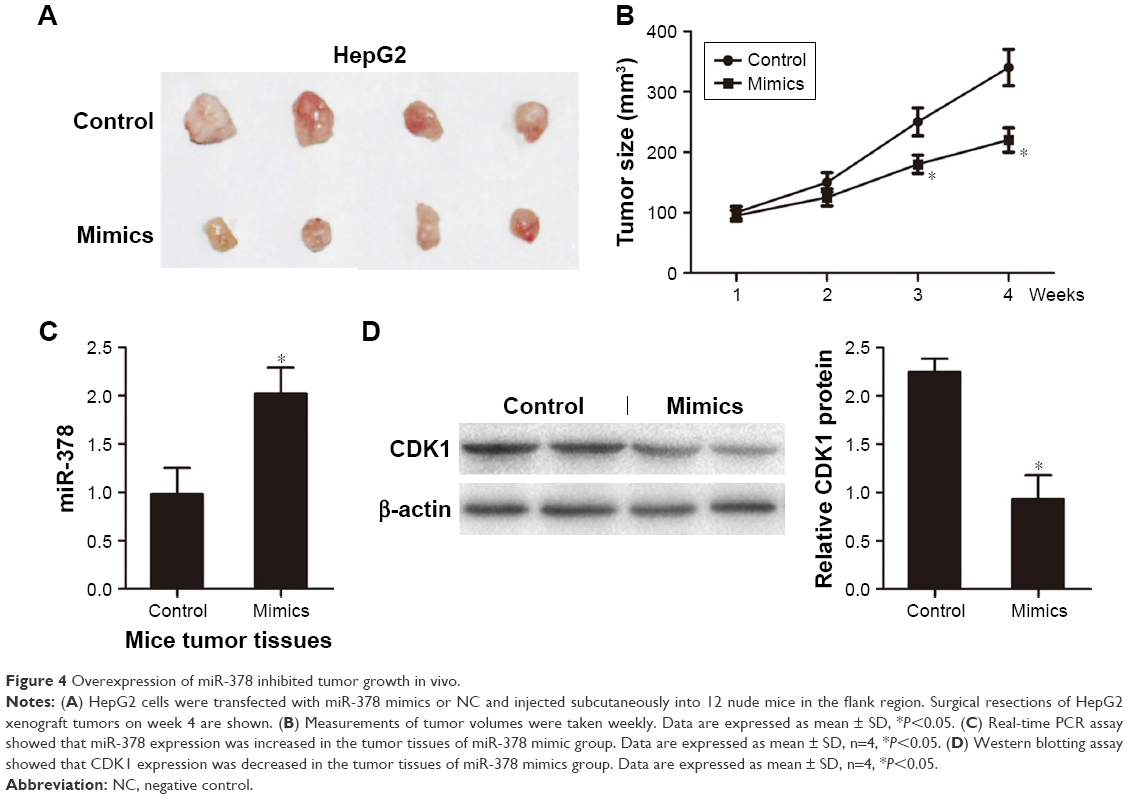 | Figure 4 Overexpression of miR-378 inhibited tumor growth in vivo. |
Metformin inhibited CDK1 expression and cell proliferation via promoting miR-378 level in HCC cells
In order to determine whether metformin inhibited CDK1 expression and cell proliferation via promoting miR-378 level in HCC cells, the CDK1 expression and cell proliferation were examined in HepG2 cells after miR-378 inhibitor transfection for 12 h and metformin pre-treatment for 24 h. The miR-378 inhibitor reversed the decreasing expression of CDK1 caused by metformin in HepG2 cells (Figure 5A). Similarly, the inhibition of cell proliferation caused by metformin treatment was also reversed by miR-378 inhibitor in HepG2 cells (Figure 5B). To confirm whether the repression of cell proliferation was due to G2/M cell cycle arrest, cell cycle distribution was determined by flow cytometry analysis. Certainly, the miR-378 inhibitor blocked G2/M arrest (19%–10.4%) caused by metformin in HepG2 cells (Figure 5C). To examine the effects of metformin-induced cell apoptosis on the repression of cell proliferation, cell apoptosis was determined by Annexin V/PI staining and flow cytometry assays. However, the miR-378 inhibitor could not inhibit metformin-induced cell apoptosis (Figure 5D). Taken together, the findings demonstrated that metformin inhibited CDK1 expression and cell proliferation via increasing miR-378 expression in HCC cells.
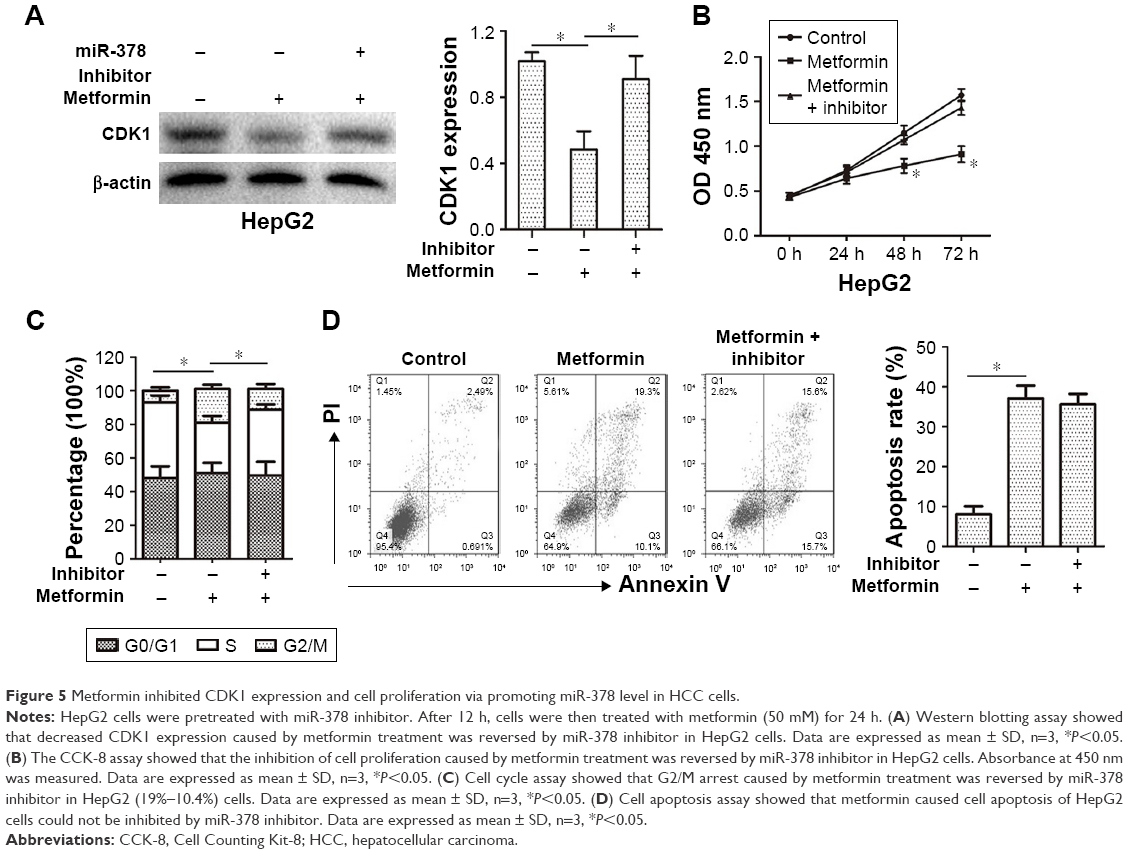 | Figure 5 Metformin inhibited CDK1 expression and cell proliferation via promoting miR-378 level in HCC cells. |
Discussion
HCC has become one of the most frequently occurring malignancies worldwide, but effective treatment options for HCC are limited.17 Recently, metformin has been shown to exert anti-neoplastic effects in many cancer cells including cancers of the breast, liver, colon, pancreas, and skin.18–20 Moreover, recent clinical data suggests that metformin impacts the miRNA profile in cancer. The anticancer activity of metformin is mediated through a direct regulation of miRNAs, which further modulates several downstream genes in metabolic or preoncogenic pathways.21 For example, metformin targets miRNA-222 in inhibiting lung cancer proliferation.7 Metformin targets miRNA-145 and miRNA-21 in treating recurrent colon cancer.22 Metformin targets miRNA-193b in treating triple-negative breast cancer.23 Metformin targets miRNA-26 through suppressing HMGA1 oncogene in treating human pancreatic cancer.24 Furthermore, animal studies have shown that metformin appears to have a direct anti-HCC effect.25
In this study, we evaluated the effects of metformin on the behavior of human HCC cells and found that metformin inhibited cell proliferation of HepG2 and Hep3B cells. Additionally, the molecular mechanism of cell proliferation suppression was associated with the cell cycle signal transduction system. Cell cycle analysis revealed that metformin caused significant delay of the G2/M transition. However, other studies have demonstrated that metformin in combination with curcumin inhibits the growth, metastasis, and angiogenesis of HCC.11 Metformin is a promising therapeutic agent for the elimination of tumor-initiating HCC cells.26 However, the molecular mechanisms of its anti-HCC activities remain poorly understood and warrant further investigation. Therefore, we examined the effect of metformin on miR-378 and CDK1 expression and found that metformin efficiently decreased protein expression of CDK1 and elevated the expression of miR-378. It has been reported that miR-378 may be a tumor suppressor in HCC.27 Interestingly, miR-378 also inhibited HCC cell proliferation via inducing G2/M cell cycle arrest. Furthermore, we demonstrated that a direct interaction between miR-378 and the CDK1 3′UTR by luciferase assay and overexpression of miR-378 caused a significant downregulation of both CDK1 mRNA and protein levels. In addition, overexpression of miR-378 inhibited tumor growth in xenograft nude mice models of HCC in vivo.
Therefore, we proposed that metformin inhibiting the proliferation of HCC cells might be associated with modulating miR-378/CDK1 axis. To clarify the mechanism involved in the suppression of metformin on HCC cells, we used an miR-378 inhibitor to reduce miR-378 level in metformin-treated HCC cells. With miR-378 inhibitor transfection, metformin treatment could not decrease CDK1 expression, suppress HCC cell proliferation, and induce G2/M cell cycle arrest. Inhibitor assays indicated that metformin-suppressed HCC cell proliferation was dependent on the inhibitory effect of miR-378 on CDK1 expression.
Collectively, the current results provide the first evidence, to our knowledge, that miR-378/CDK1 axis is involved in metformin modulating the proliferation of HCC cells, which suggests a novel molecular mechanism underlying the therapeutic effect of metformin on HCC.
Acknowledgment
This work is supported by National Nature Science Foundation of China (No 81670570).
Disclosure
The authors report no conflicts of interest in this work.
References
Gomes MA, Priolli DG, Tralhao JG, Botelho MF. Hepatocellular carcinoma: epidemiology, biology, diagnosis, and therapies. Rev Assoc Med Bras (1992). 2013;59(5):514–524. | ||
Herszenyi L, Tulassay Z. Epidemiology of gastrointestinal and liver tumors. Eur Rev Med Pharmacol Sci. 2010;14(4):249–258. | ||
Wang J, He XD, Yao N, Liang WJ, Zhang YC. A meta-analysis of adjuvant therapy after potentially curative treatment for hepatocellular carcinoma. Can J Gastroenterol. 2013;27(6):351–363. | ||
Chan DL, Morris DL, Chua TC. Clinical efficacy and predictors of outcomes of repeat hepatectomy for recurrent hepatocellular carcinoma – a systematic review. Surg Oncol. 2013;22(2):e23–e30. | ||
Gluer AM, Cocco N, Laurence JM, et al. Systematic review of actual 10-year survival following resection for hepatocellular carcinoma. HPB (Oxford). 2012;14(5):285–290. | ||
Zhou XD. Recurrence and metastasis of hepatocellular carcinoma: progress and prospects. Hepatobiliary Pancreat Dis Int. 2002;1(1):35–41. | ||
Wang Y, Dai W, Chu X, Yang B, Zhao M, Sun Y. Metformin inhibits lung cancer cells proliferation through repressing microRNA-222. Biotechnol Lett. 2013;35(12):2013–2019. | ||
Qu H, Yang X. Metformin inhibits angiogenesis induced by interaction of hepatocellular carcinoma with hepatic stellate cells. Cell Biochem Biophys. 2015;71(2):931–936. | ||
Hsieh SC, Tsai JP, Yang SF, Tang MJ, Hsieh YH. Metformin inhibits the invasion of human hepatocellular carcinoma cells and enhances the chemosensitivity to sorafenib through a downregulation of the ERK/JNK-mediated NF-κB-dependent pathway that reduces uPA and MMP-9 expression. Amino Acids. 2014;46(12):2809–2822. | ||
Yang X, Sun D, Tian Y, Ling S, Wang L. Metformin sensitizes hepatocellular carcinoma to arsenic trioxide-induced apoptosis by downregulating Bcl2 expression. Tumour Biol. 2015;36(4):2957–2964. | ||
Zhang HH, Zhang Y, Cheng YN, et al. Metformin incombination with curcumin inhibits the growth, metastasis, and angiogenesis of hepatocellular carcinoma in vitro and in vivo. Mol Carcinog. 2018;57(1):44–56. | ||
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. | ||
Lee DY, Deng Z, Wang CH, Yang BB. MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. Proc Natl Acad Sci U S A. 2007;104(51):20350–20355. | ||
Urbich C, Kuehbacher A, Dimmeler S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc Res. 2008;79(4):581–588. | ||
Song G, Sharma AD, Roll GR, et al. MicroRNAs control hepatocyte proliferation during liver regeneration. Hepatology. 2010;51(5):1735–1743. | ||
Miyoshi H, Kato K, Iwama H, et al. Effect of the anti-diabetic drug metformin in hepatocellular carcinoma in vitro and in vivo. Int J Oncol. 2014;45(1):322–332. | ||
Pang RW, Poon RT. From molecular biology to targeted therapies for hepatocellular carcinoma: the future is now. Oncology. 2007;72(Suppl 1):30–44. | ||
Morales DR, Morris AD. Metformin in cancer treatment and prevention. Annu Rev Med. 2015;66:17–29. | ||
Do MT, Kim HG, Choi JH, Jeong HG. Metformin induces microRNA-34a to downregulate the Sirt1/Pgc-1α/Nrf2 pathway, leading to increased susceptibility of wild-type p53 cancer cells to oxidative stress and therapeutic agents. Free Radic Biol Med. 2014;74:21–34. | ||
Jara JA, Lopez-Munoz R. Metformin and cancer: between the bioenergetic disturbances and the antifolate activity. Pharmacol Res. 2015;101:102–108. | ||
Zhou JY, Xu B, Li L. A new role for an old drug: metformin targets microRNAs in treating diabetes and cancer. Drug Dev Res. 2015;76(6):263–269. | ||
Nangia-Makker P, Yu Y, Vasudevan A, et al. Metformin: a potential therapeutic agent for recurrent colon cancer. PLoS One. 2014;9(1):e84369. | ||
Wahdan-Alaswad RS, Cochrane DR, Spoelstra NS, et al. Metformin-induced killing of triple-negative breast cancer cells is mediated by reduction in fatty acid synthase via miRNA-193b. Horm Cancer. 2014;5(6):374–389. | ||
Li W, Yuan Y, Huang L, Qiao M, Zhang Y. Metformin alters the expression profiles of microRNAs in human pancreatic cancer cells. Diabetes Res Clin Pract. 2012;96(2):187–195. | ||
Li J, Hernanda PY, Bramer WM, Peppelenbosch MP, van Luijk J, Pan Q. Anti-tumor effects of metformin in animal models of hepatocellular carcinoma: a systematic review and meta-analysis. PLoS One. 2015;10(6):e0127967. | ||
Saito T, Chiba T, Yuki K, et al. Metformin, a diabetes drug, eliminates tumor-initiating hepatocellular carcinoma cells. PLoS One. 2013;8(7):e70010. | ||
An J, Liu J, Liu L, et al. A genetic variant in primary miR-378 is associated with risk and prognosis of hepatocellular carcinoma in a Chinese population. PLoS One. 2014;9(4):e93707. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.