")
Back to Journals » OncoTargets and Therapy » Volume 9
Metformin induces apoptosis of human hepatocellular carcinoma HepG2 cells by activating an AMPK/p53/miR-23a/FOXA1 pathway
Authors Sun Y, Tao C, Huang X, He H, Shi H, Zhang Q, Wu H
Received 3 November 2015
Accepted for publication 14 March 2016
Published 12 May 2016 Volume 2016:9 Pages 2845—2853
DOI https://doi.org/10.2147/OTT.S99770
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Faris Farassati
Yunpeng Sun,1 Chonglin Tao,1 Xiaming Huang,1 Han He,1 Hongqi Shi,1 Qiyu Zhang,1 Huanhuan Wu2
1Department of Hepatobiliary Surgery, 2Department of Infectious Disease, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, People’s Republic of China
Abstract: The antidiabetic drug metformin has been shown to possess antitumor functions in many types of cancers. Although studies have revealed its beneficial effects on the prognosis of hepatocellular carcinoma (HCC), the detailed molecular mechanism underlying this event remains largely unknown. In this work, we showed that miR-23a was significantly induced upon metformin treatment; inhibition of miR-23a abrogated the proapoptotic effect of metformin in HepG2 cells. We next established forkhead box protein A1 (FOXA1) as the functional target of miR-23a, and silencing FOXA1 mimicked the effect of metformin. Moreover, the phosphorylation of AMP-activated protein kinase (AMPK) and the expression of p53 were increased upon metformin treatment, and the inhibition of p53 abrogated the induction of miR-23a by metformin, suggesting that AMPK/p53 signaling axis is responsible for the induction of miR-23a by metformin. In summary, we unraveled a novel AMPK/p53/miR-23a/FOXA1 axis in the regulation of apoptosis in HCC, and the application of metformin could, therefore, be effective in the treatment of HCC.
Keywords: metformin, miR-23a, FOXA1, p53, apoptosis
Introduction
Hepatocellular carcinoma (HCC) has been one of the main reasons of cancer-associated mortality.1 Despite the recent advances in the clinical management of liver cancer, the long-term prognosis of HCC is still dismal. Developing new treatment strategies for HCC could be one of the pressing needs.
It has been widely reported by previous studies that the antidiabetic drug, metformin, exerts critical beneficial effects against the tumor activity of several cancer types including HCC.2–4 The target of metformin, the AMP-activated protein kinase (AMPK), is the energy sensor, which is highly conserved in eukaryotic cells. Under various stressed conditions, the increased adenosine monophosphate (AMP)/adenosine triphosphate (ATP) ratio leads to the phosphorylation and activation of AMPK, which further restricts the energy consumption. Apart from energy control, AMPK is implicated in multiple processes such as apoptosis, autophagy, and senescence.5–8 However, the downstream events for AMPK control of these processes are not fully understood.
MicroRNAs are a class of critical regulators in cancer development and progression. Several microRNAs are implicated in HCC; moreover, it has been recently reported that metformin alters the expression profile of microRNAs in many cancers.9–13 MiR-23a has been established as a tumor suppressor, and it is involved in many pathways related to cell cycle regulation, apoptosis, and epithelial–mesenchymal transition.14–16 Particularly, it has been demonstrated that miR-23a enhances chemosensitivity in HCC,17 addressing its important role in apoptosis regulation. Although several models have been proposed by these studies, the mechanisms underlying the antitumor effects of miR-23a are still incompletely understood.
Forkhead box protein A1 (FOXA1), also known as hepatocyte nuclear factor 3-alpha (HNF-3A), is a critical regulator for liver differentiation during organogenesis and plays pivotal roles in the carcinogenesis of other cancers.18,19 A recent study has shown that FOXA1 serves as a direct target of miR-212 to mediate its antitumor effect in HCC,20 but the molecular mechanism for the regulation of FOXA1 is poorly understood.
In this work, we aimed to investigate whether miR-23a is involved in the metformin-induced apoptotic program. We found that miR-23a was significantly induced upon metformin treatment, and it served as a mediator of the proapoptotic action. Mechanistically, we identified that FOXA1 was functionally targeted by miR-23a. Moreover, we demonstrated that miR-23a was regulated by the upstream AMPK/p53 signaling upon metformin treatment. Our work thus revealed a novel signaling axis that comprises AMPK, p53, miR-23a, and FOXA1 in the regulation of apoptosis in HCC by metformin and may have translational relevance in developing the treatment strategies for HCC.
Materials and methods
Cell culture
HCC cell lines HepG2 (carrying wild-type p53) and Hep3B (p53 deficient) were purchased from American Type Culture Collection (Manassas, VA, USA). The cells were maintained in Dulbecco’s Modified Eagle’s Medium containing 10% fetal bovine serum. Cells grown in culture flask or plates were incubated in a humid atmosphere containing 5% CO2 at 37°C. A total of 50 U/mL penicillin and 100 μg/mL streptomycin were added to the medium to avoid contamination in cases other than transfection. Metformin and Compound C were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). This study did not use human tissues or the primary cultured tumor cells, only cell lines were used, thus, such permission was not required, according to The First Affiliated Hospital of Wenzhou Medical University review board.
Transfection
Small nucleotides such as miR-23a, anti-miR-23a, and their negative controls (NCs) were purchased from GenePharma (Shanghai, People’s Republic of China). The small interfering RNAs (siRNAs) for p53 and FOXA1 were commercially available from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Transfections were performed using Lipofectamine RNAiMAX (Thermo Fisher Scientific, Waltham, MA, USA) according to the protocol provided by the manufacturer.
Cell viability assay
The number of living cells was detected based on the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, cells cultured in 96-well plates were treated with different concentrations of metformin. Cells were cultured for 72 hours, and then the MTT reagent (Sigma-Aldrich Co.) was added to each well. After 4-hour incubation, 200 μL dimethyl sulfoxide was added to each well to detect formazan. The absorbance value at 470 nm was taken using a spectrophotometer.
Terminal deoxynucleotidyl transferase assay
Cells cultured on coverslips were grown in 24-well plates. After the desired treatment, cells were fixed with 4% paraformaldehyde and penetrated by 1% Triton X-100 on ice. The cells were then incubated with terminal deoxynucleotidyl transferase assay (TUNEL) reagent (Hoffman-La Roche Ltd, Basel, Switzerland) at 37°C for 1 hour according to the instructions provided by the manufacturer. Counterstaining was performed with 4′,6-diamidino-2-phenylindole (Hoffman-La Roche Ltd). For quantification of apoptosis, five nonoverlapping fields were taken from each slide and five biological repeats were performed in each group.
Real-time polymerase chain reaction
Total RNA was extracted by TRizol Reagent (Thermo Fisher Scientific). The first strand of complementary DNA was synthesized by a reverse transcriptase kit (Promega Corporation, Madison, WI, USA) using the predesigned commercial stem-loop primer set for miR-23a (GenePharma). U6 was used as an internal control, and the first strand for U6 was reverse transcribed by random primers provided in the reverse transcriptase kit. Amplification was performed using a 7500 Fast Real-Time PCR System (Thermo Fisher Scientific).
Western blot
Cells were homogenized by radioimmunoprecipitation assay buffer (Beyotime, Shanghai, People’s Republic of China), and the lysates were heat denatured in a sodium dodecyl sulfate loading buffer prior to electrophoresis. After the proteins were separated, proteins were transferred on to a polyvinylidene fluoride membrane, and the membranes were blocked with 5% skimmed milk. The milk-blocked membranes were incubated overnight with primary antibody. Secondary antibodies were incubated on the following day after a series of washing with phosphate-buffered saline–Tween 20. The band was detected with ECL detection system (Beyotime). The antibody sources were as follows: the primary antibodies for cleaved-caspase-3, AMPK, and p-AMPK were from Cell Signaling Technologies Inc. (Beverly, MA, USA), and the primary antibodies for p53, FOXA1, BCL-2, BAX, PARP, and Actin were from Santa Cruz Biotechnology Inc. All the secondary antibodies that are conjugated with horseradish peroxidase were also purchased from Santa Cruz Biotechnology Inc.
Luciferase activity assay
We cloned a fragment of the 3′ untranslated region (3′UTR) sequence from the FOXA1 messenger RNA. The sequence was then inserted into pMiR-reporter to establish the mutant 3′UTR; the seed sequence was mutated to AGGCUCUGC. Wild-type and mutant FOXA1 3′UTR plasmids were transfected along with miR-23a into HEK293 cells for 24 hours. The luciferase activity was detected according to the instructions of Dual luciferase activity assay kit (Promega Corporation). For adjusting the luciferase activity, Renilla luciferase, which is carried by TK plasmid, was used as an internal control.
Statistical analysis
Data were expressed as mean ± standard error of the mean. One-way analysis of variance was used for multiple comparisons; pairwise comparisons were performed by Student–Newman–Keuls post hoc test. For comparisons between two groups, independent t-test was used. A two-sided P<0.05 was taken as statistical significance.
Results
Metformin induces apoptosis in HCC cells
To evaluate the effect of metformin on HCC cells, we first treated HepG2 cells with various concentrations of metformin. As shown in Figure 1A, we observed a dose-dependent reduction of cell viability. Importantly, as we assessed the expression level of critical molecules in apoptosis, we found that the caspase substrates PARP were cleaved in a dose-dependent manner (Figure 1B and C). BAX and BCL-2 are two important molecules in apoptosis initiation; we found that proapoptotic BAX was gradually increased and the antiapoptotic BCL-2 was decreased with the increase of metformin dose (Figure 1B, D, and E). These results suggest that metformin induces apoptosis in HCC cell line HepG2.
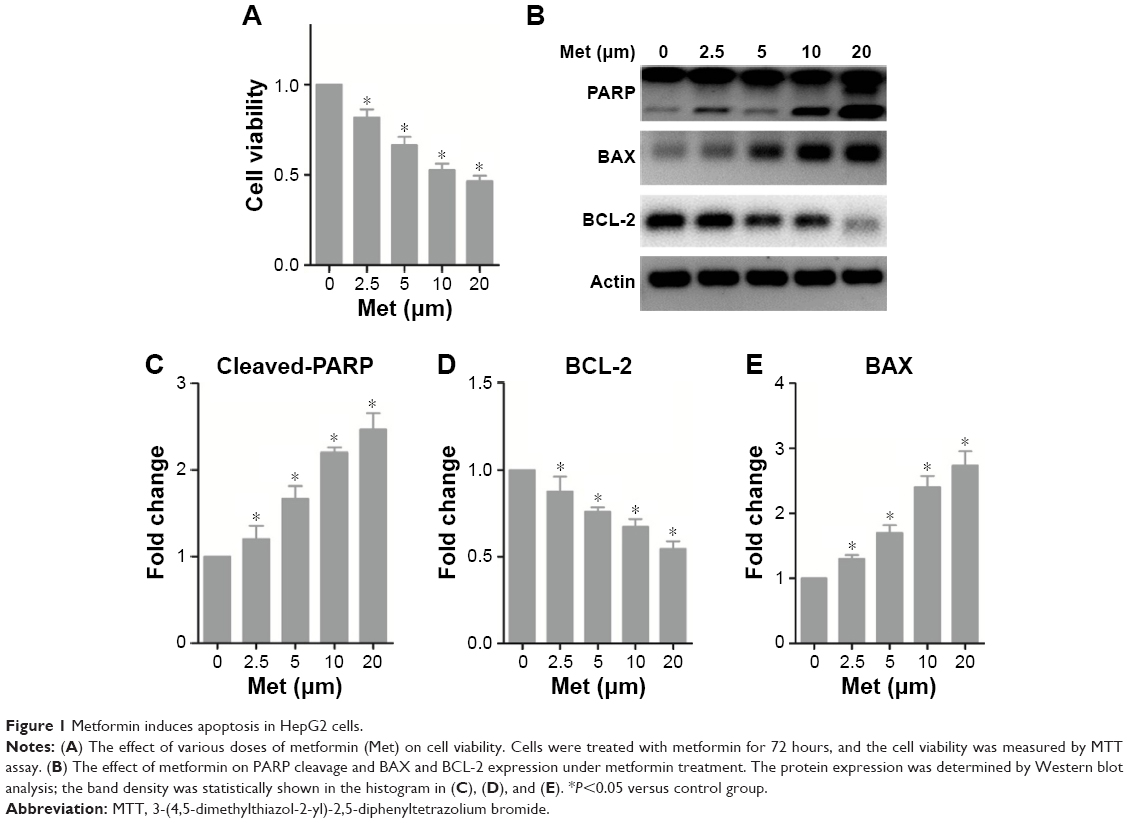 | Figure 1 Metformin induces apoptosis in HepG2 cells. |
Metformin induces apoptosis through activating miR-23a
We assessed miR-23a expression levels and found that miR-23a was significantly induced by metformin (Figure 2A). To test whether miR-23a plays an important role in the event of metformin-caused apoptosis, we manipulated endogenous miR-23a levels by transfecting its exact antisense nucleotide. Results of TUNEL assay showed that metformin significantly increased TUNEL-positive cells in NC-transfected cells; in contrast, the apoptosis was just slightly changed under miR-23a inhibition condition (Figure 2B). Western blot analysis of apoptosis markers PARP and cleaved-caspase-3 showed similar results; both PARP and caspase-3 underwent cleavage significantly under NC transfection condition; however, this effect was efficiently blocked by miR-23a inhibition (Figure 2C and D).
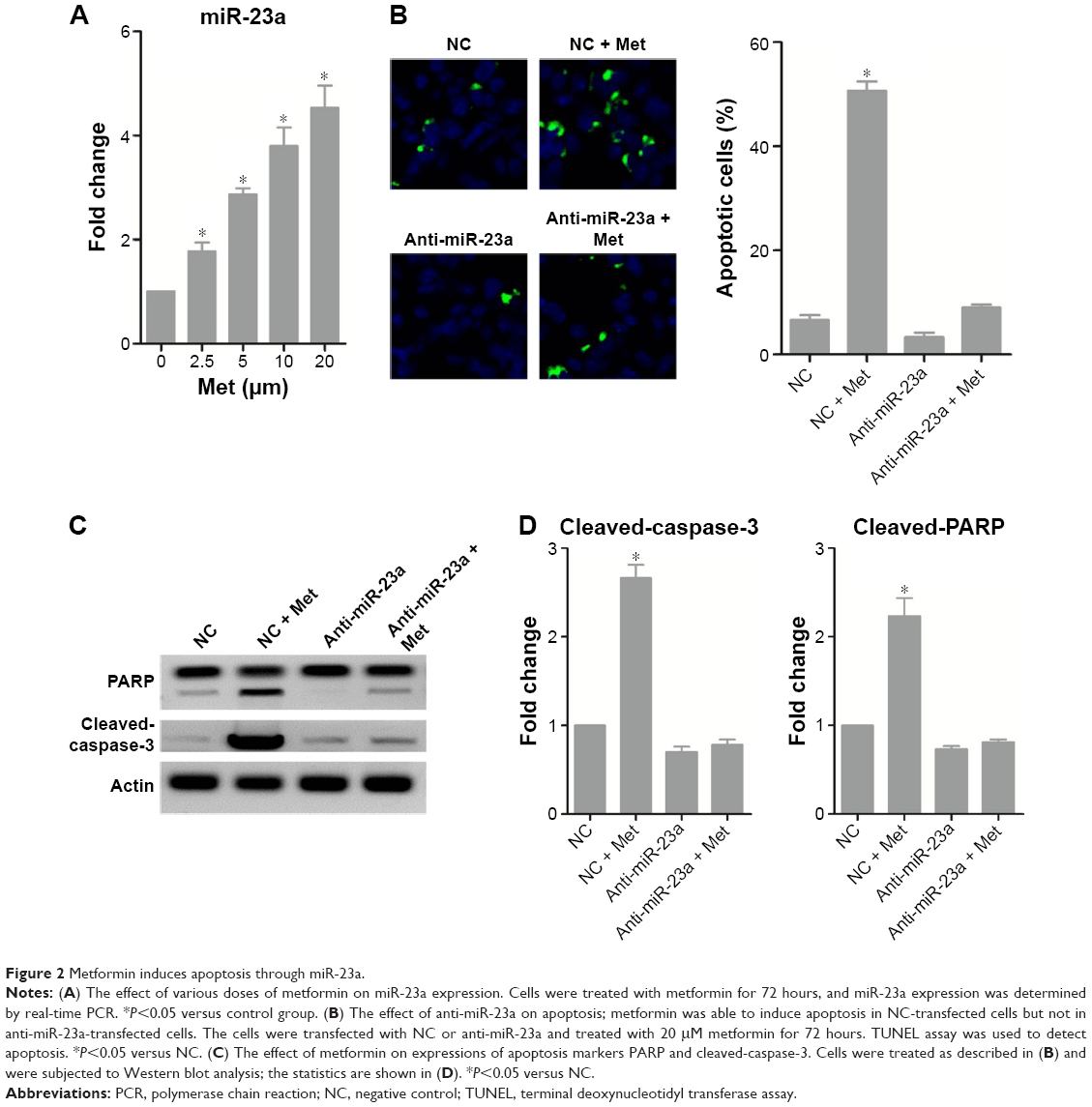 | Figure 2 Metformin induces apoptosis through miR-23a. |
FOXA1 is a functional target of miR-23a
To elucidate the molecular mechanism of the proapoptotic action of miR-23a, we searched the miRanda database and found that FOXA1 was potentially targeted by miR-23a (Figure 3A). Transfection of anti-miR-23a increased the protein expression of FOXA1 (Figure 3B), and in contrast, overexpression of miR-23a dramatically reduced FOXA1 expression (Figure 3C). Luciferase assay revealed that miR-23a was able to reduce luciferase activity in wild-type 3′UTR but not in mutant 3′UTR (Figure 3D), suggesting the direct interaction between miR-23a and FOXA1 messenger RNA.
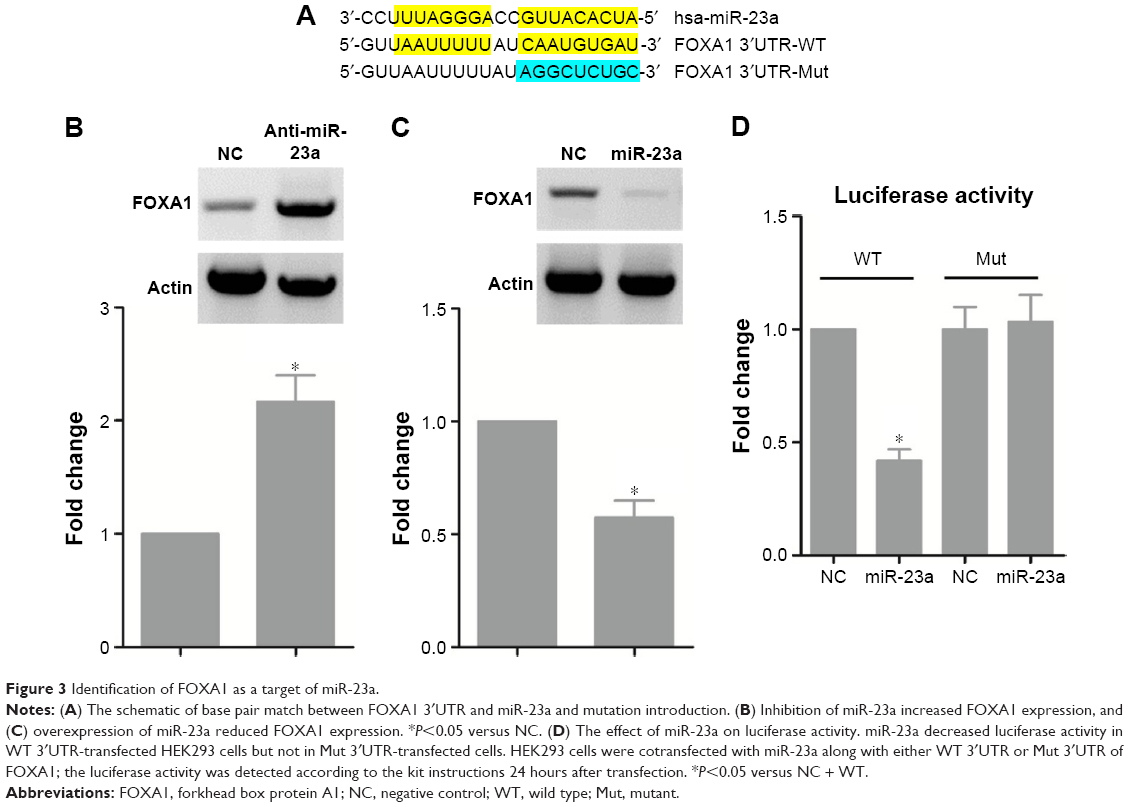 | Figure 3 Identification of FOXA1 as a target of miR-23a. |
Inhibition of FOXA1 mimics the proapoptotic effect of metformin
We next showed whether FOXA1 mediated the effect of miR-23a. Western blot analysis confirmed the knockdown efficiency of FOXA1 siRNA (Figure 4A), and transfection of the specific siRNA for FOXA1 significantly promoted PARP and caspase-3 cleavage, which mimicked the effect of metformin, and treatment with metformin only slightly enhanced the effect of si-FOXA1 alone (Figure 4A and B). Moreover, silencing FOXA1 decreased cell viability and increased TUNEL-positive cells significantly, all of which showed similar effects with metformin. Importantly, these proapoptotic effects of metformin were attenuated under FOXA1 knockdown condition (Figure 4C and D). These results confirmed that metformin-induced miR-23a upregulation at least partially functions through FOXA1.
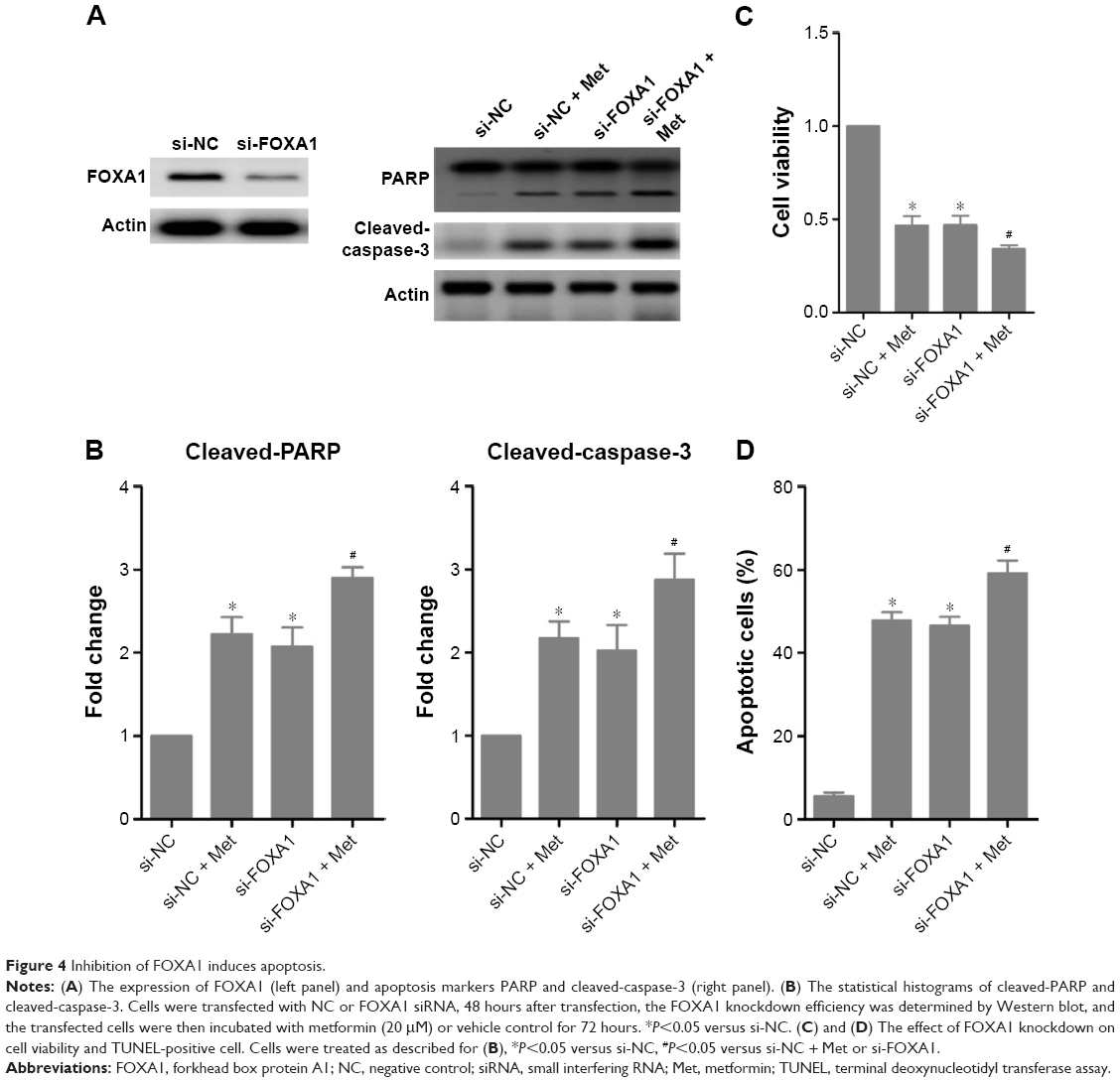 | Figure 4 Inhibition of FOXA1 induces apoptosis. |
MiR-23a induced by metformin is regulated by AMPK/p53 signaling
Through which mechanism is miR-23a regulated? It is reported that AMPK activates p53 signaling; we, therefore, tested the potential transcriptional program that involves AMPK and p53 and found that metformin activated AMPK and increased the expression of p53 (Figure 5A and B). Importantly, silencing p53 abrogated the inductive role of metformin on miR-23a in HepG2 cells (Figure 5C and D). Moreover, the AMPK inhibitor Compound C reduced miR-23a induction by metformin (Figure 5E). We next treated p53-deficient Hep3B cells with metformin and found that it failed to induce miR-23a expression (Figure 5F), which suggested that miR-23a induced by metformin requires p53 activity. These data indicated that AMPK/p53 signaling acts upstream of miR-23a.
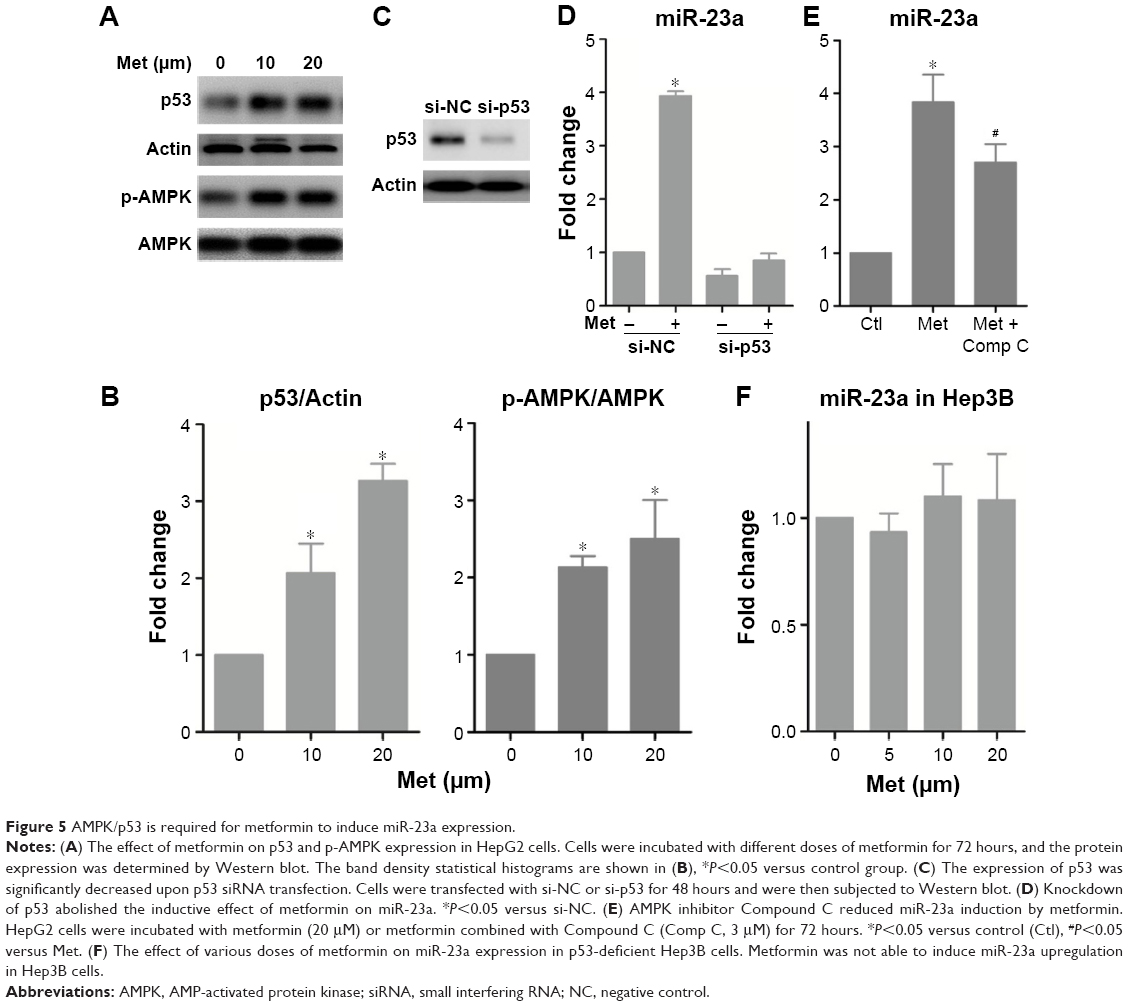 | Figure 5 AMPK/p53 is required for metformin to induce miR-23a expression. |
Discussion
As a classic antidiabetic drug, metformin has been recently shown to exert anticancer effects in many cancers by retrospective studies and experimental validations.21–23 Although some of these studies have proposed several possible mechanisms, the detailed molecular basis in downstream signaling remains largely unknown. As such, we focused on the downstream signaling events related to the anticancer action of metformin. We found that the apoptotic level and the expression of miR-23a are significantly increased in HepG2 cells upon metformin treatment, and modulation of the levels of miR-23a has critical impact on apoptosis. Further evidence showed that FOXA1 functions downstream of miR-23a and that the AMPK/p53 functions upstream of miR-23a. Therefore, we established a signaling axis that involves AMPK, p53, miR-23a, and FOXA1 in the regulation of apoptosis in HCC (Figure 6); our study may highlight the potential application of metformin in HCC treatment.
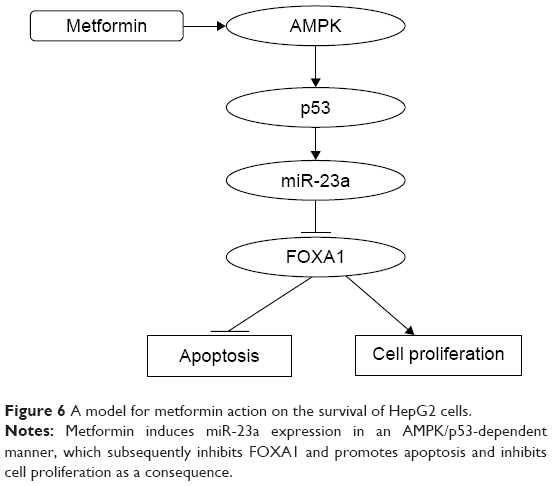 | Figure 6 A model for metformin action on the survival of HepG2 cells. |
MicroRNAs have evolutionarily conserved roles in regulating many aspects of cellular functions. The role of microRNAs in the development of progression of HCC has been extensively studied. Recent studies have shown that miR-23a plays a proapoptotic role in many cancer types;15,24 however, the reports did not reach consistency. For example, miR-23a contributes to increased radiosensitivity in nasopharyngeal carcinoma cells by altering the interleukin-8/signal transducer and activator of transcription 3 (STAT3) pathway.25 In contrast, miR-23a promoted proliferation and repressed apoptosis in pancreatic cancer cells.26 Most importantly, in HCC, it has been shown to mediate the cell responses to anticancer drugs.16,17 Consistent with most of the studies, we showed that miR-23a exerted proapoptotic function in HCC cells. Multiple genes can be targeted by a single microRNA. In our work, gain or loss of function of miR-23a exerted opposite effects in the protein level of FOXA1; combined with the data obtained from the luciferase activity assay, we identified FOXA1 as a novel target of miR-23a for the first time.
FOXA1, or HNF-3A, has a prominent role in liver development, and it is a critical transcriptional activator of liver-specific genes such as albumin. FOXA1 has been identified as a marker for breast cancer, as its expression is highly correlated with the hormone receptors such as estrogen receptor and progesterone receptor.27 Moreover, mutations of FOXA1 are often observed in prostate cancers.19 Despite its critical role in hepatocyte differentiation and other cancers, its functions in liver carcinogenesis and progression are seldom reported. Previous studies have demonstrated that FOXA factors are essential for sexual dimorphism in liver cancer,28 and its interplay with estrogen receptor has been discussed.29 A more recent finding demonstrates that FOXA1 acts as a downstream target of miR-212 and is a potential prognostic marker of HCC.20 In this study, we showed for the first time that FOXA1 is involved in the proapoptotic effect of metformin, and silencing FOXA1 mimicked the proapoptotic effect of metformin, suggesting that FOXA1 is an oncogene for HCC development. Moreover, we established a direct interaction between FOXA1 and miR-23a; our findings provided new understandings in the pathogenesis and treatment of HCC.
Metformin was initially applied to diabetes mellitus by restricting energy production and consumption. The main mechanism of this action was highly dependent upon AMPK activation. Recent retrospective data found that metformin shows beneficial effect in tumor prevention, which greatly broadened its potential application. As energy metabolism is critical in cancer development, AMPK might be a promising target for cancer treatment. Zheng et al4 reported that in clinical samples, the phosphorylation status of AMPK is overall lower in HCC tissues compared with that in normal tissues. And low p-AMPK expression correlated with poor prognosis, suggesting the potential treatment strategy of AMPK activation by metformin.4 At present, a wide spectrum of signaling pathways has been identified regulated by metformin apart from AMPK. Miyoshi et al13 recently reported that receptor tyrosine kinases such as epidermal growth factor receptor and angiogenesis-related molecules are altered by metformin; in addition, they identified a spectrum of microRNAs that are altered by metformin. Consistent with their findings, we showed that miR-23a is upregulated by metformin. In searching for the detailed mechanism by which miR-23a is regulated, we tested the previously identified AMPK/p53 pathway30 and found that AMPK was significantly activated accompanied with increased p53 expression by metformin. Silencing p53 in HepG2 cells dramatically inhibited the induction of miR-23a. Moreover, miR-23a cannot be induced in p53-deficient Hep3B cells, indicating that p53 is required for metformin to induce miR-23a. However, whether miR-23a is a direct transcriptional target of p53 remains to be clarified in future works.
It is also conceivable that miR-23a–FOXA1 axis may not be the only pathway to mediate metformin-induced apoptosis. Other possible mechanisms underlying the anticancer action of metformin needs further investigation. For example, Takayama et al31 reported that another forkhead transcriptional factor, FOXO3a, was inactivated by metformin by regulating its nuclear localization rather than affecting its expression. Moreover, FOXO3a was found to be functionally connected with p53 and AMPK,32,33 and it plays a critical role in hepatocarcinogenesis.34,35 It would be interesting to test whether it cooperates synergistically with FOXA1, p53, and AMPK in HCC upon metformin treatment. Intriguingly, previous finding suggested that miR-23a targets FOXO3a in heart;36 whether miR-23a–FOXO3a axis is involved in metformin-induced HepG2 cell apoptosis needs to be elucidated. It has been shown that STAT3 and nuclear factor-kappa B signaling was also significantly affected by metformin treatment, both of which play important roles in maintaining the tumor activity.4 In future, the downstream signaling events of STAT3 and nuclear factor-kappa B may be well characterized.
Conclusion
Our present work identified a novel signaling pathway that involves AMPK, p53, miR-23a, and FOXA1 in metformin-caused apoptosis in HCC cells. Our study provided a novel insight into a critical role of microRNA regulation in apoptosis by chemical drugs and indicated the potential application of metformin in the treatment of HCC.
Disclosure
The authors report no conflicts of interest in this work.
References
Bosch FX, Ribes J, Diaz M, Cleries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127(5 suppl 1):S5–S16. | ||
Fujimori T, Kato K, Fujihara S, et al. Antitumor effect of metformin on cholangiocarcinoma: in vitro and in vivo studies. Oncol Rep. 2015;34(6):2987–2996. | ||
Chai X, Chu H, Yang X, Meng Y, Shi P, Gou S. Metformin increases sensitivity of pancreatic cancer cells to gemcitabine by reducing CD133(+) cell populations and suppressing ERK/P70S6K signaling. Sci Rep. 2015;5:14404. | ||
Zheng L, Yang W, Wu F, et al. Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin Cancer Res. 2013;19(19):5372–5380. | ||
Li N, Huang D, Lu N, Luo L. Role of the LKB1/AMPK pathway in tumor invasion and metastasis of cancer cells (Review). Oncol Rep. 2015;34(6):2821–2826. | ||
Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. | ||
Burkewitz K, Zhang Y, Mair WB. AMPK at the nexus of energetics and aging. Cell Metab. 2014;20(1):10–25. | ||
Queiroz EA, Puukila S, Eichler R, et al. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS One. 2014;9(5):e98207. | ||
Tanaka R, Tomosugi M, Horinaka M, Sowa Y, Sakai T. Metformin causes G1-phase arrest via down-regulation of MiR-221 and enhances TRAIL sensitivity through DR5 Up-regulation in pancreatic cancer cells. PLoS One. 2015;10(5):e0125779. | ||
Yang FQ, Wang JJ, Yan JS, et al. Metformin inhibits cell growth by upregulating microRNA-26a in renal cancer cells. Int J Clin Exp Med. 2014;7(10):3289–3296. | ||
Bao B, Azmi AS, Ali S, Zaiem F, Sarkar FH. Metformin may function as anti-cancer agent via targeting cancer stem cells: the potential biological significance of tumor-associated miRNAs in breast and pancreatic cancers. Ann Transl Med. 2014;2(6):59. | ||
Cioce M, Valerio M, Casadei L, et al. Metformin-induced metabolic reprogramming of chemoresistant ALDHbright breast cancer cells. Oncotarget. 2014;5(12):4129–4143. | ||
Miyoshi H, Kato K, Iwama H, et al. Effect of the anti-diabetic drug metformin in hepatocellular carcinoma in vitro and in vivo. Int J Oncol. 2014;45(1):322–332. | ||
Zheng H, Li W, Wang Y, et al. miR-23a inhibits E-cadherin expression and is regulated by AP-1 and NFAT4 complex during Fas-induced EMT in gastrointestinal cancer. Carcinogenesis. 2014;35(1):173–183. | ||
Nie M, Yu S, Peng S, Fang Y, Wang H, Yang X. miR-23a and miR-27a promote human granulosa cell apoptosis by targeting SMAD5. Biol Reprod. 2015;93(4):98. | ||
Wang N, Zhu M, Wang X, Tan HY, Tsao SW, Feng Y. Berberine-induced tumor suppressor p53 up-regulation gets involved in the regulatory network of MIR-23a in hepatocellular carcinoma. Biochim Biophys Acta. 2014;1839(9):849–857. | ||
Wang N, Zhu M, Tsao SW, Man K, Zhang Z, Feng Y. MiR-23a-mediated inhibition of topoisomerase 1 expression potentiates cell response to etoposide in human hepatocellular carcinoma. Mol Cancer. 2013;12(1):119. | ||
Kouros-Mehr H, Slorach EM, Sternlicht MD, Werb Z. GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell. 2006;127(5):1041–1055. | ||
Barbieri CE, Baca SC, Lawrence MS, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44(6):685–689. | ||
Dou C, Wang Y, Li C, et al. MicroRNA-212 suppresses tumor growth of human hepatocellular carcinoma by targeting FOXA1. Oncotarget. 2015;6(15):13216–13228. | ||
Sui X, Xu Y, Wang X, Han W, Pan H, Xiao M. Metformin: a novel but controversial drug in cancer prevention and treatment. Mol Pharm. 2015;12(11):3783–3791. | ||
Marks AR, Pietrofesa RA, Jensen CD, Zebrowski A, Corley DA, Doubeni CA. Metformin use and risk of colorectal adenoma after polypectomy in patients with type 2 diabetes mellitus. Cancer Epidemiol Biomarkers Prev. 2015;24(11):1692–1698. | ||
Cheng G, Lanza-Jacoby S. Metformin decreases growth of pancreatic cancer cells by decreasing reactive oxygen species: Role of NOX4. Biochem Biophys Res Commun. 2015;465(1):41–46. | ||
Zhang XW, Liu N, Chen S, et al. Upregulation of microRNA-23a regulates proliferation and apoptosis by targeting in laryngeal carcinoma. Oncol Lett. 2015;10(1):410–416. | ||
Qu JQ, Yi HM, Ye X, et al. MiR-23a sensitizes nasopharyngeal carcinoma to irradiation by targeting IL-8/Stat3 pathway. Oncotarget. 2015;6(29):28341–28356. | ||
Liu N, Sun YY, Zhang XW, et al. Oncogenic miR-23a in pancreatic ductal adenocarcinogenesis via inhibiting APAF1. Dig Dis Sci. 2015;60(7):2000–2008. | ||
Ross-Innes CS, Stark R, Teschendorff AE, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481(7381):389–393. | ||
Li Z, Tuteja G, Schug J, Kaestner KH. Foxa1 and Foxa2 are essential for sexual dimorphism in liver cancer. Cell. 2012;148(1–2):72–83. | ||
Zhao Y, Li Z. Interplay of estrogen receptors and FOXA factors in the liver cancer. Mol Cell Endocrinol. 2015;418(pt 3):334–339. | ||
He G, Zhang YW, Lee JH, et al. AMP-activated protein kinase induces p53 by phosphorylating MDMX and inhibiting its activity. Mol Cell Biol. 2014;34(2):148–157. | ||
Takayama H, Misu H, Iwama H, et al. Metformin suppresses expression of the selenoprotein P gene via an AMP-activated kinase (AMPK)/FoxO3a pathway in H4IIEC3 hepatocytes. J Biol Chem. 2014;289(1):335–345. | ||
Wang F, Marshall CB, Yamamoto K, et al. Structures of KIX domain of CBP in complex with two FOXO3a transactivation domains reveal promiscuity and plasticity in coactivator recruitment. Proc Natl Acad Sci U S A. 2012;109(16):6078–6083. | ||
Wang F, Marshall CB, Yamamoto K, et al. Biochemical and structural characterization of an intramolecular interaction in FOXO3a and its binding with p53. J Mol Biol. 2008;384(3):590–603. | ||
Xie C, Xie DY, Lin BL, et al. Interferon-beta gene-modified human bone marrow mesenchymal stem cells attenuate hepatocellular carcinoma through inhibiting AKT/FOXO3a pathway. Br J Cancer. 2013;109(5):1198–1205. | ||
Xie C, Song LB, Wu JH, et al. Upregulator of cell proliferation predicts poor prognosis in hepatocellular carcinoma and contributes to hepatocarcinogenesis by downregulating FOXO3a. PLoS One. 2012;7(7):e40607. | ||
Wang K, Lin ZQ, Long B, Li JH, Zhou J, Li PF. Cardiac hypertrophy is positively regulated by MicroRNA miR-23a. J Biol Chem. 2012;287(1):589–599. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.