")
Back to Journals » Research and Reports in Urology » Volume 11
Metastatic Perivascular Epithelioid Cell Tumor Of The Kidney: A Case Report With Emphasis On Response To The Tyrosine-Kinase Inhibitor Sunitinib
Authors AlAzab RS, Alorjani MS , Sahawneh FE , Al-Sukhun S
Received 5 August 2019
Accepted for publication 24 October 2019
Published 13 November 2019 Volume 2019:11 Pages 311—317
DOI https://doi.org/10.2147/RRU.S226005
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jan Colli
Rami S AlAzab,1 Mohammed S Alorjani,2 Firas E Sahawneh,1 Sana Al-Sukhun3
1Urology Division, Department of Surgery, Faculty of Medicine, Jordan University of Science and Technology, Irbid, Jordan; 2Department of Pathology and Microbiology, Faculty of Medicine, Jordan University of Science and Technology, Irbid, Jordan; 3Oncology Practice, Hyatt Medical Center, Amman, Jordan
Correspondence: Rami S AlAzab
Urology Division, Department of Surgery, Faculty of Medicine, Jordan University of Science and Technology, P.O. Box 3030, Irbid 22110, Jordan
Tel +962799530333
Fax +962 2 7095010
Email [email protected]
Abstract: Several case reports have provided most of the information available on perivascular epithelioid cell tumors, but with no consensus regarding diagnosis or treatment paradigms. Sunitinb is a VEGFR multitargeted tyrosine-kinase inhibitor that is regarded as a first-line treatment of clear cell–type metastatic renal-cell carcinoma after cytoreductive surgery. Our case was a 29-year-old male who presented with a metastatic left renal tumor and underwent left partial nephrectomy followed by adjuvant tyrosine kinase–inhibitor treatment. We report this case to present the peculiarity of this rare pathological variant, its clinical and diagnostic features, and challenges regarding treatment options, since the response of this rare tumor to adjuvant tyrosine kinase–inhibitor therapy has not been well described.
Keywords: PEComa, tyrosine kinase, renal-cell carcinoma, sarcoma, everolimus
Introduction
Perivascular epithelioid cell tumors (PEComas) are mesenchymal tumors composed of histologically and immunohistochemically distinctive PECs. Related mesenchymal neoplasms include angiomyolipomas (AMLs), lymphangiomyomatosis, and clear-cell tumors of the lung. Similar lesions arise at a variety of visceral and soft tissue. The cell of origin is classified as the PEC (which has no known normal tissue counterpart), and the uterus is the most common site of involvement. Most PEComas are benign and do not recur after local radical surgical removal. Some cases have a well-described malignant or metastatic variant that has a uniformly fatal outcome.
Molecular pathophysiology studies have described aberrant mTOR signaling, which constitutes the rationale to use mTOR inhibitors (mTORIs) as a treatment option. Current guidelines recommend tyrosine-kinase inhibitors (TKIs) after cytoreductive nephrectomy in metastatic renal-cell carcinoma. Here, we describe a case of metastatic renal PEComa treated with an adjuvant TKI (sunitinb) and mTORIs. We selected sunitinib as first-line treatment because it inhibits multiple growth factors, including VEGFR1, VEGFR2, and VEGFR3, in addition to PDGFRs, KIT, and FLT3R, in addition to our institutional experience with this agent.
Case
A 39-year-old man presented in October 2015 with recurrent epigastric pain radiating to the back, without other gastrointestinal or constitutional symptoms. On physical examination, he had tenderness on palpation of the upper abdominal area. His blood investigations were unremarkable, but CRP was high at 48 and erythrocyte-sedimentation rate at 65. Gastroduodenoscopy revealed hiatal hernia. An abdominal computed tomography (CT) scan revealed a 7 cm mass in the left kidney with ipsilateral retroperitoneal lymph-node enlargement and bilateral lung lesions. We performed a left partial nephrectomy in combination with ipsilateral left-node dissection. Pathological evaluation revealed a 6.8×7×5.5 cm mass, confined to the renal capsule with negative surgical margins. The tumor showed deep pigmentation upon slicing, which gave an initial impression that it might represent pigmented clear-cell renal-cell carcinoma or metastatic malignant melanoma. Microscopically, the pigmented nodular tumor was composed of sheets, nests, and trabeculae of brown pigment–containing spindles and occasionally rounded cells (Figure 1). Tumor-cell nuclei had prominent small red nucleoli with occasional nuclear grooves. Immunohistochemically, the tumor was positive for vimentin, melanin A, HMB45, CK, and CK7, and demonstrated patchy, weak SMA and BCL2 positivity, but was negative for CK20, CD10, synatophysin, chromogranin, PAX8, desmin, CD34, and CD99. All neuroendocrine tumor markers were negative with areas of tumor necrosis and rare mitoses. These findings were consistent with pigmented epithelial and focal spindle-cell malignancy, in keeping with the diagnosis of malignant PEComa. Table 1 presents the positive and negative immunohistochemical markers.
 |
Table 1 Immunohistochemical Markers Tested |
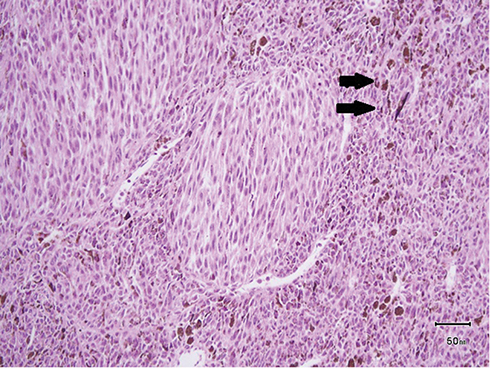 |
Figure 1 Microscopically, the pigmented nodular tumor was composed of sheets, nests, and trabeculae of brown pigment–containing spindles and occasionally rounded cells (black arrows). |
After a multidisciplinary meeting, the patient was started on sunitinib (50 mg daily for 4 weeks, followed by a 2-week break). Our choice of sunitinib was based on data available showing that TKIs are efficient as an adjuvant treatment in the setting of metastatic renal-cell carcinoma and because at that time sunitinib was covered by governmental insurance. Early CT imaging at 1 month revealed complete resection of the tumor with no evidence of local, regional, or distant progression. The patient tolerated the treatment well, apart from mild initial diarrhea, and the disease remained clinically and radiologically stable on sunitinib for 36 weeks. CT imaging at 7 months (Figure 2) and 9 months (Figure 3) after surgery confirmed no renal recurrence and stable retroperitoneal and lung metastasis, with positive regional and distant recurrence. Progression was in the form of pulmonary, mediastinal, and retroperitoneal lymph-node enlargement with new lesions in the left adrenal gland and left kidney. Figure 3 shows the mediastinal and right adrenal recurrence with a new development in the left adrenal nodule. His performance status was 1, so we elected to start him on the mTORI everolimus (10 mg daily). His disease continued to be stable for the next 9 months; however, during this period we withheld everolimus twice during treatment: once due to fatigue and mucositis, and he needed hospitalization the second time, due to noninfectious pneumonitis. After his second progression, the multidisciplinary team decided to rechallenge him with another trial of sunitinib. After one cycle, we discontinued treatment, due to severe diarrhea and poor general status. Enhanced CT scans of the chest, abdomen, and pelvis showed multiple new metastases and progression of pulmonary, pericardial, and retroperitoneal metastasis (Figure 4). Afterward, the patient had several admissions after deterioration of his general condition, during which he received best supportive therapy. Unfortunately, he succumbed to disease progression about 2 years after surgery. Institutional research board approval is not required for case reports. Written informed consent for publication of the case details and accompanying images was supplied by the patient’s older brother.
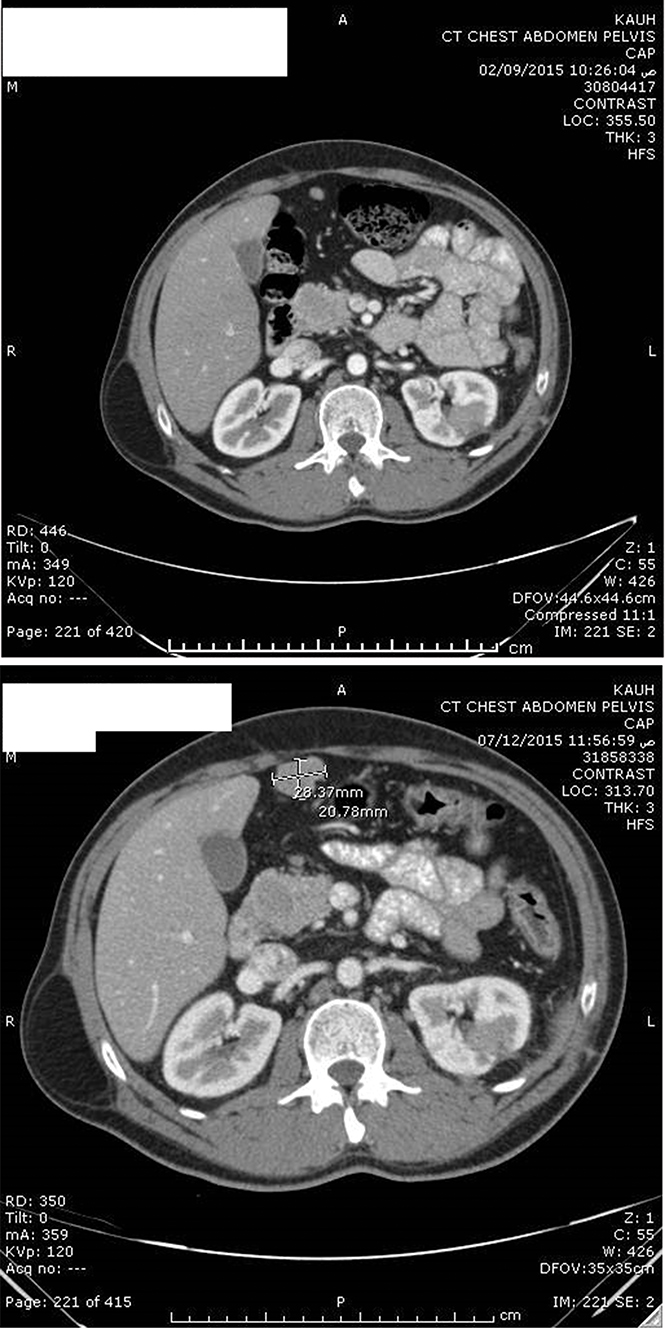 |
Figure 2 Postoperative enhanced CT scan showing stable right upper retroperitoneal metastasis after 7 months (upper image) and 9 months (lower image) after resection of the left renal tumor. |
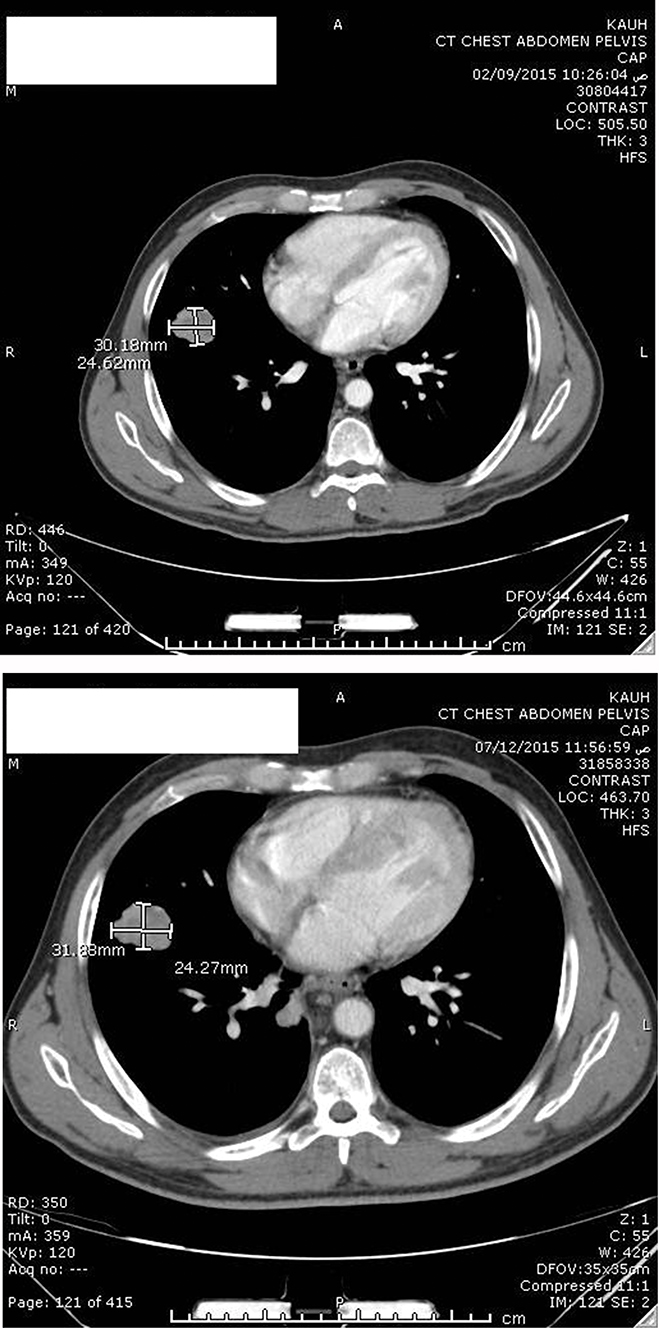 |
Figure 3 Postoperative enhanced CT scan showing stable right pulmonary metastasis after 7 months (upper image) and 9 months (lower image) after resection of the left renal tumor. |
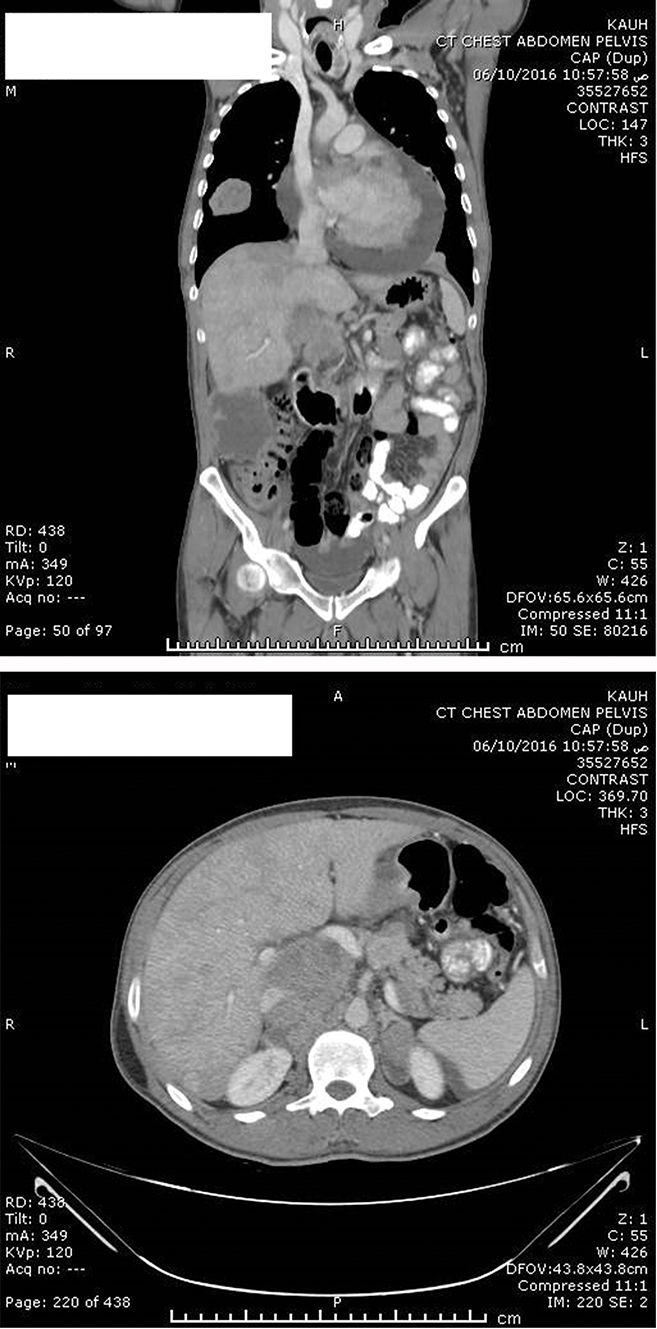 |
Figure 4 Mediastinal recurrence with pericardial effusion, bilateral adrenal and right intraperitoneal recurrences (upper image). Right adrenal recurrence with a new development of left adrenal nodule (lower image). |
Discussion
PEComas are defined by the World Health Organization Classification as “mesenchymal tumors composed of histologically and immunohistochemically distinctive perivascular epithelioid cells”.1 PEComa of the kidney is a broad term that includes many entities. AML is the most common, but rare entities like microscopic AML (microhamartoma), intraglomerular lesions, cystic AML, epithelioid AML, oncocytoma-like AML, and lymphangiomyomatosis of the renal sinus have all been described.
Many authors have disputed the entity, since there is no clear cell of origin for these tumors. The presence of melanosome proteins suggests neural crest origin, supported by the presence of epithelioid cells with clear to eosinophilic cytoplasm in the perivascular areas, expressing melanocytic and myogenic markers.1 As such, histopathological diagnosis of PEComa may be challenging.
Pathologically, differential diagnoses may include epithelioid leiomyosarcoma, stromal sarcoma, and pigmented renal-cell carcinoma. Almost all PEComas test positive for melanocytic (HMB45 and/or melan A) and smooth-muscle (actin and/or desmin) immunohistochemical markers, regardless of their organ of origin.2,3 Renal-cell carcinoma and clear-cell “sugar” tumors or epithelioid AMLs of the kidney are microscopically difficult to differentiate. Reports of heavily pigmented PEComa of the kidney are rare in the literature. Interestingly, among ten reported cases, eight occurred in women, with a wide age distribution (12–73 years). HMB45 was distinctly positive in these cases. Three cases reported metastasis, with uniformly rapid deterioration and mortality.4–9
PEComa behavior is variable and unpredictable, but as part of the sarcoma family, its size, growth pattern, mitotic rate, nuclear grade, and lymphovascular invasion are predictors of aggression and subsequent response to treatment. The rarity of this entity and the wide range of organs it affects have made it difficult to formulate guidelines for management, including treatment options and predictors of outcome. mTORIs have been included in the treatment plan of recently reported cases, while chemotherapy was ineffective in almost all reported cases.9
AMLs are more common than other PEComa entities. Pathological characteristics are heterogeneous proliferation composed of blood vessels, smooth-muscle cells, adipocytes, and peculiar myoid cells with variable melanin synthesis. The epithelioid variant has higher potential for aggressive behavior. Nese et al reported that factors strongly associated with poor prognosis of renal PEComa were stage ≥III (extrarenal tumors) and/or tumors extending into the renal vein and a carcinoma-like growth pattern.10 Though epithelioid AML is histopathologically related to PEComa, its natural course and response to treatment are variable, and reported predictors of epithelioid AML aggression are mainly related to cellular features (atypia, mitotic figures, atypical mitotic figures).11
Regarding our surgical approach to the case, we elected to perform partial rather than radical nephrectomy for several reasons. The surgery was palliative rather than curative in the presence of multiple local and distant metastases. Moreover, we anticipated that the patient would require adjuvant therapy, for which we preferred to maximize renal function reserve. Technically, the tumor was exophytic and amenable to resection with decent safety margins.
Prior case reports have described the benefit of adjuvant therapy with surgical excision of PEComas. In 2006, Parfitt et al described a primary PEComa of the urinary bladder treated with adjuvant IFNα immunotherapy after wide local excision.12 The value of mTORIs as effective palliative therapy is valid in many cases. The molecular pathophysiology of aberrant mTOR signaling provides a scientific basis for targeting this pathway. Gennatas et al reported the successful treatment of a patient with PEComa with the mTORI everolimus. In this case, the first-line treatment was the TKI imatinib, followed by the mTORI everolimus as second line treatment when the patient showed evidence of disease progression.4 Italiano et al replicated this strategy, and observed transient improvement in two patients with malignant PEComa treated with temsirolimus.4,13,14 TSC1/2 inactivation and mTOR hyperactivation in non-TSC AMLs and extrarenal PEComas support the effectiveness of this group of agents.14
Conclusion
Pathological diagnosis and characterization of pigmented renal PEComa can be challenging, especially with aspecific imaging features, usually mimicking renal-cell carcinoma. Biological behavior is undetermined, and thus it is important to share the experience regarding diagnosis and therapy.
Disclosure
We are reporting this case for academic purposes, and have received no external funding in support of our work. The submission has no commercial interests. The authors report no conflicts of interest in this work.
References
1. Fletcher CDM, Bridge JA, Hagedorn PCW, Mertens F. WHO Classification of Tumours of Soft Tissue and Bone. Lyon: IARC Press; 2013:306–309.
2. Hornick JL, Fletcher CDM. PEComa: what do we know so far? Histopathology. 2006;48:75–82. doi:10.1111/his.2006.48.issue-1
3. Huang Y, Lu G, Quan J, et al. Primary perivascular epithelioid cell tumor of the bladder. Ann Diagn Pathol. 2011;15:427–430. doi:10.1016/j.anndiagpath.2010.07.006
4. Gennatas C, Michalaki V, Kairi PV, Kondi-Paphiti A, Voros D. Successful treatment with the mTOR inhibitor everolimus in a patient with perivascular epithelioid cell tumor. World J Surg Oncol. 2012;10:181. doi:10.1186/1477-7819-10-181
5. Kollmannsberger C, Bjarnason G, Burnett P, et al. Sunitinib in metastatic renal cell carcinoma: recommendations for management of non-cardiovascular toxicities. Oncologist. 2011;16:543–553. doi:10.1634/theoncologist.2010-0263
6. Martignoni G, Pea M, Reghellin D, Zamboni G, Bonetti F. PEComas: the past, the present and the future. Virchows Arch. 2008;452:119–132. doi:10.1007/s00428-007-0509-1
7. Folpe AL, Kwiatkowski DJ. Perivascular epithelioid cell neoplasms: pathology and pathogenesis. Hum Pathol. 2010;41:1–15. doi:10.1016/j.humpath.2009.05.011
8. Chang H, Jung W, Kang Y, Jung WY. Pigmented perivascular epithelioid cell tumor (PEComa) of the kidney: a case report and review of the literature. Korean J Pathol. 2012;46:499–502. doi:10.4132/KoreanJPathol.2012.46.5.499
9. Saito K, Fujii Y, Kasahara I, Kobayashi N, Kasuga T, Kihara K. Malignant clear cell “sugar” tumor of the kidney: clear cell variant of epithelioid angiomyolipoma. J Urol. 2002;168:2533–2534. doi:10.1016/S0022-5347(05)64188-2
10. Bleeker JS, Quevedo JF, Folpe AL. Malignant perivascular epithelioid cell neoplasm: risk stratification and treatment strategies. Sarcoma. 2012;2012:541626. doi:10.1155/2012/541626
11. Nese N, Martignoni G, Fletcher CD, et al. Pure epithelioid PEComas (so-called epithelioid angiomyolipoma) of the kidney: a clinicopathologic study of 41 cases: detailed assessment of morphology and risk stratification. Am J Surg Pathol. 2011;35:161–176. doi:10.1097/PAS.0b013e318206f2a9
12. Brimo F, Robinson B, Guo C, Zhou M, Latour M, Epstein JI. Renal epithelioid angiomyolipoma with atypia: a series of 40 cases with emphasis on clinicopathologic prognostic indicators of malignancy. Am J Surg Pathol. 2010;34:715–722. doi:10.1097/PAS.0b013e3181d90370
13. Parfitt JR, Bella AJ, Wehrli BM, Izawa JI. Primary PEComa of the bladder treated with primary excision and adjuvant interferon-alpha immunotherapy: a case report. BMC Urol. 2006;6:20. doi:10.1186/1471-2490-6-20
14. Italiano A, Delcambre C, Hostein I, et al. Treatment with the mTOR inhibitor temsirolimus in patients with malignant PEComa. Ann Oncol. 2010;21(5):1135–1137. doi:10.1093/annonc/mdq044
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.