")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 11
Metastasis inhibition in breast cancer by targeting cancer cell extravasation
Authors Cominetti MR , Altei WF , Selistre-de-Araujo HS
Received 5 September 2018
Accepted for publication 4 March 2019
Published 18 April 2019 Volume 2019:11 Pages 165—178
DOI https://doi.org/10.2147/BCTT.S166725
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pranela Rameshwar
Márcia R Cominetti,1 Wanessa F Altei,2 Heloisa Sobreiro Selistre-de-Araujo2
1Department of Gerontology, Federal University of São Carlos, São Carlos, SP, Brazil; 2Department of Physiological Sciences, Federal University of São Carlos, São Carlos, SP, Brazil
Abstract: The spread of cells from primary tumors toward distant tissues and organs, also known as metastasis, is responsible for most cancer-associated deaths. The metastasis cascade comprises a series of events, characterized by the displacement of tumor cells (TCs) from the primary tumor to distant organs by traveling through the bloodstream, and their subsequent colonization. The first step in metastasis involves loss of cell-cell and cell-matrix adhesions, increased invasiveness and migratory abilities, leading to intravasation of TCs into the blood or lymphatic vessels. Stationary TCs must undergo the process of epithelial-mesenchymal transition in order to achieve this migratory and invasive phenotype. Circulating tumor cells that have survived in the circulation and left the blood or lymphatic vessels will reach distant sites where they may stay dormant for many years or grow to form secondary tumors. To do this, cells need to go through the mesenchymal-epithelial transition to revert the phenotype in order to regain epithelial cell-to-cell junctions, grow and become a clinically relevant and detectable tumor mass. This work will review the main steps of the metastatic cascade and describe some strategies to inhibit metastasis by reducing cancer cell extravasation presenting recent studies in the context of breast cancer.
Keywords: breast cancer, metastasis, extravasation, circulating tumor cells
Introduction
Cancer is one of the most serious human diseases due to its high morbidity and mortality, preceded only by cardiovascular diseases. Despite therapeutic advances in the area, there were about 2.1 million newly diagnosed female breast cancer cases in 2018 worldwide, accounting for almost 25% of cancer cases among women.1 These high numbers are due not only to an increase in the population but also to population aging.2 Breast cancer comprises a group of tumors that present high clinical, morphological and biological heterogeneity.3 Accordingly, breast tumors with similar histology and clinical features may present different prognoses and variable therapeutic responses.4 Breast cancers are classified according to several criteria. One of the most common is the classification into subtypes based on the presence or not of hormone receptors such as the estrogen receptors (ER), the progesterone receptors (PR) and the oncogene ERBB2 (HER2), namely, ER+, HER2+ (ER−/PR−/HER2+) and triple-negative (TN) (ER−PR−/HER2−) for the most common tumors.5,6
ER+ tumors, also described as LUM-A and LUM-B luminal subtypes, represent ~70% of all breast cancer cases. They typically express high levels of ER and PR, are sensitive to estrogen withdrawal and are treated with hormonal therapy. LUM-B tumors tend to result in higher tumor grades and worse prognosis compared to LUM-A tumors.7 The HER2 subtype, representing 10–15% of all breast cancer cases, is characterized by the presence of the receptor tyrosine kinase oncogene ERBB2. HER2+ tumors usually present ERBB2 gene amplification/overexpression and are good responders to anti-HER2 therapy. Finally, TN breast cancers (15–20%) represent the most aggressive breast tumors for which there is no available targeted treatment. Patients with TN tumors are mainly treated with cytotoxic chemotherapy.8 Several subtypes of TN tumors were also described that respond differently to standard or new chemotherapy.9
The dissemination of cells from primary tumors toward distant tissues followed by their subsequent growth at these secondary sites is known as metastasis, a highly complex and multistep process. The metastatic cascade comprises a series of events that can be divided into three main distinct phases: the displacement of tumor cells (TCs) from the primary tumor to distant spots; entrance into and exit from the circulatory system (namely, intravasation and extravasation, respectively); and the subsequent adhesion and growth in a different tissue (Figure 1).10
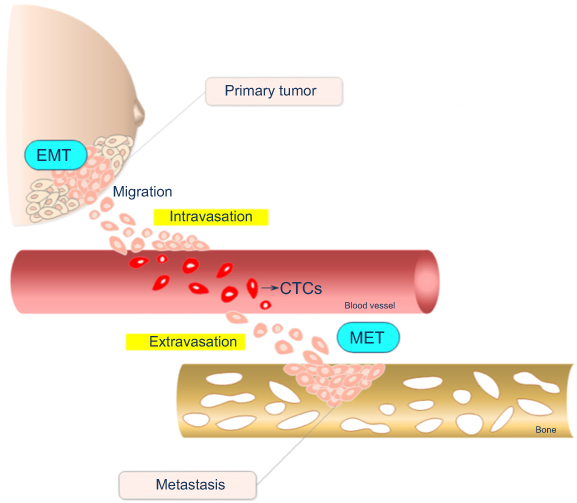 | Figure 1 Schematic representation of the metastatic cascade steps in breast tumors. Cells in the primary tumor undergo epithelial-mesenchymal transition (EMT), acquiring migratory and invasive properties. After leaving the primary tumor, cells enter into circulatory or lymphatic vessels until extravasation to a distant metastatic secondary site represented in the scheme by a bone. Once established at the secondary site, cells suffer an inverse EMT process called mesenchymal-epithelial transition (MET). The drawing has no anatomical proportions. Abbreviation: CTC, circulating tumor cells. |
The first phase involves loss of cell–cell and cell–matrix adhesions, acquisition of an invasive and migratory phenotype due to genetic and epigenetic alterations, and hypoxia-induced tumor angiogenesis, leading to intravasation of TCs into the blood or lymphatic vessels.11,12 After entering the circulation, TCs must survive several stressors before moving to a new site, where they may remain dormant for long periods before they begin to proliferate. The intravasation and extravasation processes will be described in detail later on in this review.
Metastasis is the main cause of deaths in patients with cancer.13 Unfortunately, for this condition, there are very few therapeutic options. Metastasis was first considered to develop from advanced lesions; however, recent reports have demonstrated that TCs disseminate very early to the bloodstream, even when the tumor is not detectable.14–16 Therefore, understanding the molecular mechanisms underlying all the phases of the metastatic cascade will significantly contribute to developing more efficient therapies.
The environment in primary tumors
Within the primary tumor, the hypoxic microenvironment triggers the proliferation and migration of endothelial cells to provide the tumor with new vascular networks, which will allow growth and further dissemination to secondary sites.12 In the tumor tissue, oxygen demand is higher than oxygen supply, and as the distance between cells and the existing vasculature increases, oxygen diffusion is hampered, therefore contributing to the development of a hypoxic environment.17 This low-oxygen condition activates many intracellular signaling pathways including the hypoxia-inducible factor (HIF) pathway, which is initiated with the activation of a family of transcriptional regulators, also called the HIF family. HIF target gene members mediate the adaptation of cells/tissues to chronic or acute hypoxia, including upregulation of angiogenic and hematopoietic growth factor genes.17,18 The angiogenic switch, supported by a set of distinct cell types and molecules present in the tumor microenvironment, modulates the transition from a quiescent to a proliferative vasculature. This switch requires the overexpression of pro-angiogenic growth factors, including the vascular endothelial growth factor (VEGF).19 VEGF is a critical pro-angiogenic factor that is proteolytically released from the tumor matrix by matrix metalloproteases (MMPs), predominantly by the MMP-9 secreted into the tumor microenvironment by both TCs and tumor-infiltrating leukocytes.20,21 High levels of VEGF also increase local vascular permeability, which in turn would help TCs to both enter and exit the bloodstream22–24
The angiogenic switch in hypoxic tumors frequently results in abnormal vasculature regarding its structure and function.25 An irregular branching network, lack of directional flow and compressed walls among other defects result in a hostile milieu characterized by low pH and high interstitial fluid pressure.26 This abnormal leaky vasculature facilitates the entry of TC into the bloodstream, therefore contributing to the development of metastasis. In addition, it also impairs the influx of immune cells or even drug delivery to the primary tumor.26 Due to these reasons, a new approach called vessel normalization has become an interesting strategy to reduce metastatic dissemination and increase the response to treatments. The main target for vascular normalization is VEGF and its receptors, VEGFR-1, VEGFR-2 or its co-receptors.25 Inhibition of VEGF signaling induced vessel normalization, reduced interstitial fluid pressure, and increased oxygenation and drug delivery. Due to its positive results, anti-VEGF therapy with bevacizumab is currently approved for treatment of several types of metastatic cancers including breast cancer.27
Epithelial-mesenchymal transition (EMT) program
The tumor-derived mesenchymal cells must acquire a migratory and invasive phenotype to spread from the primary tumor into the circulation, which is a crucial step in the metastatic process. A crucial step in promoting TCs to migrate and invade is the EMT program.28 EMT is a highly organized, complex and dynamic process consisting of several overlapping signaling pathways that can simultaneously exert their regulatory branches to orchestrate downstream events. Other less common types of changes in TC plasticity by morphological and phenotypical conversions include collective to amoeboid transition (CAT)29 and the mesenchymal to amoeboid transition (MAT).30
EMT hallmarks include loss of apical–basal polarity and adherence junctions, acquisition of a mesenchymal phenotype, as well as motility and invasive properties.31 Epithelial cells interact with each other through adhesion molecules such as E-cadherin and cytokeratin within tight junctions, adherent junctions, desmosomes and gap junctions. Apical–basal polarity is also a key epithelial characteristic. In response to different extracellular cell- and tissue-specific EMT-inducing signals, epithelial cells increase the expression of several transcription factors to orchestrate several morphological, cellular and molecular changes that occur during EMT. Hence, activation of the EMT program during tumorigenesis requires signaling between cancer cells, stromal cells and infiltrating cells32 such as fibroblasts, granulocytes, macrophages, mesenchymal stem cells and leukocytes, which create an inflammatory stroma that results in the release of EMT-inducing signals. In this inflammatory microenvironment, the tumor growth factor beta (TGF-β) is a cytokine considered a primary inducer of EMT.28 Inflammatory cytokines such as TNF-α33 and IL-6;34 hypoxia via HIF-1α;35 extracellular matrix (ECM) stiffness;36 adhesion receptors; and a myriad of growth factors such as VEGF, hepatocyte growth factor, fibroblast growth factor and platelet-derived growth factor are also EMT inducers.37
In response to the above-cited EMT-inducing signals, EMT-related transcription factors are activated to orchestrate the EMT program. These transcription factors include the SNAIL family zinc finger transcription factors (SNAIL1 and SNAIL2),38,39 the TWIST family basic helix-loop-helix transcription factors (TWIST1 and TWIST2),35,40–42 and the zinc finger E-box binding homeobox proteins (ZEB1 and ZEB2).43 The activation of these EMT-related transcription factors leads to downregulation of genes encoding epithelial junction proteins, thus disassembling adherent junctions, desmosomes and tight junctions, promoting loss of apical-basal polarity, hereby allowing cells to acquire a migratory and invasive phenotype.44
Concomitantly, there is an upregulation of mesenchymal genes, such as N-cadherin, fibronectin and vimentin, and a deconstruction of the cytoskeleton anchored to desmosomes in epithelial cells, in addition to vimentin upregulation.45 This transition to a mesenchymal state favors cell motility and the formation of invadopodia and actin stress fibers, which contributes to matrix degradation by MMPs and cell contractility, resulting in an invasive behavior.46 EMT is also associated to the prevention of apoptosis and senescence, to suppression of immune response and to the acquisition of chemotherapy resistance.32,47
Cell detachment from the ECM would lead to cell death by anoikis; however, before starting the apoptotic program, cells initiate the process of autophagy, which leads to cell survival for some time until they can find a new place to attach.48 Most healthy cells will die by anoikis upon prolonged cell detachment. In contrast, some TCs can develop anoikis resistance, a mechanism that allows survival regardless of cell–matrix interactions and increases migration, intravasation and survival during circulation in the bloodstream.49 Therefore, the process of autophagy has become an interesting target to prevent cancer from spreading. Currently, several inhibitors of autophagy are being tested in clinical trials to treat various types of solid tumors with significant increases in the median overall survival.50
Intravasation
The blood system is considered as the main route of the metastatic dissemination, but the lymphatic system also plays a key role in TC spread.51 Dissemination through lymphatic capillaries is facilitated due to some of their characteristics, such as the lack of a cover by smooth muscle cells and basement membrane in contrast to blood capillaries. In addition, lymphatic vessels are thin-walled single layers of endothelial cells lacking inter-endothelial tight junctions,52 which facilitate the traffic of interstitial fluids, macromolecules and even bacteria through valve-like openings.37
Cells that had recently left the primary tumor and that are circulating in the blood or lymphatic vessels are called circulating tumor cells (CTCs). These invasive and motile cells can enter the circulation long before the tumor is diagnosed. In the bloodstream, cancer cells are exposed to significant shear forces, innate immunity and oxidative stress.53 A recent study provided strong evidence for the role of shear forces in determining the place in the body where CTCs will extravasate from the blood. Using an in vivo model in zebrafish and a combination of microfluidics and optical tweezers, a critical adhesion force of 80 pN was identified whereby CTCs need to initiate stable adhesions to endothelial cells. Once adhered, TCs may resist to flow forces that exceed 200 pN; however, increased shear stress will induce extravasation at the site of arrest or even after the capillary bed. In addition, it was demonstrated that endothelial cell remodeling helps TC transmigration by engulfing adhered CTCs.54,
In an attempt to protect themselves during transit in the circulatory system, CTCs can associate with platelets55 and undergo reversible metabolic changes to increase their ability to antagonize oxidative stress.56 Accordingly, it was demonstrated that antioxidant supplementation increased lymph node metastases of human melanoma cells xenografted into mice.57 The cooperation with platelets also helps CTCs to avoid the immune system surveillance. In addition, the association with fibrin depots helps CTCs to make strong adhesions and transmigrate through the endothelial barrier.58,59 Currently, several studies highlight targeting platelets as a strategy for cancer treatment' however, there have been no breakthroughs in this area so far.60
It was reported that the direct release of clusters of TCs from primary tumors might produce microemboli. These CTCs clusters may range from 2 to 50 cells and clog capillaries resulting in metastatic foci at the sites of endothelium binding of clogged capillaries.61 Indeed, clusters of CTCs were isolated from the patients’ blood with different types of cancer.62 Aggregates of CTCs were previously related to deaths by thromboembolism and increased metastatic efficiency.61,63
A new mechanism of metastatic spreading by which TCs do not even need to enter the bloodstream was recently described.64 The authors from this paper present strong evidence of a “pericyte-like” behavior of some TCs that were previously called pericyte mimicry or extravascular migratory metastasis.65,66 This process was previously thought of as one way to facilitate the intravasation; however, the authors present evidence for a new mechanism by which TCs spread around the capillaries and compete with pericytes for a position in the interaction with the endothelial basal lamina. This process is dependent on the activation of the L1CAM/ILK/YAP (cell adhesion molecule L1/integrin-linked kinase/Yes-associated protein) signaling axis of mechanotransduction.64
Extravasation and secondary tumor formation
After intravasation, the CTCs that have survived in the circulation might undergo an opposite phenomenon, the extravasation to a secondary site to form metastasis. Once TCs have reached the secondary dissemination sites, they must revert back to the epithelial phenotype in order to regain epithelial cell-to-cell junctions, proliferate and become a clinically relevant and detectable tumor mass.47,67 This is reinforced by histologically examined metastases, which appear epithelial in phenotype and resemble the primary tumor.68 This reversal process is called mesenchymal-epithelial transition (MET) and allows TCs to proliferate to form the secondary tumor.69 These secondary lesions are primarily responsible for most cancer deaths, either by directly impairing the organ or through complications because of the lack of efficient treatments. Extravasation, therefore, is a crucial event in the metastatic cascade, and the search for new therapeutic targets for this process is urgently needed.
Taking into account all these steps, the metastatic cascade can be considered a highly inefficient process.70 In fact, only a few TCs succeed in producing metastasis despite the fact that primary tumors can release thousands of cells into the bloodstream.58 By using experimental models of intravascular injection of TCs, many studies have reported that the capillary network typically traps CTCs within few minutes after injecting, where they become adhered to the endothelium and subject to flow forces and to cells from the immune system including neutrophils and natural killer (NK) cells. This phase may last from a few hours to three days. Considering these hindrances, it was reported that only about 0.1% of disseminated TCs successfully develop metastasis.71 About 85% of melanoma cells were reported to survive in the circulation and extravasate, but only 2.5% of the surviving cells will form micrometastases. From these micrometastases, about 1% develop into macrometastases,72 suggesting that extravasation, colonization and the outgrowth of macrometastases are the most rate-limiting steps in the metastatic cascade. It is important to note, however, that in the study by Luzzi and coworkers,72 the melanoma cells were injected via intraportal vein to target the liver. Therefore, these cells did not circulate significantly within the animal’s bloodstream, which may have contributed to the high percentage of surviving cells. These cells experienced an extremely limited transit within the liver vessels and a much higher percentage of CTCs may have perished in the bloodstream than reported. It is likely that surviving within the bloodstream is another limiting step. Moreover, solitary TCs that have reached the secondary site may remain dormant for months or even years as undetectable cancer cell populations due to either a cell cycle arrest or a balance between proliferation and apoptosis.71 In the presence of appropriated signals, these cells might eventually start proliferating to become clinically detectable metastases.73
Breast cancer organ-specific metastases
Most breast cancer metastasis is found in the lungs, liver, bones and brain resulting in unfavorable prognosis.74 More than 120 years ago, Stephen Paget, analyzing postmortem data from hundreds of breast cancer patients, observed that the organ distribution of metastases happened in a nonrandom way.75 He then coined the “seed and soil” hypothesis, which postulates that TCs (seeds) are widely disseminated in the body and progress to form metastases only in specific organs (soil), whose proper settlement depends on a pre-metastatic niche microenvironment.76,77 Conversely, an alternative “anatomical hypothesis” claims that CTCs can be physically trapped in the first organ in which they encounter vessels whose diameter is too small to allow their passage.59 It seems that depending on the tumor type examined, either or both hypotheses are applicable.78 Circulation patterns guide CTCs through the capillary bed. In most organs, the venous circulation leads to the right cardiac ventricle and then to the lungs, whereas the mesenteric venous system from the gut first drains into the liver. The resulting retention of CTCs in the lungs or liver, respectively, can explain the high metastatic incidence in these organs.43,79 Some CTCs, however, are able to bypass these first filters to reach all other organs through the arterial circulation. The structure of the vascular walls is an important characteristic by which a CTC eventually extravasates to colonize a secondary organ The capillaries in the liver and bone marrow are lined with fenestrated endothelial cells and a discontinuous basal lamina,80 which represent gaps that may facilitate the extravasation of CTCs and contribute to the high incidence of liver and bone metastasis.81 On the other hand, the endothelium of lung capillaries has tight junctions and a basement membrane, and the brain has a blood–brain barrier whose capillary walls are additionally reinforced by pericytes and astrocyte processes.80 The surgical resection of the primary tumor was demonstrated to potentiate the growth of residual neoplastic disease by enhancing the expression of genes that mediate breast cancer metastasis to the lung, including some genes involved in TC adhesion, invasion and angiogenesis.82
TCs also release exosomes that are involved in the preparation of the pre-metastatic niche. Exosomes are small vesicles with sizes ranging from 40 to 100 nm, derived from the endosomal membranes of TC that can interfere in every step of the metastatic cascade due to their capacity to interact with either the tumor microenvironment or distant tissues. These vesicles are considered as “mirrors” of the origin cell and carry bioactive cargoes, including growth factors, angiogenic factors, transmembrane receptors, proteinases, ECM molecules and RNAs.83 They are believed to drive organotropic metastasis, interfering with the content of extracellular matrix deposit through the interaction of their integrin content with the target tissue84 by some processes which are not yet fully understood. Exosomal proteomic data revealed distinct integrin expression patterns, where integrins α6β4 and α6β1 are associated with lung metastasis and αvβ5 with liver metastasis.85
Regarding brain metastasis, only 10–16% of metastatic breast cancer patients developed this neoplasia. The incidence appears to be increasing, however, especially among HER2+ and TN subsets of breast cancers.86,87 This is the result, at least in part, of the development of therapeutic agents that successfully treat visceral, but not brain metastases,88 leaving this tissue an easy target to be colonized as a secondary metastatic organ. Moreover, glia is able to induce a significant increase in metastatic cell proliferation, suggesting that brain tissue may produce contributing factors to TC growth.88
Several genes mediating specialized extravasation or colonization functions of breast cancer cells required for breast-derived bone, pulmonary or brain metastasis have been identified. These mediators include Fascin-1 and other components of invadopodia,89 autocrine enhancers of TC motility such as epiregulin and WNT ligands,90 mediators of endothelial disjunction and vascular permeability including angiopoietin-like 4, VEGF, cyclooxygenase 2, MMP1 and osteonectin.45,82–85,92 All these factors might be considered in the search for new targets for treatments against metastasis.
In summary, defining the specific site of metastatic colonization depends on the combination of preparation signals from the TCs and the tumor stroma, the type of CTCs, circulation anatomical patterns mostly from the primary site, and TC-autonomous functions.53
Reduction of cancer cell extravasation as a strategy to decrease metastatic colonization
All efforts made in cancer research until now have made it possible to understand why it is so difficult to prevent or treat metastasis. By looking at the steps of the metastatic cascade, it became clear that several of them must be simultaneously addressed in order to treat or prevent metastasis (Figure 2). High-throughput models for assessing several steps of the metastatic cascade are needed in order to test sets of new or old compounds. Thus, microfluidic systems have been designed to mimic the endothelial barrier and the ECM under a flow simulating the blood shear stress.93
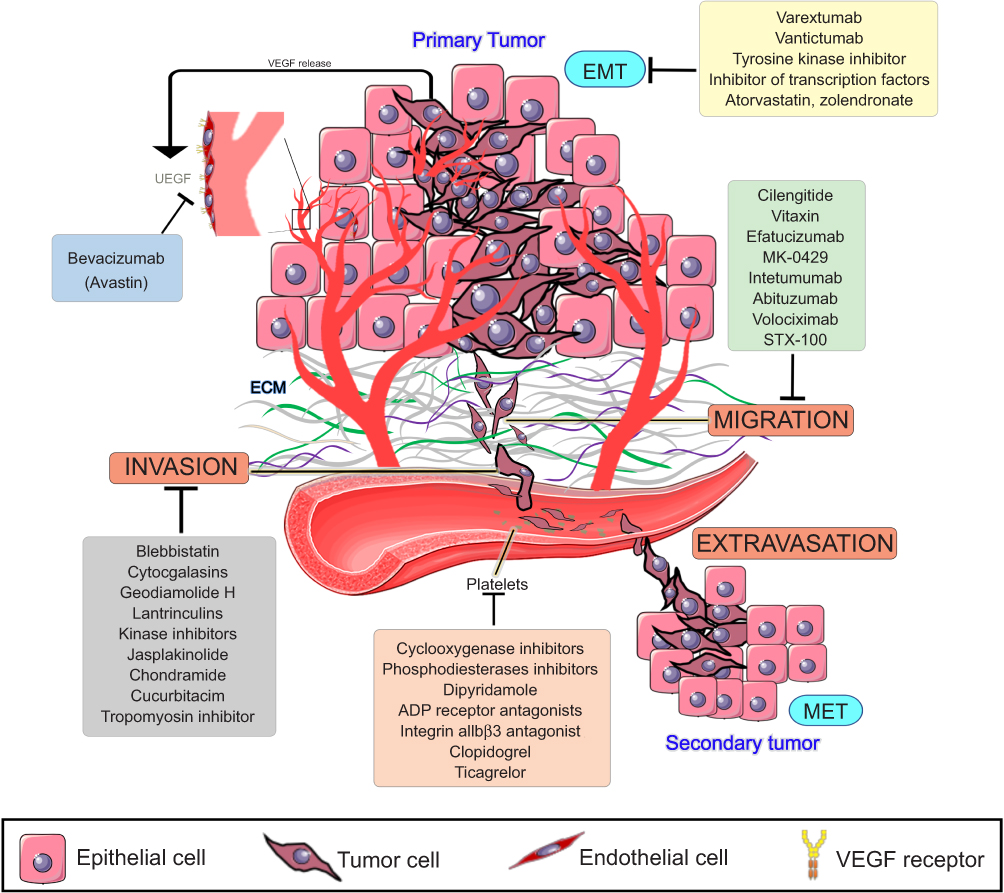 | Figure 2 Metastasis cascade and the currently explored targets and their inhibitors. Data from Wojtukiewicz et al,60 Marcucci et al,94 Raab-Westphal et al,95 Rosel et al,96 and Sini et al.97 Bevacizumab and antiplatelets compounds are commercially available, while drugs targeting EMT and migration are in clinical trials (clinicaltrials.gov). Anti-invasion drugs, known as migrastatics, are under experimental investigations. Abbreviations: VEGF, vascular endothelial growth factor, EMT, epithelial-mesenchymal transition, MET, mesenchymal-epithelial transition. |
Inhibitors of adhesion and migration key receptors such as the integrins are under current clinical trials with modest results.98,99 Cilengitide, a specific nanomolar inhibitor of αvβ3 and α5β1 integrins, has entered in almost 30 clinical trials, mostly in combination with other treatments, for several types of cancer, including one breast cancer study. The results presented so far indicated that cilengitide is well tolerated, with minor side effects but only small benefits in survival median time.98,99 However, other adhesion key receptors such as α4β1 integrin and P- and L-selectins have emerged as potential targets and perhaps their inhibition would lead to better results.58 Thus, the anticoagulants heparin or the low-molecular-weight heparin form have been tested in a number of clinical trials showing positive results in the survival of cancer patients.100
The EMT, autophagy and anoikis resistance programs are good candidates as targets for inhibitors, and several players of these processes have been identified.101 In healthy cells, anoikis is controlled by proapoptotic factors from the B-cell lymphoma-2 (BCL2) protein family members. However, metastatic cancer cells develop resistance mechanisms to the loss of cell attachment that is independent of BCL2 control.102,103 TNBC cells are particularly dependent on autophagy for survival after detachment from the matrix, by a mechanism controlled IL6/STAT3 activity.104
Good candidates to treat metastasis seem to be the compounds to keep extravasated cells in dormancy. In addition to inhibiting tumor angiogenesis, VEGF-A inhibition prevents early micrometastatic growth and induced dormancy in lung cancer cells.66 Several clinical trials were performed targeting tumor angiogenesis by inhibition of secreted VEGF or its receptors.105 Currently, bevacizumab, a humanized monoclonal antibody to VEGF (Avastin®), is commercially available for treatment of some types of advanced cancer; however, due to the lack of effect, it is not recommended for metastatic breast cancers.
The development of secondary lesions does not depend only on the characteristics of TCs, but it also depends on a large set of interactions between tumor and host cells, the extracellular matrix, vessel morphology and soluble compounds produced by a myriad of cells, among other factors. The association of the blood coagulation system, inflammation and cancer development has been recognized for many years, and the potential of antiplatelet therapy in cancer treatment has also been addressed. Since TCs remain only from a few hours to a few days in the bloodstream, the interaction with blood cells or platelets can be inhibited with antiplatelet drugs. Thus, aspirin and other cyclooxygenase inhibitors have been tested in many in vitro and in vivo models, as well as in clinical trials with positive results in metastasis prevention.106–109
Another proposed target is the antiviral protein BST-2, highly expressed in aggressive breast tumors. Covalent dimerization of cysteine residues of BST-2 leads to anoikis resistance and cell survival by promoting a proteasome-mediated degradation of a BIM-a key pro-apoptotic factor.110 In addition, a set of microRNAs has been proposed to control EMT, such as miR-29,111 miR-300,112 miR-655113 and miR-373,114 each one controlling distinct intracellular pathways. Despite the diversity of regulatory mechanisms, miRNAs may be interesting as targets for drug design due to the relative simplicity of the molecules and forms of delivery.
“State of the art” of breast cancer metastasis therapies
According to The Global Oncology Trends,115 fourteen new anticancer drugs were approved in 2018. All these molecules are targeted-based therapies, and two of them were designed for breast cancer treatment. The USA is the largest investor and responsible for the newest developed drugs.
Abemaciclib,116 palbociclib117,118 and ribociclib119 are new molecules which were approved for breast cancer metastasis over the last five years.120 They inhibit CDK 4/6, serine/threonine kinases and essential regulators of cell cycle progression, upregulated in many TCs.121,122 These drugs are recommended to treat metastatic breast cancer in hormone receptor–positive (HR+) and HER2-negative (HER2−) patients and are considered a successful innovation in the cancer pharmaceutical industry due to their oral usage and good pharmacological responses.116,119
In Brazil, hormone therapy is still widely recommended to treat breast cancer, followed by anti-HER2 agents, such as Pertuzumabe, approved in 2018 by the Agência Nacional de Vigilancia Sanitaria (ANVISA).123 Additionally, cytotoxic therapy is widely used in patients with advanced, metastatic or recurrent breast cancer, mainly in TN.124 Ribociclib was also approved for use in association with the aromatase inhibitor Everolimus; however, CDK4/6 inhibitors are still not available in the public health system.125,126
Despite investments in new researches, patients with breast cancer HR− and HER2− still depend on cytotoxic chemotherapy as a systemic treatment, and bevacizumabe can be used in some cases. The most used agents are taxanes, topoisomerase II inhibitors, platinum-based drugs, vinca alkyloids, and other antitubulins and antimetabolites.127,128
The National Cancer Institute provides 63 commercially available drugs approved by the USA Food and Drug Administration for breast cancer treatment to be used alone or in combination. The main ones are presented in Table 1. From these, sixteen are based on the presence of hormone receptors (ER and PR) and/or HER2 protein expression. With the exception of CDK4/6 inhibitors, most of the available drugs still rely on mechanisms of action related to cell cytotoxicity and cell division inhibition, which are nonselective molecules, causing low life quality for patients.129
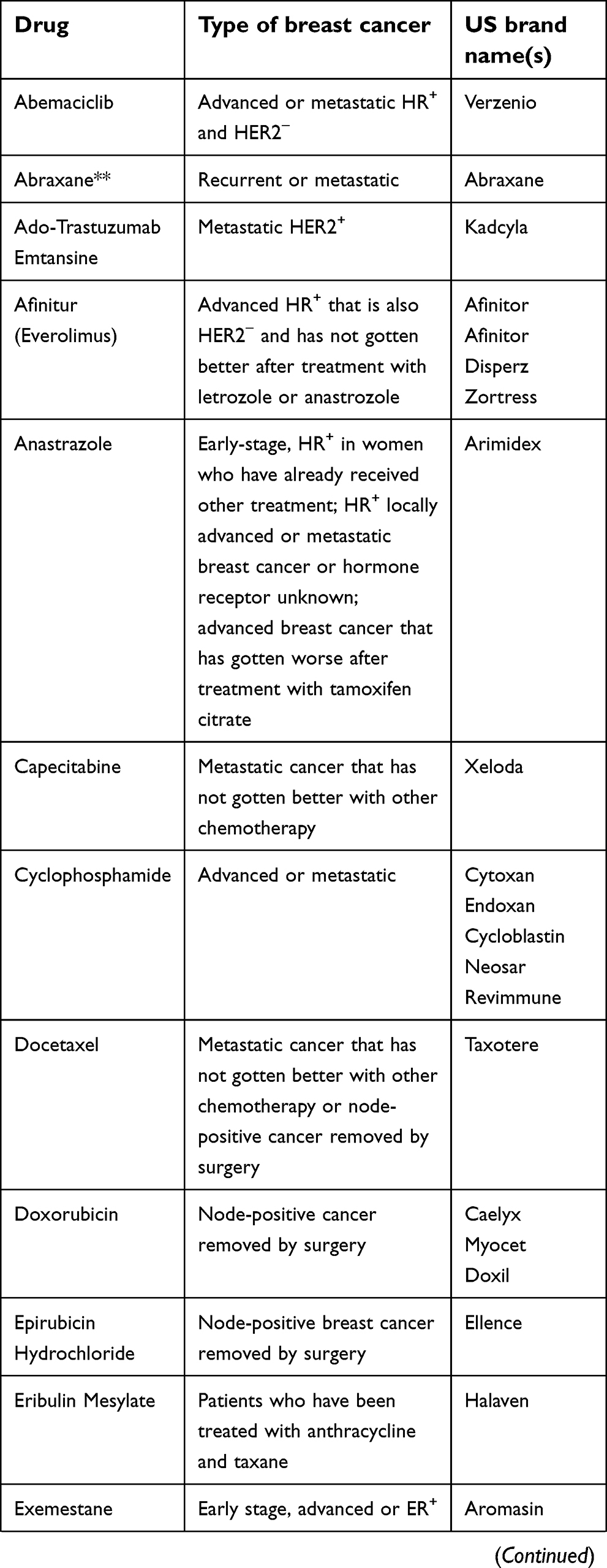 | Table 1 Breast cancer drugs approved by the US Food and Drug Administration* |
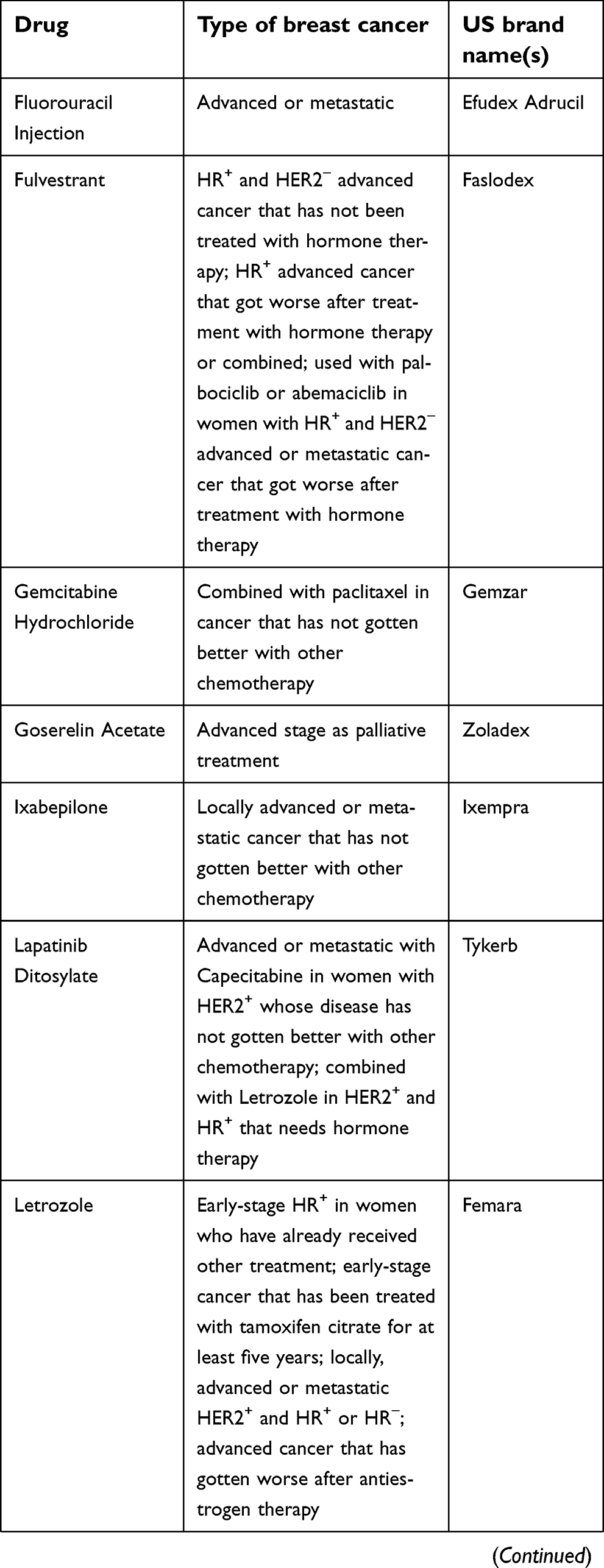 | Table 1 (Continued). |
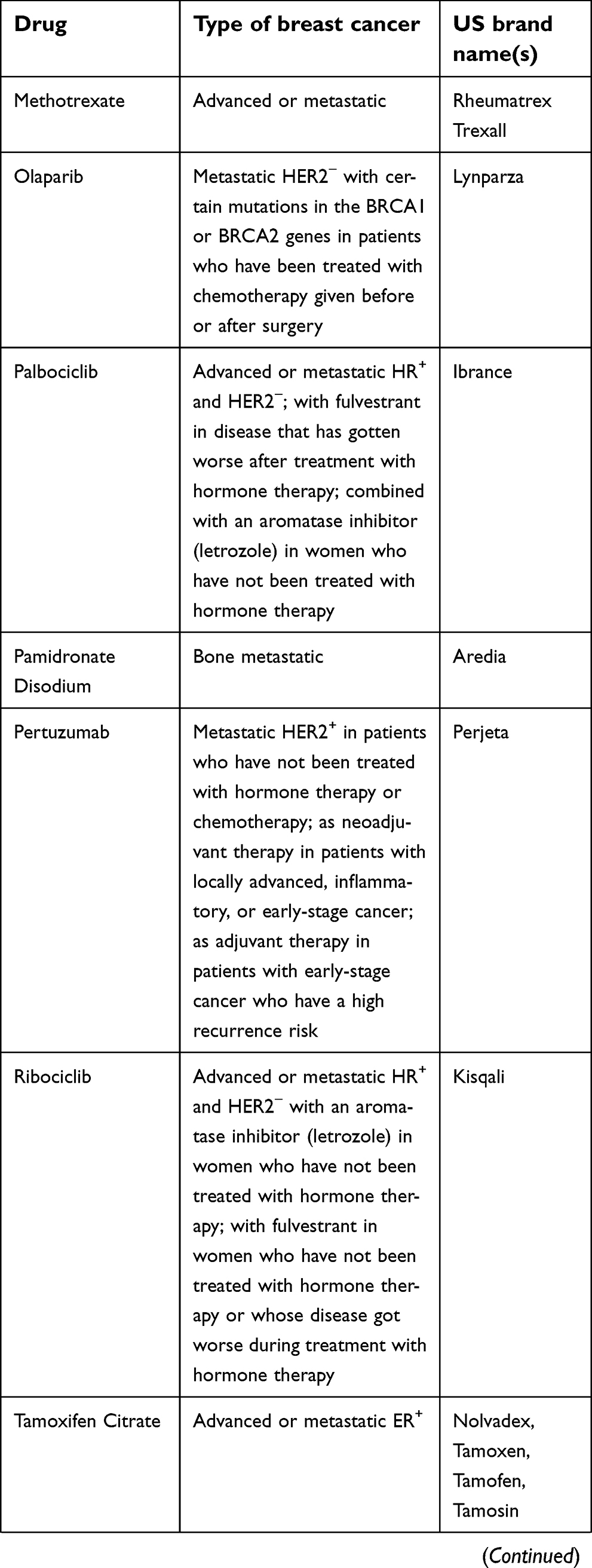 | Table 1 (Continued). |
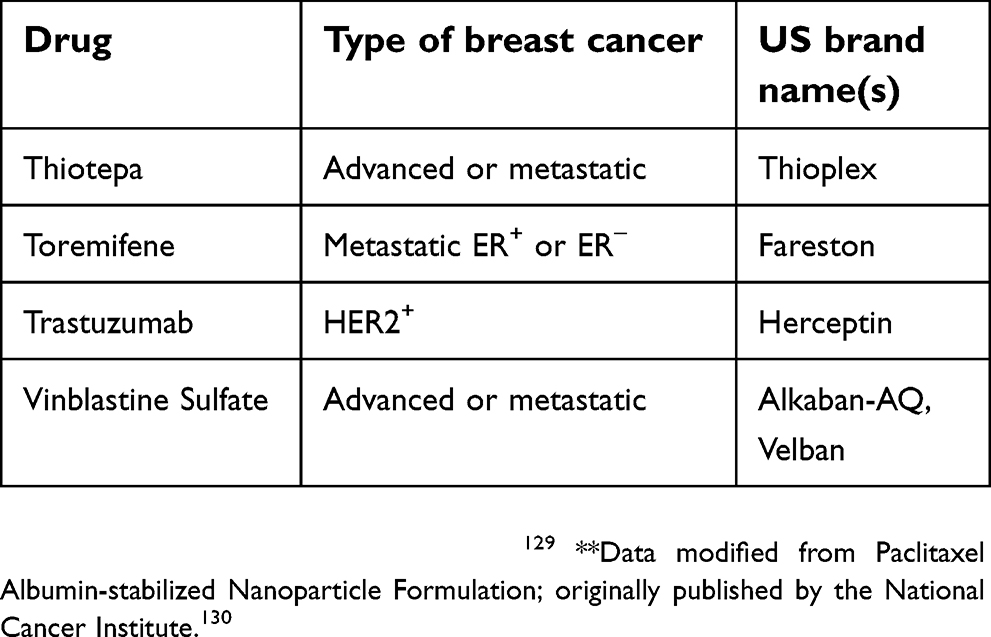 | Table 1 (Continued). |
In summary, despite all efforts of anticancer drug discovery programs, few compounds with innovative modes of action have been developed. Thus, investigations on the basic cancer cell biology from different perspectives could lead to new answers and solutions. There are no available drugs that target cell extravasation to impair the metastatic process, therefore looking at the cell surroundings and their communication with the microenvironment, as well as with other cells can be considered a promising approach for anti-metastatic drug development. Many preclinical and clinical studies have proposed an extensive set of new targets, and most of them are promising to treat or prevent metastasis. Efforts to develop high throughput models to test these targets and the libraries of new and known compounds are urgently needed, in order to avoid the time-consuming process of the development of new pharmaceuticals.
Acknowledgments
The authors would like to thank Lia Mara Grosso Neves for preparing Figure 1. This work was supported by FAPESP (São Paulo Research Foundation, grants #2013/00798-2, 2015/24940-8, and 2014/1 8747-8), CAPES (Coordination for the Improvement of Higher Education Personnel), and CNPq (National Council for Scientific Research).
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work. All authors contributed equally to the writing of the manuscript. WFA prepared Figure 2.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
2. Fitzmaurice C, Allen C, Barber RM, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study global burden. JAMA Oncol. 2017. doi:10.1001/jamaoncol.2016.5688
3. Cianfrocca M, Gradishar W. New molecular classifications of breast cancer. CA Cancer J Clin. 2009. doi:10.3322/caac.20029
4. Stricker TP, Brown CD, Bandlamudi C, et al. Robust stratification of breast cancer subtypes using differential patterns of transcript isoform expression. PLoS Genet. 2017. doi:10.1371/journal.pgen.1006589
5. Koboldt DC, Fulton RS, McLellan MD, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012. doi:10.1038/nature11412
6. Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000. doi:10.1038/35021093
7. Sledge GW, Mamounas EP, Hortobagyi GN, Burstein HJ, Goodwin PJ, Wolff AC. Past, present, and future challenges in breast cancer treatment. J Clin Oncol. 2014. doi:10.1200/JCO.2014.55.4139
8. Jin X, Mu P. Targeting breast cancer metastasis. Breast Cancer Basic Clin Res. 2015. doi:10.4137/BCBCR.S25460
9. Sharma P. Biology and management of patients with triple-negative breast cancer. Oncologist. 2016. doi:10.1634/theoncologist.2016-0067
10. Bersini S, Jeon JS, Moretti M, Kamm RD. In vitro models of the metastatic cascade: from local invasion to extravasation. Drug Discov Today. 2014. doi:10.1016/j.drudis.2013.12.006
11. Fidler IJ. The pathogenesis of cancer metastasis: the “seed and soil” hypothesis revisited. Nat Rev Cancer. 2003. doi:10.1038/nrc1098
12. Rankin EB, Nam JM, Giaccia AJ. Hypoxia: signaling the metastatic cascade. Trends in Cancer. 2016. doi:10.1016/j.trecan.2016.05.006
13. Verma V, Lautenschlaeger T. MicroRNAs in non-small cell lung cancer invasion and metastasis: from the perspective of the radiation oncologist. Expert Rev Anticancer Ther. 2016. doi:10.1080/14737140.2016.1191950
14. Dasgupta A, Lim AR, Ghajar CM. Circulating and disseminated tumor cells: harbingers or initiators of metastasis? Mol Oncol. 2017. doi:10.1002/1878-0261.12022
15. Grzelak CA, Ghajar CM. Metastasis ‘systems’ biology: how are macro-environmental signals transmitted into microenvironmental cues for disseminated tumor cells? Curr Opin Cell Biol. 2017. doi:10.1016/j.ceb.2017.06.002
16. Hosseini H, Obradovic MMS, Hoffmann M, et al. Early dissemination seeds metastasis in breast cancer. Nature. 2016. doi:10.1038/nature20785
17. Semenza GL. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit Rev Biochem Mol Biol. 2000. doi:10.1080/10409230091169186
18. Zimna A, Kurpisz M. Hypoxia-Inducible factor-1 in physiological and pathophysiological angiogenesis: applications and therapies. Biomed Res Int. 2015. doi:10.1155/2015/549412
19. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. 2017. doi:10.1038/nrc.2017.51
20. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010. doi:10.1016/j.cell.2010.03.015
21. Cf M, Cl S-P, Ju R, et al. Blocking αvβ3 integrin by a recombinant RGD disintegrin impairs VEGF signaling in endothelial cells. Biochimie. 2012. doi:10.1016/j.biochi.2012.04.020
22. Subhani S, Vavilala DT, Mukherji M. HIF inhibitors for ischemic retinopathies and cancers: options beyond anti-VEGF therapies. Angiogenesis. 2016. doi:10.1007/s10456-016-9510-0
23. Jiang M, Qin C, Han M. Primary breast cancer induces pulmonary vascular hyperpermeability and promotes metastasis via the VEGF-PKC pathway. Mol Carcinog. 2016. doi:10.1002/mc.22352
24. Ferrara N. Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev. 2010. doi:10.1016/j.cytogfr.2009.11.003
25. Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001. doi:10.1038/nm0901-987
26. Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011. doi:10.1038/nrd3455
27. Crawford Y, Ferrara N. VEGF inhibition: insights from preclinical and clinical studies. Cell Tissue Res. 2009. doi:10.1007/s00441-008-0675-8
28. Yeung KT, Yang J. Epithelial-mesenchymal transition in tumor metastasis. Mol Oncol. 2017. doi:10.1002/1878-0261.12017
29. Hegerfeldt Y, Tusch M, Bröcker EB, Friedl P. Collective cell movement in primary melanoma explants: plasticity of cell-cell interaction, β1-integrin function, and migration strategies. Cancer Res. 2002;62(7):2125–2130.
30. Wolf K, Mazo I, Leung H, et al. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J Cell Biol. 2003. doi:10.1083/jcb.200209006
31. Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013;80. doi:10.1126/science.1234850.
32. Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008. doi:10.1016/j.devcel.2008.05.009
33. Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of Snail by NF-κB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009. doi:10.1016/j.ccr.2009.03.016
34. Lo HW, Hsu SC, Xia W, et al. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007. doi:10.1158/0008-5472.CAN-07-0575
35. Yang MH, Wu MZ, Chiou SH, et al. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat Cell Biol. 2008. doi:10.1038/ncb1691
36. Wei SC, Fattet L, Tsai JH, et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat Cell Biol. 2015. doi:10.1038/ncb3157
37. Van Zijl F, Krupitza G, Mikulits W. Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res - Rev Mutat Res. 2011. doi:10.1016/j.mrrev.2011.05.002
38. Peinado H, Olmeda D, Cano A. Snail ZEB and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007. doi:10.1038/nrc2131
39. Cano A, Pérez-Moreno MA, Rodrigo I, et al. The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000. doi:10.1038/35000025
40. Tania M, Khan MA, Fu J. Epithelial to mesenchymal transition inducing transcription factors and metastatic cancer. Tumor Biol. 2014. doi:10.1007/s13277-014-2163-y
41. Tran DD, Corsa CAS, Biswas H, Aft RL, Longmore GD. Temporal and spatial cooperation of Snail1 And Twist1 during epithelial-mesenchymal transition predicts for human breast cancer recurrence. Mol Cancer Res. 2011. doi:10.1158/1541-7786.MCR-11-0371
42. Li CW, Xia W, Huo L, et al. Epithelial-mesenchymal transition induced by TNF-α requires NF-κB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012. doi:10.1158/0008-5472.CAN-11-3123
43. Wong TS, Gao W, Chan JYW. Transcription regulation of E-cadherin by zinc finger E-box binding homeobox proteins in solid tumors. Biomed Res Int. 2014. doi:10.1155/2014/921564
44. Qin Y, Capaldo C, Gumbiner BM, Macara IG. The mammalian scribble polarity protein regulates epithelial cell adhesion and migration through E-cadherin. J Cell Biol. 2005. doi:10.1083/jcb.200506094
45. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014. doi:10.1038/nrm3758
46. Leong HS, Robertson AE, Stoletov K, et al. Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 2014. doi:10.1016/j.celrep.2014.07.050
47. Thiery JP. Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002. doi:10.1038/nrc822
48. Vlahakis A, Debnath J. The interconnections between autophagy and integrin-mediated cell adhesion. J Mol Biol. 2017. doi:10.1016/j.jmb.2016.11.027
49. Towers CG, Thorburn A. Therapeutic targeting of autophagy. EBioMedicine. 2016. doi:10.1016/j.ebiom.2016.10.034
50. Wang C, Hu Q, Shen HM. Pharmacological inhibitors of autophagy as novel cancer therapeutic agents. Pharmacol Res. 2016. doi:10.1016/j.phrs.2016.01.028
51. Brown P. Lymphatic system: unlocking the drains. Nature. 2005. doi:10.1038/436456a
52. Mauri C, Wang G, Schulte-Merker S. From fish embryos to human patients: lymphangiogenesis in development and disease. Curr Opin Immunol. 2018;53:167–172. doi:10.1016/J.COI.2018.05.003
53. Massagué J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016. doi:10.1038/nature17038
54. Follain G, Osmani N, Azevedo AS, et al. Hemodynamic forces tune the arrest, adhesion, and extravasation of circulating tumor cells. Dev Cell. 2018. doi:10.1016/j.devcel.2018.02.015
55. Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011. doi:10.1016/j.ccr.2011.09.009
56. Le Gal K, Ibrahim MX, Wiel C, et al. Antioxidants can increase melanoma metastasis in mice. Sci Transl Med. 2015. doi:10.1126/scitranslmed.aad3740
57. Piskounova E, Agathocleous M, Murphy MM, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015. doi:10.1038/nature15726
58. Labelle M, Hynes RO. The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov. 2012. doi:10.1158/2159-8290.CD-12-0329
59. Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer. 2011. doi:10.1038/nrc3004
60. Wojtukiewicz MZ, Hempel D, Sierko E, Tucker SC, Honn KV. Antiplatelet agents for cancer treatment : a real perspective or just an echo from the past? Cancer Metastasis Rev. 2017;36(2):305–329. doi:10.1007/s10555-017-9683-z.
61. Aceto N, Bardia A, Miyamoto DT, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014. doi:10.1016/j.cell.2014.07.013
62. Cho EH, Wendel M, Luttgen M, et al. Characterization of circulating tumor cell aggregates identified in patients with epithelial tumors. Phys Biol. 2012. doi:10.1088/1478-3975/9/1/016001
63. Glinsky VV, Glinsky GV, Glinskii OV, et al. Intravascular metastatic cancer cell homotypic aggregation at the sites of primary attachment to the endothelium. Cancer Res. 2003;63(13):3805–3811.
64. Er EE, Valiente M, Ganesh K, et al. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat Cell Biol. 2018. doi:10.1038/s41556-018-0138-8
65. Barnhill RL, Lugassy C. Angiotropic malignant melanoma and extravascular migratory metastasis: description of 36 with emphasis on a new mechanism of tumour spread. Pathology. 2004. doi:10.1080/00313020412331282708
66. Kienast Y, Von Baumgarten L, Fuhrmann M, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med. 2010. doi:10.1038/nm.2072
67. Banyard J, Bielenberg DR. The role of EMT and MET in cancer dissemination. Connect Tissue Res. 2015. doi:10.3109/03008207.2015.1060970
68. Chui MH. Insights into cancer metastasis from a clinicopathologic perspective: epithelial-mesenchymal transition is not a necessary step. Int J Cancer. 2013. doi:10.1002/ijc.27745
69. Gunasinghe NPAD, Wells A, Thompson EW, Hugo HJ. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev. 2012. doi:10.1007/s10555-012-9377-5
70. Irmisch A, Huelsken J. Metastasis: new insights into organ-specific extravasation and metastatic niches. Exp Cell Res. 2013. doi:10.1016/j.yexcr.2013.02.012
71. MacK GS, Marshall A. Lost in migration. Nat Biotechnol. 2010. doi:10.1038/nbt0310-214
72. Luzzi KJ, MacDonald IC, Schmidt EE, et al. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. 1998. doi:10.1016/S0002-9440(10)65628-3
73. Willis L, Alarcón T, Elia G, et al. Breast cancer dormancy can be maintained by small numbers of micrometastases. Cancer Res. 2010. doi:10.1158/0008-5472.CAN-09-3144
74. Chen MT, Sun HF, Zhao Y, et al. Comparison of patterns and prognosis among distant metastatic breast cancer patients by age groups: A SEER population-based analysis. Sci Rep. 2017. doi:10.1038/s41598-017-10166-8
75. Fidler IJ, Poste G. The “seed and soil” hypothesis revisited. Lancet Oncol. 2008. doi:10.1016/S1470-2045(08)70201-8
76. Guise T. Examining the Metastatic Niche: targeting the Microenvironment. Semin Oncol. 2010. doi:10.1053/j.seminoncol.2010.10.007
77. Carlini MJ, De Lorenzo MS, Puricelli L. Cross-talk between tumor cells and the microenvironment at the metastatic niche. Curr Pharm Biotechnol. 2011(11):1900–1908. doi:BSP/CPB/E-Pub/000182-12-12 [pii].
78. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011. doi:10.1016/j.cell.2011.09.024
79. Denève E, Riethdorf S, Ramos J, et al. Capture of viable circulating tumor cells in the liver of colorectal cancer patients. Clin Chem. 2013. doi:10.1373/clinchem.2013.202846
80. Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007. doi:10.1161/01.RES.0000255691.76142.4a
81. Budczies J, von Winterfeld M, Klauschen F, et al. The landscape of metastatic progression patterns across major human cancers. Oncotarget. 2015. doi:10.18632/oncotarget.2677
82. Al-Sahaf O, Wang JH, Browne TJ, Cotter TG, Redmond HP. Surgical injury enhances the expression of genes that mediate breast cancer metastasis to the lung. Ann Surg. 2010. doi:10.1097/SLA.0b013e3181efc635
83. Sung BH, Ketova T, Hoshino D, Zijlstra A, Weaver AM. Directional cell movement through tissues is controlled by exosome secretion. Nat Commun. 2015. doi:10.1038/ncomms8164
84. Steinbichler TB, Dudás J, Riechelmann H, Skvortsova II. The role of exosomes in cancer metastasis. Semin Cancer Biol. 2017. doi:10.1016/j.semcancer.2017.02.006
85. Hoshino A, Costa-Silva B, Shen TL, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015. doi:10.1038/nature15756
86. Palmieri D, Bronder JL, Herring JM, et al. Her-2 overexpression increases the metastatic outgrowth of breast cancer cells in the brain. Cancer Res. 2007. doi:10.1158/0008-5472.CAN-06-3316
87. Santarelli JG, Sarkissian V, Hou LC, Veeravagu A, Tse V. Molecular events of brain metastasis. Neurosurg Focus. 2007. doi:10.3171/foc.2007.22.3.2
88. Fitzgerald DP, Palmieri D, Hua E, et al. Reactive glia are recruited by highly proliferative brain metastases of breast cancer and promote tumor cell colonization. Clin Exp Metastasis. 2008. doi:10.1007/s10585-008-9193-z
89.
90. Matsuda Y, Schlange T, Oakeley EJ, Boulay A, Hynes NE. WNT signaling enhances breast cancer cell motility and blockade of the WNT pathway by sFRP1 suppresses MDA-MB-231 xenograft growth. Breast Cancer Res. 2009. doi:10.1186/bcr2317
91. Padua D, Zhang XHF, Wang Q, et al. TGFβ primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008. doi:10.1016/j.cell.2008.01.046
92. Gupta GP, Nguyen DX, Chiang AC, et al. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature. 2007. doi:10.1038/nature05760
93. Chen J, Xue C, Zhao Y, Chen D, Wu MH, Wang J. Microfluidic impedance flow cytometry enabling high-throughput single-cell electrical property characterization. Int J Mol Sci. 2015. doi:10.3390/ijms16059804
94. Marcucci F, Stassi G, De Maria R. Epithelial–mesenchymal transition: a new target in anticancer drug discovery. Nat Publ Gr. 2016;15(5):311–325. doi:10.1038/nrd.2015.13
95. Raab-Westphal S, Marshall JF, Goodman SL. Integrins as therapeutic targets : successes and cancers. Cancers (Basel). 2017;9(9):pii:E110. doi:10.3390/cancers9090110
96. Rosel D, Fernandes M, Veselý P, et al. Migrastatics — anti-metastatic and anti-invasion drugs : promises and challenges. Trends Cancer. 2017;3(6):391–406. doi:10.1016/j.trecan.2017.04.008
97. Sini V, Cassano A, Corsi D, et al. Bevacizumab as first-line treatment in HER2-negative advanced breast cancer: pros and cons. Tumori J. 2016;102(5):472–480. doi:10.5301/tj.5000555
98. Chinot OL. Cilengitide in glioblastoma: when did it fail? Lancet Oncol. 2014. doi:10.1016/S1470-2045(14)70403-6
99. Stupp R, Hegi ME, Gorlia T, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071–22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014. doi:10.1016/S1470-2045(14)70379-1
100. Kakkar AK, Levine MN, Kadziola Z, et al. Low molecular weight heparin, therapy with dalteparin, and survival in advanced cancer: the fragmin advanced malignancy outcome study (FAMOUS). J Clin Oncol. 2004. doi:10.1200/JCO.2004.10.002
101. Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008. doi:10.1016/j.cell.2008.03.027
102. Galante JM, Mortenson MM, Bowles TL, Virudachalam S, Bold RJ. ERK/BCL-2 pathway in the resistance of pancreatic cancer to anoikis. J Surg Res. 2009. doi:10.1016/j.jss.2008.05.017
103. Oudenaarden CRL, van de Ven RAH, Derksen PWB. Re-inforcing the cell death army in the fight against breast cancer. J Cell Sci. 2018;131. doi:10.1242/jcs.212563.
104. Lieblein JC, Ball S, Hutzen B, et al. STAT3 can be activated through paracrine signaling in breast epithelial cells. BMC Cancer. 2008. doi:10.1186/1471-2407-8-302
105. Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006. doi:10.1038/ncponc0403
106. Algra AM, Rothwell PM. Effects of daily aspirin in secondary prevention of stroke on cancer mortality and non-vascular death: analysis of individual patient data from randomised controlled trials. Cerebrovasc Dis. 2013. doi:10.1159/000353129
107. Algra AM, Rothwell PM. Effects of regular aspirin on long-term cancer incidence and metastasis: A systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012. doi:10.1016/S1470-2045(12)70112-2
108. Rothwell PM, Wilson M, Elwin CE, et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20 year follow-up of five randomised trials. Lancet. 2010. doi:10.1016/S0140-6736(10)61543-7
109. Li N. Platelets in cancer metastasis: to help the “villain” to do evil. Int J Cancer. 2016. doi:10.1002/ijc.29847
110. Mahauad-Fernandez WD, Okeoma CM. Cysteine-linked dimerization of BST-2 confers anoikis resistance to breast cancer cells by negating proapoptotic activities to promote tumor cell survival and growth. Cell Death Dis. 2017. doi:10.1038/cddis.2017.68
111. Rostas JW, Pruitt HC, Metge BJ, et al. MicroRNA-29 negatively regulates EMT regulator N-myc interactor in breast cancer. Mol Cancer. 2014. doi:10.1186/1476-4598-13-200
112. Yu J, Xie F, Bao X, Chen W, Xu Q. MiR-300 inhibits epithelial to mesenchymal transition and metastasis by targeting Twist in human epithelial cancer. Mol Cancer. 2014. doi:10.1186/1476-4598-13-121
113. Lv ZD, Kong B, Liu XP, et al. miR-655 suppresses epithelial-to-mesenchymal transition by targeting Prrx1 in triple-negative breast cancer. J Cell Mol Med. 2016. doi:10.1111/jcmm.12770
114. Chen D, Dang B-L, Huang J, et al. MiR-373 drives the epithelial-to-mesenchymal transition and metastasis via the miR-373-TXNIP-HIF1α-TWIST signaling axis in breast cancer. Oncotarget. 2015. doi:10.18632/oncotarget.4702
115.
116. Goetz MP, Toi M, Campone M, et al. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol. 2017;35(32):3638–3646. doi:10.1200/JCO.2017.75.6155
117. Steger GG, Gnant M, Bartsch R. Palbociclib for the treatment of postmenopausal breast cancer – an update. Expert Opin Pharmacother. 2016;17(2):255–263. doi:10.1517/14656566.2016.1133590
118. McShane TM, Wolfe TA, Ryan JC. Updates on managing advanced breast cancer with palbociclib combination therapy. Ther Adv Med Oncol. 2018;10:175883591879384. doi:10.1177/1758835918793849
119. Lopez-Tarruella S, Jerez Y, Marquez-Rodas I, Echavarria I, Martin M. Ribociclib for the treatment of advanced hormone receptor-positive, HER2-negative breast cancer. Future Oncol. 2017;13(24):2137–2149. doi:10.2217/fon-2017-0183
120. Kwapisz D. Cyclin-dependent kinase 4/6 inhibitors in breast cancer: palbociclib, ribociclib, and abemaciclib. Breast Cancer Res Treat. 2017;166(1):41–54. doi:10.1007/s10549-017-4385-3
121. Lamb R, Lehn S, Rogerson L, Clarke RB, Landberg G. Cell cycle regulators cyclin D1 and CDK4/6 have estrogen receptor-dependent divergent functions in breast cancer migration and stem cell-like activity. Cell cycle. 2013;12(15):2384–2394. doi:10.4161/cc.25403
122. Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):1–13. doi:10.1186/bcr2419
123. Ministério da Saúde. Portaria Conjunta Nº 19 de 3 de Julho ed 2018. Aprova as Diretrizes Diagnósticas e Terapêuticas do Carcinoma de Mama [Joint ordinance number 19, July 3, 2018. Approves the diagnostic and therapeutic breast carcinoma guidelines];2018. Available from:
124.
125.
126.
127. Gadi VK, Davidson NE. Practical approach to triple-negative breast cancer. J Oncol Pract. 2017;13(5):293–300. doi:10.1200/JOP.2017.022632
128. Jitariu A, Cîmpean AM, Ribatti D, Raica M. Triple negative breast cancer: the kiss of death. Oncotarget. 2017;8(28):46652–46662. doi:10.18632/oncotarget.16938
129.
130.
131.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.