")
Back to Journals » OncoTargets and Therapy » Volume 12
Metabolic reprogramming results in abnormal glycolysis in gastric cancer: a review
Authors Liu Y , Zhang Z, Wang J, Chen C, Tang X , Zhu J, Liu J
Received 4 October 2018
Accepted for publication 11 January 2019
Published 13 February 2019 Volume 2019:12 Pages 1195—1204
DOI https://doi.org/10.2147/OTT.S189687
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Yuanda Liu,1 Ze Zhang,2 Junyang Wang,1 Chao Chen,1 Xiaohuan Tang,1 Jiaming Zhu,1,* Jingjing Liu1,*
1Department of Gastrointestinal Surgery, The Second Hospital of Jilin University, Changchun 130041, China; 2Department of General Surgery, The First Hospital of Jilin University, Changchun 130021, China
*These authors contributed equally to this work
Abstract: The Warburg effect in tumor cells involves the uptake of high levels of glucose, enhanced glycolysis, and the metabolism of pyruvate to lactic acid rather than oxidative phosphorylation to generate energy under aerobic conditions. This effect is closely related to the occurrence, invasion, metastasis, drug resistance, and poor prognosis of gastric cancer (GC). Current research has further demonstrated that the Warburg effect in GC cells is not only mediated by the glycolysis pathway, but also includes roles for mitochondria, noncoding RNAs, and other proteins that do not directly regulate metabolism. As a result, changes in the glycolysis pathway not only lead to abnormal glucose metabolism, but they also affect mitochondrial functions, cellular processes such as apoptosis and cell cycle regulation, and the metabolism of lipids and amino acids. In this review, we discuss metabolic reprogramming in GC based on glycolysis, a possible link between glucose metabolism, lipid metabolism, and amino acid metabolism, and we clarify the role of mitochondria. We also examine recent studies of metabolic inhibitors in GC.
Keywords: gastric cancer, glycolysis, mitochondria, metabolic reprogramming, Helicobacter pylori
Introduction
Gastric cancer (GC) is currently one of the most common malignant tumors diagnosed, and it is the second leading cause of mortality due to cancer.1 GC originates in gastric epithelial cells and similar to other types of tumors, it exhibits a Warburg effect. This effect involves a high uptake of glucose, enhanced glycolysis, and the metabolization of a large amount of pyruvate to lactic acid rather than oxidative phosphorylation to provide energy under aerobic conditions.2 The metabolic changes dominated by the Warburg effect have recently been referred to as metabolic reprogramming, and studies of these changes have provided a deeper understanding of tumor cell metabolism. For example, it has been demonstrated that GC cells and normal cells exhibit metabolic differences not only in glucose metabolism, but also in the metabolism of lipids and amino acids. Accordingly, a large number of specific biomarkers of GC have been identified. Metabolic reprogramming of GC has also been recognized as a novel approach for the study of the biological characteristics of GC.
In this review, we focus on a discussion of recent insights into the glycolysis pathway in GC and we summarize the complex relationship that exists between glucose metabolism, lipid metabolism, and amino acid metabolism. The role and impact of mitochondrial function in each of these aspects are also considered.
Helicobacter pylori promotes the genesis of GC by inducing metabolic reprogramming
Infection by Helicobacter pylori (Hp) is the most important primary cause of GC. However, progression from a Hp infection to atrophic gastritis and eventually GC is a long-term process.3 In vitro, Hp-infected gastric epithelial cells have exhibited increased glycolysis and increased expression of Lon protease 1 (Lonp1), a protein that activates the mitochondrial unfolded protein response and maintains mitochondrial function. Correspondingly, knockdown of Lonp1 has been shown to reverse alterations in metabolism that are caused by Hp,4 thereby suggesting that aerobic glycolysis and mitochondrial dysfunction correlate with the genesis of GC. Hp-induced GC is also characterized by higher expression levels of the M2 isoform of pyruvate kinase, PKM2, among other factors that are induced in GC and that affect mitochondrial function.5,6 Cytotoxin-associated gene A (CagA) has been shown to upregulate expression of PKM2 and pyruvate dehydrogenase kinase (PDK1). Moreover, when CagA localizes to mitochondria, it inhibits the activity of sirtuin 3 (SIRT3) and promotes stability of hypoxia-inducible factor 1α (HIF-1α).7 Vacuolating cytotoxin A (VacA) is another Hp protein, and it has been shown to trigger mitochondrial dysfunction, promote mitochondrial division, and reduce mitochondrial DNA (mtDNA) copy number.8–10 Taken together, these findings support a model in which Hp induces GC by promoting glycolysis and mitochondrial dysfunction (Table 1).
 | Table 1 Specific Hp proteins that are associated with metabolic reprogramming in GC |
Molecular factors that affect metabolic reprogramming in GC cells
Metabolic reprogramming that occurs in GC mainly involves changes in the following molecular factors: 1) key enzymes in glycolysis, 2) mitochondrial proteins, 3) noncoding RNAs, and 4) proteins that regulate these factors. The changes manifested by these molecules include significant differences in their levels between GC tissues and normal tissues. Moreover, interventions targeting these molecules have been found to not only affect the metabolic profile of GC cells, but also to affect the genesis, proliferation, invasion, and metastasis of GC cells as we describe below.
Changes in key glycolysis enzymes during metabolic reprogramming in GC
The glycolysis profile of GC is unique and is based on the ability of GC cells to maintain their growth under hypoxic conditions, to provide raw materials for cell biosynthesis and cell division, and to maintain intracellular redox homeostasis.11 These functions also correlate with the genesis, proliferation, invasion, and metastasis of GC cells. The following six proteins have been identified as key glycolytic enzymes that are affected by metabolic reprogramming in GC (Figure 1).
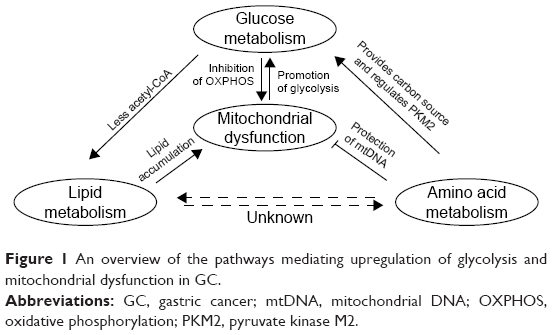 | Figure 1 An overview of the pathways mediating upregulation of glycolysis and mitochondrial dysfunction in GC. |
Hexokinase II
The hexokinase (HK) family of proteins catalyzes the conversion of glucose to glucose 6-phosphate (G6P) as the first rate-limiting step in glycolysis. G6P also participates in the pentose phosphate pathway and the process of gluconeogenesis. Among the HK family of proteins, HKII has been found to be consistently overexpressed in most tumors, including GC. HKII is also associated with poor prognosis of GC in most cases.12 HKII binds to voltage-dependent anion channels (VDACs) on the outer membrane of mitochondria to obtain ATP that is required for glycolysis.13 However, HKII can also bind to the outer membrane of mitochondria, specifically via the antiapoptotic protein, Bcl-2, and the proapoptotic protein, Bax, to inhibit apoptosis.14,15 Thus, HKII is an excellent target for blocking glucose flux.
Pyruvate kinase M2
Pyruvate kinase (PK) is a key enzyme in the final step of glycolysis, which catalyzes the conversion of phosphoenolpyruvate to pyruvate. There are four members of the PK protein family, PKM1, PKM2, PKL, and PKR.16 PKM1 and PKM2 are encoded by PKM, while PKL and PKR are encoded by the PKLR gene.16 In addition, PKM1 exhibits PK activity, yet PKM2 does not. Tumor cells generally express high levels of PKM2 and low levels of PKM1, thereby promoting glycolysis and inhibiting mitochondrial oxidative phosphorylation.16 When PKM2 was knocked out in GC cells, the PI3K/AKT/mTOR pathway and autophagy were inhibited, thereby leading to a decrease in the proliferation and invasive phenotype of GC cells.17,18 PKM2 can also translocate to the nucleus and promote transcription of HIF-1α and Bcl-xl to further enhance glycolysis.19 Moreover, interactions between PKM2, β-catenin, and octamer-binding transcription factor 4 (OCT4) have been shown to maintain the stemness quality of cells.20,21 In mitochondria, PKM2 interacts with and activates Bcl-2 to inhibit apoptosis.22 Correspondingly, overexpression of PKM2 promotes mitochondrial fusion, fewer copies of mtDNA, and the expression and degradation of p53. Overexpression of PKM2 also reduces levels of electron transport chain complex proteins I, III, and V.23 Taken together, these studies indicate that PKM2 promotes glycolysis and contributes to the dysfunction of mitochondria.
Pyruvate dehydrogenase kinase
The PDK family of proteins includes four isoforms. Many studies have recently focused on PDK1, which is generally expressed at high levels in tumors and is associated with tumor proliferation, metastasis, and poor prognosis.24 PDK1 inhibits the activity of pyruvate dehydrogenase (PDH) to promote the metabolization of pyruvate to lactic acid, and it helps regulate the AKT/NF-κB pathway.6 The ability of PDK to inhibit PDH activity also leads to a decrease in the level of acetyl-CoA to influence the de novo synthesis of lipids.25
Enolase
Enolase (ENO1) catalyzes the conversion of phosphoglycerol to phosphoenolpyruvate in glycolysis and is highly expressed in GC. Knockdown of ENO1 has been shown to inhibit glycolysis and increase the sensitivity of GC cells to cisplatin. Conversely, overexpression of ENO1 enhances the proliferation and metastasis of GC cells.26,27 In a proteomic analysis, ENO1 was found to be closely related to heat shock protein beta-1 (also known as Hsp27), while it has also been found to affect the regulation of anti-stress pathways.28
Glucose transporter
As implied by their name, glucose transporters (GLUTs) 1–4 are responsible for the transport of glucose into cells, and in GC, where GLUT1 and GLUT4 are highly expressed. When GLUT1 was knocked out in GC cells in vitro, metabolic reprogramming was significantly reversed and apoptosis was triggered.29 Levels of HK2 and PKM2 also declined in the absence of GLUT1.29 Conversely, upregulation of GLUT4 by p38 mitogen-activated protein kinase affects myocyte enhancer factor 2α and promotes glycolysis.30
Lactate dehydrogenase
Lactate dehydrogenase (LDH) catalyzes the conversion of pyruvate to lactic acid and is a key enzyme in the metabolic reprogramming of tumors. LDH is highly expressed in GC and promotes glycolysis. In GC, the transcription factor, fork head/winged-helix 1, upregulates the M isoform of LDH, LDHA.31 Meanwhile, downregulation of LDHA by OCT4 has been associated with a good prognosis in GC.32
Association between mitochondria and metabolic reprogramming in GC
Mitochondria are the energy factories for cells and their functions are often compromised in tumors. While dysfunctional mitochondria have been recently shown to promote tumor survival, the underlining mechanism(s) remain to be elucidated.33 Here, we describe three aspects of mitochondrial function in GC.
MtDNA
Low levels of mtDNA have been shown to have an important effect on the biogenesis and homeostasis of mitochondria.34 In 62.5% of GC patients, mutations in mitochondrial genes have been identified. A decrease in mtDNA copy number has also been observed in 54.8% of GC patients, especially patients affected by advanced GC.15,35 Furthermore, it has recently been reported that curcumin suppresses the growth of GC by increasing the production of ROS to mediate depletion of DNA polymerase gamma. These changes affect mitochondrial oxidative phosphorylation and inhibit cell biosynthesis.36
Mitochondrial topoisomerase I
Replication of mtDNA requires topoisomerases, except for mitochondrial topoisomerase I (TOP1MT). The latter exerts a negative effect on the regulation on mtDNA replication, yet plays an important role in the formation and maintenance of D-LOOP regions of mtDNA and in maintaining mitochondrial homeostasis.37,38 Thus, low levels of TOP1MT are generally expressed in GC. In the absence of TOP1MT, oxidative stress and mitochondrial autophagy are induced,37 glycolysis is promoted, the epithelial–mesenchymal transition (EMT) is accelerated, and the proliferation, invasion, and metastasis of GC cells are enhanced.39 Expression of LDHA and glycolysis are also increased in the absence of TOP1MT, thereby indicating that TOP1MT affects cell proliferation by modulating glycolysis.39 In clinical studies of GC, TOP1MT has been associated with poor prognosis.
SIRT3
SIRT3 localizes to mitochondria and is mainly involved in regulating oxidative stress. However, SIRT3 has also been shown to upregulate manganese-dependent superoxide dismutase, inhibit mitochondrial ROS levels, regulate the unfolded protein response of mitochondria, and maintain mitochondrial homeostasis.7,40,41 Thus, SIRT3 is considered a tumor suppressor gene. However, there are conflicting data regarding the role of SIRT3 in GC. For example, expression of SIRT3 in GC tissues has been found to be lower in some studies, and higher in others, than in normal tissues.42 Similarly, overexpression of SIRT3 has been associated with a significant reduction in cell proliferation,43 and conversely, has also been associated with enhanced cell proliferation.42 When SIRT3 was overexpressed in GC cells, promotion of ATP production and glycolysis were observed.42 Moreover, SIRT3 has been shown to acetylate LDHA to enhance its LDH activity.42 In the clinic, patients with high levels of SIRT3 expression have been associated with good prognosis.44 Thus, the role of SIRT3 in GC remains an active area of research, and inconsistencies among reported results remain to be resolved.
Despite advances in characterizing mitochondrial factors and their roles in metabolic reprogramming in GC, there have been very few studies that have examined the relationship between mitochondria and metabolic reprogramming. However, as described above, the localization of glycolysis-related proteins to mitochondria can affect the functions of mitochondria, while mitochondrial proteins can also affect glycolysis. Thus, it appears that mitochondria are tightly linked with metabolic reprogramming that occurs, and this complex relationship remains to be thoroughly characterized.
The effect of other molecules on metabolic reprogramming in GC
There are other types of molecules which can regulate glycolysis and mitochondrial function in GC, and we introduce some of these molecules below. These representative molecules demonstrate how metabolic reprogramming can correlate with the EMT, cell cycle, and other cellular processes.
Snail
Snail is a key molecule in the EMT process and also regulates glycolysis. When Snail is overexpressed in GC cells, glucose intake is increased while activity of fructose bisphosphatase 1 is inhibited.45 In GC, overexpression of Snail has been shown to correlate with metastasis and poor prognosis.46
Hypoxia-inducible factor 1α
HIF-1α is a key factor for cell survival under hypoxic conditions. In GC, HIF-1α is highly expressed and is closely related to poor prognosis and drug resistance.47 The level of HIF-1α expression in GC metastases has also been found to be higher than that in primary tumors, and it appears to promote GC metastasis by inhibiting integrin 5α.48
Thymidine phosphorylase
Thymidine phosphorylase (TP) catalyzes the conversion of thymidine to thymine and 2-deoxy-D-ribose 1-phosphate. In GC, TP activates the NF-κB pathway to promote angiogenesis, lymphangiogenesis, and metastasis.49 Under conditions of nutritional deprivation, thymidine provides a carbon source for glycolysis, and a high level of thymidine catabolism has been observed in GC.49
Monocarboxylic acid transporter
The monocarboxylic acid transporter (MCT) family of proteins has 14 members. In GC, MCT1, MCT2, and especially MCT4, are highly expressed. These proton pumps transport monocarboxylic acids, pyruvic acid, lactic acid, ketone bodies, and other substances to maintain high glucose flux. They also play an important role in cell survival under highly acidic conditions in tumor cells.50 Moreover, expression of MCT4 has also been associated with advanced tumor size-lymph nodes-metastases staging of GC.51
p53
p53 is a tumor suppressor protein which plays an important role in cell cycle arrest, apoptosis, DNA repair, and glycolysis.52 Currently, it is widely accepted that wild-type (WT) p53 inhibits glycolysis and promotes oxidative phosphorylation. For example, WT p53 downregulates GLUT1/4 and GLUT3 by inhibiting NF-κB signaling, thereby reducing the intracellular flux of glucose.53,54 WT p53 can also inhibit expression of HKII at the transcriptional level and upregulate the protein, parkin,55 to induce ubiquitination of PKM2 and inhibit glycolysis.56 In addition, WT p53 is able to inhibit glycolysis by upregulating TP53-induced glycolysis and the apoptosis regulator, TIGAR.54 In humans, p53 mutations have been detected in >50% of cancer patients.57 Mutant p53 proteins have also been shown to mediate functions that oppose those of WT p53. For example, mutant p53 proteins have been shown to promote glycolysis in tumors via upregulation of GLUT1, GLUT4, and HKII.58 In GC, mutant p53 inhibits TIGAR, thereby enhancing glycolysis, and it correlates with poor prognosis.59,60
Effect of noncoding RNAs on metabolic reprogramming in GC
Noncoding RNAs, including microRNAs and long noncoding RNAs (lncRNAs), mediate complex regulatory effects on the structure, function, and processes of cells.61 It has been reported that microRNAs and lncRNAs regulate key molecules involved in the metabolism of glucose to influence the proliferation, invasion, and metastasis of GC.62,63
Effect of microRNAs on metabolic reprogramming in GC
Various microRNAs have been shown to regulate glycolysis in GC. For example, Mir-22 upregulates ENO1.26 Mir-186 inhibits the activity of HIF-1α, thereby leading to an increase in glucose uptake, lactate production, ATP/ADP, and a decrease in NAD+/NADH.64 Mir-let-7a inhibits GC progression by inhibiting PKM2,65 while Mir-133b inhibits polypyrimidine tract-binding protein 1 to reduce levels of PKM2.66 Mir-181b reduces glucose intake and increases ATP production via inhibition of HKII expression.67 Furthermore, Mir-448 maintains glycolysis levels by inhibiting KDM2B,68 Mir-148 enhances glycolysis by upregulating GLUT1,69 Mir-375 inhibits expression of PDK1,70 and Mir-128b binds the 3′ UTR of PDK1 to inhibit expression of PDK1.71
Effects of lncRNAs on metabolic reprogramming in GC
Various lncRNAs have also been shown to regulate glycolysis in GC. For example, the lncRNA, MACC1-AS1, has been shown to enhance MET-related transcription factor 1 (MACC1), activate the AMPK-LIN28 pathway, enhance glycolysis and the antioxidant capacity of GC cells, and play a cancer-promoting role in GC.72 Correspondingly, MACC1 is associated with poor prognosis in GC. Recent studies have further shown that MACC1 contributes to resistance to trastuzumab in GC via activation of the PI3K/AKT signaling pathway.73 Under conditions of nutritional deprivation, AMPK upregulates levels of MCAA1, while levels of HKII, PDK1, and LDH are also upregulated under these conditions.74 Lnc00152 promotes GC by inhibiting miR-139-5p, although miR-139-5p is able to inhibit glycolysis by upregulating AMP-activated protein kinase 1α (PRKAA1).75 Finally, lncTINCR is a molecular sponge of miR-375, and it promotes expression of PDK1 and proliferation and metastasis in GC.76
The effects of lipid metabolism and amino acid metabolism on metabolic reprogramming in GC
Intracellular pathways for the metabolism of lipids, amino acids, and glucose are needed for life, and they are closely linked with each other. Correspondingly, in cancer cells, abnormal glucose metabolism is often accompanied by changes in the metabolism of lipids and amino acids.
Effect of lipid metabolism on metabolic reprogramming in GC
In GC, levels of lipids and triglycerides generally increase.77 In a metabolomic analysis of GC cells, low levels of 3-hydroxybutanoic acid (the end product of fatty acid β-oxidation) and inhibition of cholesterol synthesis were detected.28 A metabolic analysis of a rat GC model also showed that oxidative stress-related pathways, fatty acid degradation, and amino acid metabolism were all blocked.78
Endoscopic explorations have revealed a novel ultrastructure for GC, which includes a white opaque substance (WOS). Histology studies further revealed that the interior of this WOS consists of a large number of lipid drops. This novel structure is considered to be a hallmark of GC, and its formation is consistent with observations that lipid degradation is inhibited and lipid synthesis is enhanced in GC. The latter processes also contribute to an accumulation of lipids intracellularly.79,80 Meanwhile, if lipid metabolism is compromised, low levels of mitochondrial oxidative phosphorylation are observed, and this may be related to poor prognosis in GC.81 Correspondingly, when a ketogenic diet consisting of average protein content, low levels of carbohydrates, and enriched omega-3 fatty acid and medium-chain triglyceride content was fed to a nude mouse GC metastasis model, reduced glucose uptake and significant inhibition of GC growth were observed.82
The effect of amino acid metabolism on metabolic reprogramming in GC
Metabolomic analyses have shown that the concentration of leucine, a ketogenic amino acid, is significantly reduced in GC, while levels of glycine, phenylalanine, and arginine are significantly increased.28 In addition, the amino acid transporter gene, LAT1, has been shown to be upregulated in GC cell membranes.83 In GC cells subjected to glucose deprivation, nonessential amino acids can prevent activation of the mitochondrial apoptosis pathway by maintaining mitochondrial homeostasis and a stable mtDNA copy number.84,85 The ability of PKM2 to act as a sensor of amino acids has also recently been demonstrated. In particular, phenylalanine, alanine, tryptophan, methionine, valine, and proline are able to inhibit it, and histidine and serine are able to activate PKM2.86,87 These findings suggest that amino acids can influence glucose metabolism in a more complex manner than previously anticipated, and a further study of this relationship is needed.
Novel metabolic inhibitors for treatment of GC
There are many new drugs that have been designed to target the glycolysis pathway, and many of these induce cell apoptosis. Below, we describe various metabolic inhibitors that have been developed to target four of the six glycolysis targets listed in Section “Molecular factors that affect metabolic reprogramming in GC cells”, as well as HIF-1α.
Targeting of key glycolysis enzymes
Targeting HKII
HKII binds to VDACs or Bcl-2 in combination with Bax on the outer membrane of mitochondria to obtain ATP for glycolysis, or to inhibit apoptosis, respectively. Both 3-BrPA, an inhibitor of HKII, and SCT, an inhibitor of phosphate fructose kinase,13,88 downregulate Bcl-2 and upregulate Bax, to inhibit glycolysis and enhance mitochondria-associated apoptosis.89,90 Meanwhile, 2-deoxy-D-glucose has been shown to competitively combine with HKII to block glucose flux.91 Glycolysis activity is also reduced when baicalein inhibits HKII, PDK1, and LDHA, and baicalein has been shown to reduce the resistance of stomach adenocarcinoma cells to fluorouracil.92 Furthermore, licorice chalcone A is a novel HKII inhibitor, as well as an inhibitor of the AKT pathway,93 which has been shown to reduce glucose consumption and lactate production in GC cells, with the latter condition inducing apoptosis.
Targeting PKM2
As described above, PKM2 has various roles in a cell. Thus, inhibitors that target this protein exhibit various effects. For example, inhibition of PKM2 in GC cells by LY294002 induces apoptosis.94 Meanwhile, pantoprazole (PPZ), a third-generation proton pump inhibitor and a newly identified PKM2 inhibitor, inhibits the Akt/GSK-β/β-catenin pathway and reverses sensitivity to chemotherapy in GC cells.95,96 PPZ has also been shown to reverse chemotherapy resistance in SGC7901 cells by downregulating V-ATPases/mTOR/HIF-1α/P-gp and MRP1 signaling pathways.97 Furthermore, inhibition of PKM2 by resveratrol induces endoplasmic reticulum stress in tumor cells, promotes mitochondrial division, and leads to apoptosis in tumor cells. Accordingly, overexpression of PKM2 has been shown to relieve the effect of resveratrol.98
Targeting of PDK1 and LDH
Dichloroacetate has been shown to inhibit PDK1 and activate mitochondrial oxidative phosphorylation in GC cells, thereby resulting in an accumulation of mitochondrial ROS and apoptosis.99 Meanwhile, oxamate is an LDH inhibitor, which inhibits mTOR activity, increases levels of ROS, and induces apoptosis in GC cells.100
Targeting of HIF-1α
YC-1 is an inhibitor of HIF-1α, which reduces glycolysis and increases oxidative phosphorylation and intracellular ROS levels in GC cells. Under hypoxic conditions in vitro, treatment of GC cells with YC-1 has led to apoptosis.101 Similarly, rosmarinic acid is another inhibitor of HIF-1α, which reduces glycolysis and induces apoptosis in GC cells.102
While none of these drugs are currently available for clinical use, they have the potential to facilitate in vitro and/or in vivo studies of glycolysis to elucidate mechanistic details.
Conclusion
It is clear that a complex relationship exists among the pathways responsible for the metabolism of lipids, amino acids, and glucose, although the components, interactions, and regulatory mechanisms associated with these fundamental pathways remain to be fully characterized. In addition, it has been demonstrated that mitochondria play an important role in metabolic reprogramming in GC, not only by regulating oxidative phosphorylation, but also by regulating the metabolism of lipids and amino acids (Figure 2). An important observation regarding the studies conducted to date involves differences in the results of in vitro vs in vivo studies. For example, in vitro studies involving low-glucose media have exhibited higher degrees of malignancy and drug resistance than in vivo studies.103 Therefore, it is important for future studies to identify and optimize in vitro experimental conditions so that they are consistent with in vivo conditions. As a result, effects that are cell-specific and/or pathology-specific could be distinguished from those that represent fundamental aspects of glycolysis.
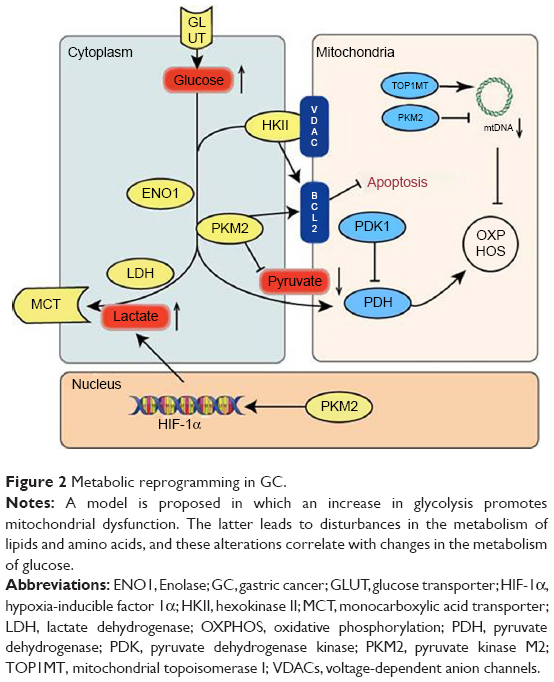 | Figure 2 Metabolic reprogramming in GC. |
We hypothesize that alterations in mitochondrial functions may initiate metabolic reprogramming that can occur in GC, despite this aspect being largely unexplored in studies of GC to date. Thus, we would advocate that future research of GC should include investigations of mitochondria and their functions in order to obtain a better understanding of the genesis and development of GC. In addition, the significance of changes observed in various metabolites remains to be clarified beyond the identification that these changes are manifestations of metabolic reprogramming. It is anticipated that further studies of these metabolites and their roles could facilitate the development of effective biomarkers for the diagnosis of GC, as well as for prognosis assessments and evaluation of treatment response.
Acknowledgments
We thank Professor Sun Liankun (Department of Pathophysiology, School of Basic Medical Sciences, Jilin University) for providing a mitochondria-related study method and Medjaden Bioscience Limited for editing and proofreading. This work was supported by grants from the Jilin Province Science and Technology Development Project (20180101169JC), the Jilin Provincial Health Department Project (2016Q023), the Jilin Provincial Development and Reform Commission Project (2016CO51-2), the National Natural Science Foundation of China (81772794, 81672948), the Jilin Provincial Research Foundation for the Development of Science and Technology Projects (20170623021TC, 20160414005 GH), the Jilin University Bethune Plan B Projects (2015222), and the 13th Five-Year Science and Technology Project of Jilin Provincial Education Department (JJKH20170834KJ, JJKH20170825KJ).
Disclosure
The authors report no conflicts of interest in this work.
References
Bray F, Ferlay J, Soerjomataram I, et al. GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. | ||
Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41(3):211–218. | ||
Sitarz R, Skierucha M, Mielko J, Offerhaus GJA, Maciejewski R, Polkowski WP. Gastric cancer: epidemiology, prevention, classification, and treatment. Cancer Manag Res. 2018;10:239–248. | ||
Luo B, Wang M, Hou N, et al. ATP-dependent Lon protease contributes to Helicobacter pylori-induced gastric carcinogenesis. Neoplasia. 2016;18(4):242–252. | ||
Shiroki T, Yokoyama M, Tanuma N, et al. Enhanced expression of the M2 isoform of pyruvate kinase is involved in gastric cancer development by regulating cancer-specific metabolism. Cancer Sci. 2017;108(5):931–940. | ||
Zhang BG, Hu L, Zang MD, et al. Helicobacter pylori CagA induces tumor suppressor gene hypermethylation by upregulating DNMT1 via AKT-NFκB pathway in gastric cancer development. Oncotarget. 2016;7(9):9788–9800. | ||
Lee DY, Jung DE, Yu SS, Lee YS, Choi BK, Lee YC. Regulation of SIRT3 signal related metabolic reprogramming in gastric cancer by Helicobacter pylori oncoprotein CagA. Oncotarget. 2017;8(45):78365–78378. | ||
Moyat M, Velin D. Use of VacA as a vaccine antigen. Toxins. 2016;8(6):181. | ||
Rigoli L, Caruso RA. Mitochondrial DNA alterations in the progression of gastric carcinomas: unexplored issues and future research needs. World J Gastroenterol. 2014;20(43):16159–16166. | ||
Rassow J, Meinecke M. Helicobacter pylori VacA: a new perspective on an invasive chloride channel. Microbes Infect. 2012;14(12):1026–1033. | ||
Guaragnella N, Giannattasio S, Moro L. Mitochondrial dysfunction in cancer chemoresistance. Biochem Pharmacol. 2014;92(1):62–72. | ||
Wu J, Hu L, Wu F, Zou L, He T. Poor prognosis of hexokinase 2 overexpression in solid tumors of digestive system: a meta-analysis. Oncotarget. 2017;8(19):32332–32344. | ||
Pedersen PL. Voltage dependent anion channels (VDACs): a brief introduction with a focus on the outer mitochondrial compartment’s roles together with hexokinase-2 in the “Warburg effect” in cancer. J Bioenerg Biomembr. 2008;40(3):123–126. | ||
Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002;277(9):7610–7618. | ||
Rho M, Kim J, Jee CD, et al. Expression of type 2 hexokinase and mitochondria-related genes in gastric carcinoma tissues and cell lines. Anticancer Res. 2007;27(1A):251–258. | ||
Israelsen WJ, vander Heiden MG. Pyruvate kinase: function, regulation and role in cancer. Semin Cell Dev Biol. 2015;43:43–51. | ||
Wang C, Jiang J, Ji J, et al. PKM2 promotes cell migration and inhibits autophagy by mediating PI3K/Akt activation and contributes to the malignant development of gastric cancer. Sci Rep. 2017;7(1):2886. | ||
Kitayama K, Yashiro M, Morisaki T, et al. Pyruvate kinase isozyme M2 and glutaminase might be promising molecular targets for the treatment of gastric cancer. Cancer Sci. 2017;108(12):2462–2469. | ||
Hamanaka RB, Chandel NS. Cell biology. Warburg effect and redox balance. Science. 2011;334(6060):1219–1220. | ||
Lee J, Kim HK, Han YM, Kim J. Pyruvate kinase isozyme type M2 (PKM2) interacts and cooperates with OCT-4 in regulating transcription. Int J Biochem Cell Biol. 2008;40(5):1043–1054. | ||
Yang W, Xia Y, Ji H, et al. Corrigendum: nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature. 2017;480(7674):142. | ||
Liang J, Cao R, Wang X, et al. Mitochondrial PKM2 regulates oxidative stress-induced apoptosis by stabilizing Bcl2. Cell Res. 2017;27(3):329–351. | ||
Wu H, Yang P, Hu W, et al. Overexpression of PKM2 promotes mitochondrial fusion through attenuated p53 stability. Oncotarget. 2016;7(47):78069–78082. | ||
Stacpoole PW. Therapeutic targeting of the pyruvate dehydrogenase Complex/Pyruvate dehydrogenase kinase (PDC/PDK) axis in cancer. J Natl Cancer Inst. 2017;109(11):djx071. | ||
Jeoung NH. Pyruvate dehydrogenase kinases: therapeutic targets for diabetes and cancers. Diabetes Metab J. 2015;39(3):188–197. | ||
Qian X, Xu W, Xu J, et al. Enolase 1 stimulates glycolysis to promote chemoresistance in gastric cancer. Oncotarget. 2017;8(29):47691–47708. | ||
Liu YQ, Huang ZG, Li GN, et al. Effects of α-enolase (ENO1) over-expression on malignant biological behaviors of AGS cells. Int J Clin Exp Med. 2015;8(1):231–239. | ||
Leal MF, Chung J, Calcagno DQ, et al. Differential proteomic analysis of noncardia gastric cancer from individuals of northern Brazil. PLoS One. 2012;7(7):e42255. | ||
Zhang TB, Zhao Y, Tong ZX, Guan YF. Inhibition of glucose-transporter 1 (GLUT-1) expression reversed Warburg effect in gastric cancer cell MKN45. Int J Clin Exp Med. 2015;8(2):2423–2428. | ||
Liu J, Wen D, Fang X, Wang X, Liu T, Zhu J. P38MAPK signaling enhances glycolysis through the up-regulation of the glucose transporter GLUT-4 in gastric cancer cells. Cell Physiol Biochem. 2015;36(1):155–165. | ||
Jiang W, Zhou F, Li N, Li Q, Wang L. FOXM1-LDHA signaling promoted gastric cancer glycolytic phenotype and progression. Int J Clin Exp Pathol. 2015;8(6):6756–6763. | ||
Zhang Y, Zhang X, Wang X, et al. Inhibition of LDH-A by lentivirus-mediated small interfering RNA suppresses intestinal-type gastric cancer tumorigenicity through the downregulation of OCT4. Cancer Lett. 2012;321(1):45–54. | ||
Jia D, Park J, Jung K, Levine H, Kaipparettu B. Elucidating the metabolic plasticity of cancer: mitochondrial reprogramming and hybrid metabolic states. Cells. 2018;7(3):21. | ||
Kamalidehghan B, Houshmand M, Ismail P, Panahi MS, Akbari MH. Delta mtDNA4977 is more common in non-tumoral cells from gastric cancer sample. Arch Med Res. 2006;37(6):730–735. | ||
Lee HC, Yin PH, Lin JC, et al. Mitochondrial genome instability and mtDNA depletion in human cancers. Ann NY Acad Sci. 2005;1042(1):109–122. | ||
Wang L, Chen X, Du Z, et al. Curcumin suppresses gastric tumor cell growth via ROS-mediated DNA polymerase γ depletion disrupting cellular bioenergetics. J Exp Clin Cancer Res. 2017;36(1):47. | ||
Dalla Rosa I, Zhang H, Khiati S, Wu X, Pommier Y. Transcription profiling suggests that mitochondrial topoisomerase IB acts as a topological barrier and regulator of mitochondrial DNA transcription. J Biol Chem. 2017;292(49):20162–20172. | ||
Sobek S, Boege F. DNA topoisomerases in mtDNA maintenance and ageing. Exp Gerontol. 2014;56:135–141. | ||
Wang H, Zhou R, Sun L, et al. TOP1MT deficiency promotes GC invasion and migration via the enhancements of LDHA expression and aerobic glycolysis. Endocr Relat Cancer. 2017;24(11):565–578. | ||
Hirschey MD, Shimazu T, Goetzman E, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464(7285):121–125. | ||
Finley LW, Carracedo A, Lee J, et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell. 2011;19(3):416–428. | ||
Cui Y, Qin L, Wu J, et al. SIRT3 enhances glycolysis and proliferation in SIRT3-expressing gastric cancer cells. PLoS One. 2015;10(6):e0129834. | ||
Wang L, Wang WY, Cao LP. SIRT3 inhibits cell proliferation in human gastric cancer through down-regulation of Notch-1. Int J Clin Exp Med. 2015;8(4):5263–5271. | ||
Huang KH, Hsu CC, Fang WL, et al. SIRT3 expression as a biomarker for better prognosis in gastric cancer. World J Surg. 2014;38(4):910–917. | ||
Yu J, Li J, Chen Y, et al. Snail enhances glycolysis in the epithelial-mesenchymal transition process by targeting FBP1 in gastric cancer. Cell Physiol Biochem. 2017;43(1):31–38. | ||
Okubo K, Uenosono Y, Arigami T, et al. Clinical significance of altering epithelial-mesenchymal transition in metastatic lymph nodes of gastric cancer. Gastric Cancer. 2017;20(5):802–810. | ||
Zhu CL, Huang Q, Liu CH, Lin XS, Xie F. Prognostic value of HIF-1α expression in patients with gastric cancer. Mol Biol Rep. 2013;40(11):6055–6062. | ||
Mao ZJ, Tang QJ, Zhang CA, et al. Anti-proliferation and anti-invasion effects of diosgenin on gastric cancer BGC-823 cells with HIF-1α shRNAs. Int J Mol Sci. 2012;13(5):6521–6533. | ||
Tabata S, Yamamoto M, Goto H, et al. Thymidine catabolism as a metabolic strategy for cancer survival. Cell Rep. 2017;19(7):1313–1321. | ||
Pinheiro C, Longatto-Filho A, Azevedo-Silva J, Casal M, Schmitt FC, Baltazar F. Role of monocarboxylate transporters in human cancers: state of the art. J Bioenerg Biomembr. 2012;44(1):127–139. | ||
Yan P, Li YH, Tang ZJ, Shu X, Liu X. High monocarboxylate transporter 4 protein expression in stromal cells predicts adverse survival in gastric cancer. Asian Pac J Cancer Prev. 2014;15(20):8923–8929. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Tilborghs S, Corthouts J, Verhoeven Y, et al. The role of nuclear factor-kappa B signaling in human cervical cancer. Crit Rev Oncol Hematol. 2017;120:141–150. | ||
Zhao M, Zhang Z. Glucose transporter regulation in cancer: a profile and the loops. Crit Rev Eukaryot Gene Expr. 2016;26(3):223–238. | ||
Liu K, Li F, Han H, et al. Parkin regulates the activity of pyruvate kinase M2. J Biol Chem. 2016;291(19):10307–10317. | ||
Hong M, Xia Y, Zhu Y, et al. TP53-induced glycolysis and apoptosis regulator protects from spontaneous apoptosis and predicts poor prognosis in chronic lymphocytic leukemia. Leuk Res. 2016;50:72–77. | ||
Yue X, Zhao Y, Xu Y, Zheng M, Feng Z, Hu W. Mutant p53 in cancer: accumulation, gain-of-function, and therapy. J Mol Biol. 2017;429(11):1595–1606. | ||
Zhang C, Liu J, Liang Y, et al. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013;4(1):2935. | ||
Kim SH, Choi SI, Won KY, Lim S-J. Distinctive interrelation of p53 with SCO2, COX, and TIGAR in human gastric cancer. Pathol Res Pract. 2016;212(10):904–910. | ||
Tahara T, Shibata T, Okamoto Y, et al. Mutation spectrum of TP53 gene predicts clinicopathological features and survival of gastric cancer. Oncotarget. 2016;7(27):42252–42260. | ||
Zhang M, Du X. Noncoding RNAs in gastric cancer: research progress and prospects. World J Gastroenterol. 2016;22(29):6610–6618. | ||
Fan C, Tang Y, Wang J, et al. Role of long non-coding RNAs in glucose metabolism in cancer. Mol Cancer. 2017;16(1):130. | ||
Zhang LF, Jiang S, Liu MF. MicroRNA regulation and analytical methods in cancer cell metabolism. Cell Mol Life Sci. 2017;74(16):2929–2941. | ||
Liu L, Wang Y, Bai R, Yang K, Tian Z. miR-186 inhibited aerobic glycolysis in gastric cancer via HIF-1α regulation. Oncogenesis. 2016;5:e224. | ||
Tang R, Yang C, Ma X, et al. MiR-let-7a inhibits cell proliferation, migration, and invasion by down-regulating PKM2 in gastric cancer. Oncotarget. 2016;7(5):5972–5984. | ||
Sugiyama T, Taniguchi K, Matsuhashi N, et al. MiR-133b inhibits growth of human gastric cancer cells by silencing pyruvate kinase muscle-splicer polypyrimidine tract-binding protein 1. Cancer Sci. 2016;107(12):1767–1775. | ||
Li LQ, Yang Y, Chen H, Zhang L, Pan D, Xie WJ. MicroRNA-181b inhibits glycolysis in gastric cancer cells via targeting hexokinase 2 gene. Cancer Biomark. 2016;17(1):75–81. | ||
Hong X, Xu Y, Qiu X, et al. MiR-448 promotes glycolytic metabolism of gastric cancer by downregulating KDM2B. Oncotarget. 2016;7(16):22092–22102. | ||
Ding X, Liu J, Liu T, Ma Z, Wen D, Zhu J. miR-148b inhibits glycolysis in gastric cancer through targeting SLC2A1. Cancer Med. 2017;6(6):1301–1310. | ||
Xu Y, Deng Y, Yan X, Zhou T. Targeting miR-375 in gastric cancer. Expert Opin Ther Targets. 2011;15(8):961–972. | ||
Zhang L, Lei J, Fang ZL, Xiong JP. MiR-128b is down-regulated in gastric cancer and negatively regulates tumour cell viability by targeting PDK1/Akt/NF-κB axis. J Biosci. 2016;41(1):77–85. | ||
Zhao Y, Liu Y, Lin L, et al. The lncRNA MACC1-AS1 promotes gastric cancer cell metabolic plasticity via AMPK/Lin28 mediated mRNA stability of MACC1. Mol Cancer. 2018;17(1):69. | ||
Liu J, Pan C, Guo L, et al. A new mechanism of trastuzumab resistance in gastric cancer: MACC1 promotes the Warburg effect via activation of the PI3K/Akt signaling pathway. J Hematol Oncol. 2016;9(1):76. | ||
Lin L, Huang H, Liao W, et al. MACC1 supports human gastric cancer growth under metabolic stress by enhancing the Warburg effect. Oncogene. 2015;34(21):2700–2710. | ||
Sun K, Hu P, Xu F. LINC00152/miR-139-5p regulates gastric cancer cell aerobic glycolysis by targeting PRKAA1. Biomed Pharmacother. 2018;97:1296–1302. | ||
Chen Z, Liu H, Yang H, Gao Y, Zhang G, Hu J. The long noncoding RNA, TINCR, functions as a competing endogenous RNA to regulate PDK1 expression by sponging miR-375 in gastric cancer. Onco Targets Ther. 2017;10:3353–3362. | ||
Tugnoli V, Mucci A, Schenetti L, et al. Ex vivo HR-MAS magnetic resonance spectroscopy of human gastric adenocarcinomas: a comparison with healthy gastric mucosa. Oncol Rep. 2006;16(3):543–553. | ||
Gu J, Hu X, Shao W, et al. Metabolomic analysis reveals altered metabolic pathways in a rat model of gastric carcinogenesis. Oncotarget. 2016;7(37):60053–60073. | ||
Sugano K, Sato K, Yao K. New diagnostic approaches for early detection of gastric cancer. Dig Dis. 2004;22(4):327–333. | ||
Yao K, Iwashita A, Nambu M, et al. Nature of white opaque substance in gastric epithelial neoplasia as visualized by magnifying endoscopy with narrow-band imaging. Dig Endosc. 2012;24(6):419–425. | ||
Enjoji M, Kohjima M, Ohtsu K, et al. Intracellular mechanisms underlying lipid accumulation (white opaque substance) in gastric epithelial neoplasms: a pilot study of expression profiles of lipid-metabolism-associated genes. J Gastroenterol Hepatol. 2016;31(4):776–781. | ||
Otto C, Kaemmerer U, Illert B, et al. Growth of human gastric cancer cells in nude mice is delayed by a ketogenic diet supplemented with omega-3 fatty acids and medium-chain triglycerides. BMC Cancer. 2008;8(1):122. | ||
Ichinoe M, Mikami T, Yoshida T, et al. High expression of L-type amino-acid transporter 1 (LAT1) in gastric carcinomas: comparison with non-cancerous lesions. Pathol Int. 2011;61(5):281–289. | ||
Wang G, Dai L, Luo L, et al. Non-essential amino acids attenuate apoptosis of gastric cancer cells induced by glucose starvation. Oncol Rep. 2014;32(1):332–340. | ||
Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13(5):589–598. | ||
Yuan M, McNae IW, Chen Y, et al. An allostatic mechanism for M2 pyruvate kinase as an amino-acid sensor. Biochem J. 2018;475(10):1821–1837. | ||
Feksa LR, Cornelio A, Dutra-Filho CS, Wyse AT, Wajner M, Wannmacher CM. The effects of the interactions between amino acids on pyruvate kinase activity from the brain cortex of young rats. Int J Dev Neurosci. 2005;23(6):509–514. | ||
Guo X, Zhang X, Wang T, Xian S, Lu Y. 3-Bromopyruvate and sodium citrate induce apoptosis in human gastric cancer cell line MGC-803 by inhibiting glycolysis and promoting mitochondria-regulated apoptosis pathway. Biochem Biophys Res Commun. 2016;475(1):37–43. | ||
Wang TA, Zhang XD, Guo XY, Xian SL, Lu YF. 3-Bromopyruvate and sodium citrate target glycolysis, suppress survivin, and induce mitochondrial-mediated apoptosis in gastric cancer cells and inhibit gastric orthotopic transplantation tumor growth. Oncol Rep. 2016;35(3):1287–1296. | ||
Wang TA, Xian SL, Guo XY, Zhang XD, Lu YF. Combined 18F-FDG PET/CT imaging and a gastric orthotopic xenograft model in nude mice are used to evaluate the efficacy of glycolysis-targeted therapy. Oncol Rep. 2018;39(1):271–279. | ||
Xu Y, Wang Q, Zhang L, Zheng M. 2-Deoxy-D-glucose enhances TRAIL-induced apoptosis in human gastric cancer cells through downregulating JNK-mediated cytoprotective autophagy. Cancer Chemother Pharmacol. 2018;81(3):555–564. | ||
Chen F, Zhuang M, Zhong C, et al. Baicalein reverses hypoxia-induced 5-FU resistance in gastric cancer AGS cells through suppression of glycolysis and the PTEN/Akt/HIF-1α signaling pathway. Oncol Rep. 2015;33(1):457–463. | ||
Wu J, Zhang X, Wang Y, et al. Licochalcone A suppresses hexokinase 2-mediated tumor glycolysis in gastric cancer via downregulation of the Akt signaling pathway. Oncol Rep. 2018;39(3):1181–1190. | ||
Lu J, Chen M, Gao S, Yuan J, Zhu Z, Zou X. LY294002 inhibits the Warburg effect in gastric cancer cells by downregulating pyruvate kinase M2. Oncol Lett. 2018;15(4):4358–4364. | ||
Zhang B, Yang Y, Shi X, et al. Proton pump inhibitor pantoprazole abrogates adriamycin-resistant gastric cancer cell invasiveness via suppression of Akt/GSK-β/β-catenin signaling and epithelial-mesenchymal transition. Cancer Lett. 2015;356(2 Pt B):704–712. | ||
Huang S, Chen M, Ding X, Zhang X, Zou X. Proton pump inhibitor selectively suppresses proliferation and restores the chemosensitivity of gastric cancer cells by inhibiting STAT3 signaling pathway. Int Immunopharmacol. 2013;17(3):585–592. | ||
Chen M, Huang SL, Zhang XQ, et al. Reversal effects of pantoprazole on multidrug resistance in human gastric adenocarcinoma cells by down-regulating the V-ATPases/mTOR/HIF-1α/P-gp and MRP1 signaling pathway in vitro and in vivo. J Cell Biochem. 2012;113(7):2474–2487. | ||
Wu H, Wang Y, Wu C, Yang P, Li H, Li Z. Resveratrol induces cancer cell apoptosis through MiR-326/PKM2-mediated ER stress and mitochondrial fission. J Agric Food Chem. 2016;64(49):9356–9367. | ||
Xuan Y, Hur H, Ham IH, et al. Dichloroacetate attenuates hypoxia-induced resistance to 5-fluorouracil in gastric cancer through the regulation of glucose metabolism. Exp Cell Res. 2014;321(2):219–230. | ||
Zhao Z, Han F, Yang S, Wu J, Zhan W. Oxamate-mediated inhibition of lactate dehydrogenase induces protective autophagy in gastric cancer cells: involvement of the Akt-mTOR signaling pathway. Cancer Lett. 2015;358(1):17–26. | ||
Wakiyama K, Kitajima Y, Tanaka T, et al. Low-dose YC-1 combined with glucose and insulin selectively induces apoptosis in hypoxic gastric carcinoma cells by inhibiting anaerobic glycolysis. Sci Rep. 2017;7(1):12653. | ||
Han S, Yang S, Cai Z, et al. Anti-Warburg effect of rosmarinic acid via miR-155 in gastric cancer cells. Drug Des Devel Ther. 2015;9:2695–2703. | ||
Bhattacharya B, Low SH, Soh C, et al. Increased drug resistance is associated with reduced glucose levels and an enhanced glycolysis phenotype. Br J Pharmacol. 2014;171(13):3255–3267. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.